

Abstract
Mammalian Timeless is a multifunctional protein that performs essential roles in the circadian clock, chromosome cohesion, DNA replication fork protection, and DNA replication/DNA damage checkpoint pathways. The human Timeless exists in a tight complex with a smaller protein called Tipin (Timeless-interacting protein). Here we investigated the mechanism by which the Timeless-Tipin complex functions as a mediator in the ATR-Chk1 DNA damage checkpoint pathway. We find that the Timeless-Tipin complex specifically mediates Chk1 phosphorylation by ATR in response to DNA damage and replication stress through interaction of Tipin with the 34-kDa subunit of replication protein A (RPA). The Tipin-RPA interaction stabilizes Timeless-Tipin and Tipin-Claspin complexes on RPA-coated ssDNA and in doing so promotes Claspin-mediated phosphorylation of Chk1 by ATR. Our results therefore indicate that RPA-covered ssDNA not only supports recruitment and activation of ATR but also, through Tipin and Claspin, it plays an important role in the action of ATR on its critical downstream target Chk1.
Keywords: Cell Cycle, Checkpoint Control, DNA Damage, DNA-Protein Interaction, Signal Transduction
Introduction
DNA damage and replication checkpoints are controlled by cellular signal transduction pathways that recognize and respond to alterations in DNA structure by halting or delaying cell cycle progression to allow sufficient time for DNA repair and the completion of DNA replication (1). The phosphoinositide 3-kinase related kinases ATM2 and ATR play essential roles in this response by phosphorylating and activating a number of proteins that function to inhibit cell cycle progression and promote DNA repair, including p53, Chk1, and Chk2 (1). Importantly, the disruption of genes involved in the DNA damage checkpoint response is associated with a number of human diseases, including cancer (1, 2).
Although both ATM and ATR may become activated in response to different forms of DNA damage, the mechanism and kinetics of activation are thought to be related to the types of DNA lesions that are induced by DNA-damaging agents. Whereas ATM is primarily activated in response to overt double-strand breaks in DNA induced by ionizing radiation (IR) and related chemical agents (3), ATR is stimulated under a wider array of genome destabilizing conditions, such as during replication fork stalling, nucleotide excision repair, and double-strand break processing and at deprotected telomeres (1, 4, 5).
Because of the variety of genotoxic stressors that activate ATR, it has been suggested that a common DNA structural intermediate may be involved in the initiation or maintenance of the ATR signal transduction pathway (5–7). Consistent with this hypothesis, the uncoupling of DNA helicase and polymerase activities at replication forks, resection of DNA ends at double-strand breaks, and removal of bulky DNA adducts by nucleotide excision repair all generate single-stranded DNA (ssDNA) that may become bound by RPA, the major ssDNA-binding protein in eukaryotes (8, 9). Through a specific interaction of the 70-kDa subunit of RPA (RPA1) with the ATR-interacting protein ATRIP (7, 10, 11), a constitutive binding partner of ATR (12), RPA is thought to promote the stable association of ATR-ATRIP with sites of DNA damage and replication stress. However, full stimulation of ATR kinase activity requires several additional factors, including the Rad17-replication factor C complex, which loads the PCNA-like 9-1-1 clamp (Rad9-Hus1-Rad1) onto primer-template junctions that are also present at sites of DNA damage and at stalled replication forks (1). Through an interaction with the C-terminal tail of Rad9, the ATR-activating protein TopBP1 (13) may then be brought into proximity of ATR where it can stimulate ATR kinase activity (14).
Although a large number of proteins are potentially phosphorylated by ATR in response to DNA damage (15, 16), a primary checkpoint substrate is the kinase Chk1 (5, 17, 18). Phosphorylation of two residues (Ser317 and Ser345) in a C-terminal regulatory region of Chk1 activates its kinase activity (17, 18) and changes its subnuclear localization (19), enabling the phosphorylation of proteins important for DNA repair and cell cycle progression, such as the Cdc25 family of phosphatases, which directly regulate cyclin-dependent kinase activity to impact cell cycle progression (20). Chk1 therefore plays a critical role in the DNA damage checkpoint response and in promoting genome stability. Its relevance to human health and disease is further highlighted by the recent development of Chk1 inhibitors for clinical use in cancer therapies in combination with traditional chemotherapeutics that induce DNA damage (21).
The mechanism of how ATR contacts and phosphorylates Chk1 during the DNA damage response is unclear, although data from a variety of model systems have demonstrated a requirement for the Claspin protein as a mediator of this signaling event (22–26). Initially discovered as a Chk1-interacting protein in Xenopus egg extracts (26), Claspin has been shown in human cells, Xenopus egg extracts, and reconstituted ATR kinase reactions to specifically stimulate the phosphorylation of Chk1 but not the phosphorylation of other ATR substrate proteins (23, 24, 27). Importantly, like ATR, Claspin is required for Chk1 phosphorylation and DNA damage checkpoint signaling in response to agents that induce a variety of forms of DNA damage (24, 28, 29), indicating that a common mechanism exists for its recruitment and function in ATR-Chk1 signaling at sites of DNA damage and replication stress.
We initially reported that the protein Timeless also mediated Chk1 phosphorylation and DNA damage checkpoint responses in human cells exposed to UV irradiation or hydroxyurea (HU), a compound that depletes deoxynucleotide precursors and causes DNA polymerases to stall (30). In a series of recent reports, we and others confirmed that both Timeless and its binding partner Tipin (Timeless-interacting protein) (31) contribute to intra-S and G2/M checkpoint responses to UV, HU, and other agents, including IR (32–36). Although these different agents induce a variety of structural changes to DNA, including stalled replication forks, nucleotide excision repair gaps, and double-strand breaks, the observation that the Timeless-Tipin complex contributes to proper checkpoint responses to all of these types of damage indicates that a common mechanism may be involved in Timeless-Tipin function and/or recruitment to sites of DNA damage.
Our previous observation that the Timeless-Tipin complex binds to RPA (32) indicated a possible mechanism for Timeless-Tipin recruitment and function to promote ATR signaling. Here we show that the Timeless-Tipin complex specifically mediates Chk1 phosphorylation by ATR in response to DNA damage and replication stress through an interaction of Tipin with the 34-kDa subunit of RPA (RPA2), which stabilizes both the Timeless-Tipin complex and Claspin on RPA-coated ssDNA. These results therefore indicate that RPA function in ATR checkpoint signaling extends beyond recruitment and activation of ATR and also includes, through Timeless-Tipin and Claspin, a possible mechanism for facilitating Chk1 phosphorylation by ATR at sites of DNA damage.
EXPERIMENTAL PROCEDURES
Cell Lines
HeLa, HEK293T, and FlpTM-In T-RExTM-293 cells (Invitrogen) were maintained in Dulbecco's minimal essential medium supplemented with 10% fetal bovine serum and penicillin-streptomycin. Sf21 and Hi5 cells (Invitrogen) were grown in Grace's insect medium (Invitrogen) supplemented with 10% fetal bovine serum. Derivation of stable Flp-InTM T-RExTM-293 cell lines expressing FLAG-tagged Tipin was performed according to the manufacturer's protocols (Invitrogen).
Immunoblotting
Standard immunoblotting procedures were used to detect proteins in cell lysates and in pull-down assays with recombinant proteins. Antibodies against Timeless and Tipin were generously provided by Anthony Gotter and Hisao Masai (34, 36). A peptide corresponding to an N-terminal fragment of Tipin (CSPERQDGEGTEPDEESG) synthesized by the University of North Carolina Protein Sequence and Peptide Synthesis Facility was conjugated to keyhole limpet hemocyanin by Covance and used to generate an additional rabbit anti-Tipin antibody used in this work. ATR (N-19), Claspin (H-300), Chk1 (G-4), Chk2 (H-300), RPA1 (B-6), and XPB (S-19) antibodies were purchased from Santa Cruz. RPA1 and RPA2 antibodies were obtained from Calbiochem. Phospho-RPA2 (Ser33) and phospho-MCM2 (Ser108) antibodies were from Bethyl Laboratories. Anti-FLAG and anti-His antibodies were from Sigma and Abgent, respectively. ORC2 and MEK2 antibodies were purchased from BD Biosciences. ATRIP antibody was obtained from Zymed Laboratories Inc.. Phospho-Chk1 (Ser345) antibody was from Cell Signaling Technology.
Plasmids
Plasmids used in this work can be obtained from Addgene. pcDNA3-FLAG-Tipin, pcDNA4-FLAG-Timeless, and pcDNA3-FLAG-RPA2 were described previously (30, 32, 37). Vectors encoding FLAG-Tipin mutants (E185A, E190A, and L195A) were generated by site-directed mutagenesis and were cloned in pcDNA3. To generate siRNA-resistant forms of Tipin, the region of Tipin targeted by Dharmacon siRNA catalog number J-020843 (5′-AGAGGACTTCCAGCCTTA-3′) was changed to 5′-AGGGGCCTGCCGGCGTTG-3′ by site-directed mutagenesis in the pcDNA3 vector. FLAG-Tipin (WT and L195A) sequences were also subcloned into the BamHI and XhoI sites of pcDNA5/FRT/TO.
To generate pFastBac1-FLAG-Timeless, pFastBacHTb-His-FLAG-Timeless (32) was digested with XbaI/KpnI and the insert ligated into the identical sites in pFastBac1. pcDNA3-FLAG-Tipin (L195A) was cut with BamHI and XhoI and inserted into the identical sites of pFastBacHTb to generate pFastBacHTb-FLAG-Tipin (L195A). To generate pFastBac1-His-Tipin (L195A), pcDNA3-FLAG-Tipin (L195A) was used as template in a PCR with previously described primers (32) and cloned into the XbaI and KpnI sites of pFastBac1.
Tipin (WT and L195A) sequences were also cloned into the NdeI and XhoI sites of pET21b by PCR using pcDNA3-FLAG-Tipin (WT or L195A) as a template and the PCR primers 5′-GAAGCTAGAGCACTCGAGCACTGA-3′ and 5′-TCAGTGCTCGAGTGCTCTAGCTTC-3′.
Expression and Purification of Recombinant Proteins
RPA and aRPA were purified from Escherichia coli as described previously (38, 39). Baculoviruses used to express and purify proteins from insect cells (Sf21 or Hi5 cells; Invitrogen) were either previously reported (30, 32, 37, 40) or were prepared with the Bac-to-Bac baculovirus expression system (Invitrogen) using protocols recommended by the manufacturer. His-FLAG-Tipin and the FLAG-Timeless/His-Tipin heterodimer were purified from baculovirus-infected Sf21 cells as previously described (32). His-tagged forms of Tipin (WT and L195A) were also purified from E. coli (BL21-Codon Plus (DE3)-RIPL) using nickel-nitrilotriacetic acid-agarose (Qiagen).
Transfection
DNA and siRNA transfections in HeLa cells and Flp-InTM T-RExTM-293 cells employed Lipofectamine 2000 or Lipofectamine RNAiMAX (Invitrogen) using protocols provided by the manufacturer. HEK293T cells were transfected with calcium phosphate. The Timeless and Tipin siRNAs were previously described (30, 32), and the current work utilized an additional siRNA targeting Tipin (Dharmacon catalog number J-020843) and the nontargeting control siRNA 2 (Dharmacon catalog number D-001210-02).
Immobilized DNA Pulldown Assays
An 80-mer biotinylated ssDNA (37) was immobilized on streptavidin-coupled Dynabeads (M-280) as recommended by the manufacturer (Dynal), typically at 1 pmol of DNA/μl of magnetic beads. Standard reactions involved incubation of the indicated proteins in 50 μl of binding buffer (10 mm Tris, pH 7.4, 100 mm NaCl, 10% glycerol, 10 μg/ml bovine serum albumin, 0.01% Nonidet P-40) for 30 min at room temperature before collecting the beads on a magnet and washing three times with 200 μl of binding buffer. Preincubation of the ssDNA with RPA was for 20–30 min before washing and addition of the other indicated proteins. The bound proteins were eluted and boiled in 1× SDS-PAGE sample buffer (50 mm Tris, pH 6.8, 100 mm dithiothreitol, 1% SDS, 5% glycerol, 0.005% bromphenol blue), separated by SDS-PAGE, and analyzed by immunoblotting.
Immunoprecipitation
Immunoprecipitations were performed with anti-FLAG-agarose (Sigma). For immunoprecipitation reactions employing purified proteins, the indicated proteins were incubated in binding buffer containing anti-FLAG-agarose for at least 5 h and then washed three times with 500 μl of binding buffer. Bound proteins were eluted in either binding buffer containing 200 μg/ml FLAG peptide (Sigma) or 2× SDS-PAGE sample buffer. Immunoprecipitations from cell extracts involved incubation of cell lysates with anti-FLAG-agarose overnight, washes with Tris-buffered saline, and elution with FLAG peptide or 2× SDS-PAGE sample buffer.
Subcellular Fractionation
Subcellular fractionation of mammalian cells to enrich for chromatin-bound proteins was performed essentially as described (41), with the addition of 10 mm NaF, 1 mm Na3VO4, and a 1:200 dilution (v/v) of protease inhibitor mixture (Sigma) to all buffers. Cytosolic and nuclear extracts were also prepared from human cell lines, essentially as described (42).
RESULTS
Timeless-Tipin Mediates Phosphorylation of Chk1 but Not Other ATR Substrates
Although we and others previously reported a requirement for the Timeless-Tipin complex for phosphorylation of the checkpoint kinase Chk1 by ATR in response to UV irradiation and other agents that induce replication fork stalling (30, 32–36, 43), it was not determined whether Timeless-Tipin was required for phosphorylation of other ATR substrates. We therefore used RNA interference to reduce Timeless-Tipin protein levels in HeLa cells and then examined the phosphorylation status of the ATR targets Chk1, Claspin, RPA2, and MCM2, because these proteins undergo rapid, ATR-dependent phosphorylation after HU treatment and are bona fide components of the intra-S phase checkpoint (17, 18, 28, 29, 44–46). As reported previously (30, 32, 34, 36), Chk1 displayed significantly less phosphorylation on Ser345 in HU-treated cells depleted of Timeless-Tipin than in cells transfected with a nontargeting control siRNA (Fig. 1A). Importantly, the observation that total Chk1 protein levels remained unchanged under these conditions showed that this reduced Chk1 phosphorylation was not due to altered Chk1 stability, because phosphorylation of Chk1 on this residue can trigger its proteosomal degradation (47, 48). We also noticed that Claspin failed to undergo a characteristic ATR-dependent mobility shift after HU treatment in cells depleted of Timeless-Tipin. Because Claspin fails to stably associate with replication forks in the absence of Timeless-Tipin (33, 34, 43), these results indicate that abnormal regulation of Claspin in cells depleted of Timeless-Tipin is correlated with defects in Chk1 phosphorylation by ATR in response to replication stress.
FIGURE 1.
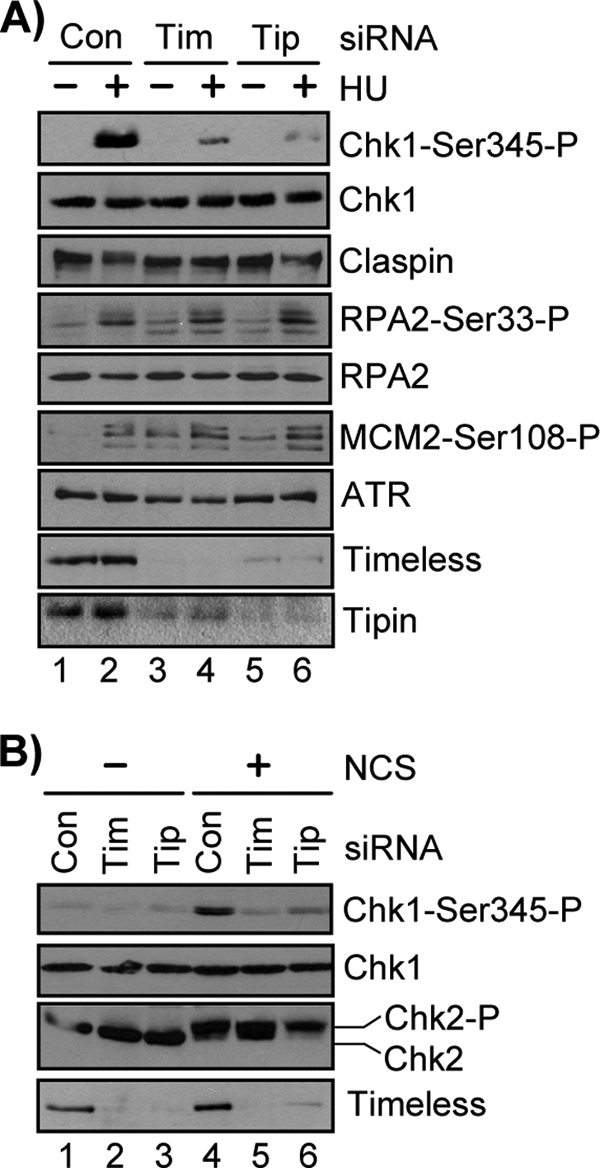
Timeless and Tipin are required to specifically mediate Chk1 phosphorylation in response to DNA damage and replication stress. A, 48 h after transfection of HeLa cells with either Control (Con), Timeless (Tim), or Tipin (Tip) siRNA, the cells were treated with 1 mm HU for 30 min. The lysates were prepared, separated by SDS-PAGE, and analyzed by immunoblotting with antibodies against the indicated proteins. B, cells were transfected and analyzed as in A, but cells were instead treated with 100 ng/ml NCS for 1 h.
In contrast, no significant defect was observed on the phosphorylation status of Ser33 of RPA2, a site that is rapidly phosphorylated by ATR after HU treatment and that is important in regulating RPA function in the intra-S phase checkpoint response (44, 45). We also observed that control siRNA-transfected and Timeless-Tipin siRNA-transfected cells exhibited similar HU-induced phosphorylation of MCM2 on Ser108, a known target of ATR (46, 49) and component of the replicative MCM helicase that functions in DNA replication and the intra-S phase checkpoint response (46, 50). Consistent with a recent report indicating a role for Timeless-Tipin in coupling DNA unwinding to DNA synthesis (51), knockdown of Timeless and Tipin led to a modest increase in the phosphorylation of RPA2-Ser33 and MCM2-Ser108 compared with cells transfected with a control siRNA (Fig. 1A). Based on the results with HU treatment, however, we conclude that Timeless-Tipin activity in ATR-dependent checkpoint signaling is required for the Claspin-Chk1 pathway but not for ATR to phosphorylate other checkpoint substrates during the response to replication stress. Because a similar phenotype has been reported for Claspin function in ATR signaling after DNA damage or replication stress (23, 24, 27) and because Claspin fails to stably associate with replication forks in the absence of Timeless-Tipin (33, 34, 43), these results indicate that Timeless, Tipin, and Claspin function together in a common pathway to mediate Chk1 phosphorylation by ATR.
Although Timeless, Tipin, and Claspin have well recognized roles in mediating Chk1 phosphorylation in response to agents that inhibit the progression of DNA polymerases, genetic studies have indicated that these genes also contribute to proper G2/M and intra-S checkpoint responses in human cells exposed to IR (28, 35), suggesting that these proteins function in mediating ATR-dependent checkpoint responses to DNA double-strand breaks. Although Claspin has been shown to be required for Chk1 phosphorylation after exposure to IR (24, 28), it was not clear whether the requirement for Timeless-Tipin in IR-induced checkpoint activation was related to a requirement for Chk1 phosphorylation. We therefore treated Timeless-Tipin-depleted HeLa cells with neocarzinostatin, an IR-mimetic that generates double-strand breaks in DNA. As shown in Fig. 1B, NCS treatment in cells depleted of Timeless-Tipin did not induce Chk1 phosphorylation to the extent observed in control cells. Importantly, no defect was observed in Chk2 phosphorylation, as visualized by a characteristic mobility shift on SDS-PAGE, indicating normal activation of ATM-Chk2 signaling in NCS-treated cells lacking Timeless-Tipin. Based on these results, we conclude that like ATR and Claspin, the Timeless-Tipin complex is required for Chk1 phosphorylation in human cells exposed to a variety of different types of DNA-damaging agents.
RPA Stabilizes Timeless, Tipin, and Claspin on ssDNA
The requirement for Timeless, Tipin, and Claspin in ATR-Chk1 signaling in response to different forms of DNA damage indicates that a common DNA intermediate may be involved in regulating their activities at sites of DNA damage and stalled replication forks. Similarly, a primary mode for recruitment and activation of the ATR kinase involves the generation of ssDNA during DNA damage processing and replication fork stalling. This ssDNA is expected to rapidly become bound by RPA, the major eukaryotic ssDNA-binding protein that coordinates a variety of DNA metabolic processes (8, 9). Through a direct interaction of the constitutively bound ATR-interacting protein ATRIP with the largest subunit of RPA, the ATR kinase is thought to be recruited to sites of DNA damage so that it can phosphorylate its checkpoint substrates (7, 10, 11). Because we recently reported that Tipin also directly binds to RPA (32), we considered that the Tipin-RPA interaction may be important for the functions of Timeless, Tipin, and Claspin at sites of DNA damage and replication stress.
To characterize the interactions of Timeless, Tipin, and Claspin with RPA and ssDNA, we purified recombinant forms of these factors using either bacterial or insect expression systems (Fig. 2A) and tested their direct interactions with one another and with ssDNA. To examine protein binding to DNA, we immobilized a biotinylated 80-mer ssDNA on streptavidin-coupled magnetic beads and then incubated the DNA with various combinations of the indicated proteins, as has been done previously to study the interactions of RPA and ATRIP on ssDNA (7, 37). The purified Timeless-Tipin complex did not stably associate with the 80-mer ssDNA (Fig. 2B, lane 3), but preincubation of the immobilized ssDNA with RPA imparted stable binding of Timeless-Tipin (Fig. 2B, lanes 4 and 5). Consistent with our previous report that Tipin mediated the association of the Timeless-Tipin complex with RPA in solution (32), Timeless was not required for Tipin association with RPA on ssDNA, because recombinant Tipin alone was capable of binding to RPA-covered ssDNA but not to ssDNA lacking RPA (Fig. 2C, lanes 4–7).
FIGURE 2.

Interaction between RPA2 and Tipin stabilizes Timeless-Tipin and Claspin on RPA-coated ssDNA. A, Coomassie Blue-stained gel of purified proteins. RPA, aRPA, and Tipin-His were purified from E. coli, and His-FLAG-Tipin (HF-Tipin), His-FLAG-Timeless/His-Tipin (HF-Tim/His-Tip), and His-FLAG-Claspin (HF-Claspin) were purified from baculovirus-infected insect cells. Note that the gel displays only the 70-kDa (RPA1) and 34-kDa (RPA2/RPA4) subunits of RPA and aRPA, because the 17-kDa (RPA3) subunit was electrophoresed off of the gel. Lane 1 contains a molecular weight ladder, where numbers indicate molecular mass in kDa. B, His-FLAG-Timeless/His-Tipin complex (Tim/Tip; 10 pmol) was incubated with 5 pmol of immobilized 80-mer ssDNA preincubated for 30 min with 0, 2.5, or 5 pmol RPA in binding buffer. Input represents 5 pmol of HF-Timeless/His-Tipin complex and 5 pmol RPA. C, ssDNA (1 pmol) was preincubated with 0, 0.2, 0.5, or 1.25 pmol of RPA or aRPA in 50 μl of binding buffer before washing and addition of 20 pmol of Tipin. Input shows 0.5 pmol of RPA or aRPA and 1 pmol of Tipin. D, His-FLAG-tagged Tipin from baculovirus-infected insect cells was immobilized on anti-FLAG-agarose and then incubated with 1 μg of either RPA or aRPA overnight at 4 °C in 100 μl of binding buffer. Input represents 5% of the binding reactions. E, Tipin-His purified from E. coli (1 μg) was incubated with anti-FLAG resin alone or resin containing His-FLAG-tagged Claspin from baculovirus-infected insect cells. The reactions were in 100 μl of binding buffer overnight at 4 °C. Input shows 5% of the indicated reactions. Heavy chain of anti-FLAG IgG is displayed as a loading control. F, immobilized ssDNA (5 pmol) was incubated with RPA (10 pmol), His-FLAG-Tipin (10 pmol), and/or His-FLAG-Claspin (10 pmol), as indicated, and bound proteins were analyzed as in B and C. IP, immunoprecipitation.
Many proteins that interact with RPA make contact with more than one subunit of the RPA heterotrimer (8), and a recent yeast-two hybrid analysis identified an interaction between Tipin and the 34-kDa RPA2 subunit of RPA (36). We therefore sought to determine the importance of the RPA2 subunit for interaction with Tipin by using a novel, alternative form of RPA (aRPA) that contains the similarly sized RPA4 protein in place of RPA2 (39, 52, 53). Although RPA4-containing aRPA did not support SV40 DNA replication in vitro (39) or cell cycle progression in human cells (53), we have recently found in reconstitution assays with purified factors that aRPA supports nucleotide excision repair (54) and the recruitment and activation of ATR-ATRIP by ssDNA,3 suggesting that aRPA may function in general genome maintenance activities but not DNA replication. Consistent with a recent report that RPA and aRPA show similar binding affinities toward ssDNA (39), we found that similar amounts of the shared RPA1 subunit of RPA and aRPA were retained on the immobilized ssDNA (Fig. 2C). However, aRPA failed to promote the association of Tipin with the immobilized ssDNA (Fig. 2C, lanes 8–11). When we incubated RPA or aRPA with a FLAG-tagged form of Tipin immobilized anti-FLAG-agarose, very little aRPA bound to Tipin (Fig. 2D). We conclude that within the context of the RPA heterotrimeric complex, the RPA2 subunit of RPA plays a primary role in its stable association with Tipin.
Because the Timeless-Tipin complex has been shown to co-immunoprecipitate with Claspin in human cell extracts and to be required for the stable association of Claspin with chromatin containing stalled replication forks in both human cells and Xenopus egg extracts (33, 34, 43), we next sought to investigate the interactions of Claspin with Tipin, RPA, and ssDNA. Using the purified proteins described above, we observed that Tipin directly bound to a FLAG-tagged form of Claspin immobilized on anti-FLAG-agarose (Fig. 2E), indicating that Timeless is not necessary for Tipin to bind to Claspin. We then examined the association of Claspin with the immobilized 80-mer ssDNA and found that Claspin can bind weakly to ssDNA (Fig. 2F), consistent with previous work (40). However, when the ssDNA was coated with RPA, Claspin failed to associate with the DNA. Also, we have never observed any direct interaction of Claspin with RPA in the absence of DNA (data not shown). Interestingly, when the RPA-coated ssDNA was first incubated with recombinant Tipin prior to addition of Claspin to the binding reaction, Claspin was now able to associate with the immobilized DNA (Fig. 2F, lane 3). These results show that Tipin is necessary and sufficient for Claspin association with RPA-coated ssDNA, a common intermediate in DNA damage processing and replication fork stalling. These data may therefore explain the requirement for Tipin in the association of Claspin with chromatin containing stalled replication forks in human cells and in Xenopus egg extracts (33, 34, 43).
Identification of a Mutant Form of Tipin That Does Not Bind RPA
It was previously noted that Tipin contains a region of amino acids that show significant similarity to a motif present in the nucleotide excision repair protein XPA and other repair proteins that is involved in contacting the RPA2 subunit of RPA (Fig. 3A) (31, 32, 55). To further investigate the importance of the RPA-Tipin interaction in DNA damage checkpoint signaling, we mutated three amino acids in Tipin that are identical in human XPA and then examined the ability of FLAG-tagged forms of the Tipin mutants to co-immunoprecipitate with RPA after expression in HEK293T cells. Whereas two mutations (E185A and E190A) did not interfere with co-precipitation of RPA, mutation of Leu195 to Ala (L195A) completely abrogated the RPA interaction (Fig. 3B). Using baculovirus-expressed FLAG-tagged Tipin-WT and Tipin-L195A immobilized on anti-FLAG resin, we observed that RPA was able to bind well to the wild type but not to the L195A mutant form of Tipin (Fig. 3C). Similarly, recombinant Tipin-L195A purified from either E. coli or baculovirus-infected insect cells failed to associate with immobilized ssDNA covered with RPA (data not shown). Importantly, although the L195A mutation altered Tipin mobility on SDS-PAGE, it did not affect the ability of Tipin to form complexes with Timeless when co-expressed in insect cells, but as expected, Timeless-Tipin complexes containing Tipin-L195A were unable to associate with RPA-coated ssDNA in vitro (Fig. 3D). Similarly, Tipin-WT and Tipin-L195A bound equivalently to FLAG-Claspin immobilized on anti-FLAG-agarose (Fig. 3E), but the Tipin-L195A mutant did not promote Claspin association with immobilized ssDNA coated with RPA (Fig. 3F). We conclude that the L195A mutation in Tipin specifically affects its ability to interact with RPA but not to either Timeless or Claspin.
FIGURE 3.

Identification and characterization of a Tipin RPA-binding mutant. A, alignment of XPA and Tipin amino acid sequences. Black shading shows amino acid identity, and gray shading highlight indicates similarity. B, FLAG-tagged forms of Tipin were transiently expressed in HEK293T cells, immunoprecipitated with anti-FLAG-agarose, and analyzed by SDS-PAGE and immunoblotting with antibodies against FLAG, RPA1, and RPA2. Note that mutation of these amino acids alters Tipin mobility on SDS-PAGE. C, His-FLAG-tagged Tipin (HF-Tipin; WT and L195A) from baculovirus-infected insect cells was immobilized on anti-FLAG-agarose and then incubated with RPA. Resin was washed, and bound proteins were analyzed by SDS-PAGE and immunoblotting. D, FLAG-Timeless/His-Tipin (Tim/Tip) complexes prepared by baculoviral co-infection and anti-FLAG-agarose purification were incubated with immobilized ssDNA containing saturating amounts of RPA. Bound proteins were analyzed by SDS-PAGE and immunoblotting. Input represents 50% of the Timeless-Tipin complex used in the binding reaction. E, His-FLAG-tagged Claspin (HF-Claspin) from baculovirus-infected insect cells and immobilized on anti-FLAG resin (lanes 1–3) was incubated overnight at 4 °C with 1.5 μg of either Tipin-His-WT or Tipin-His-L195A purified from E. coli in 100 μl of binding buffer. Anti-FLAG resin lacking Claspin was used as a negative control (lanes 4–5). Input represents 5% of the Tipin used in the binding reaction. F, immobilized 80-mer ssDNA lacking or containing saturating amounts of RPA was incubated in reactions with HF-Claspin alone or together with HF-Tipin-WT or HF-Tipin-L195A. The beads were washed, and bound proteins were analyzed by SDS-PAGE and immunoblotting. IP, immunoprecipitation.
Expression and Nuclear Localization of Timeless-Tipin
We next wished to test whether the RPA-Tipin interaction was required for Chk1 phosphorylation by ATR in genotoxin-treated human cells. Our initial experiments using transient transfection of plasmid vectors expressing siRNA-resistant forms of Tipin and siRNAs targeting endogenous Tipin were inconclusive. Because siRNA-mediated knockdown of either Timeless or Tipin affects the stability and nuclear localization of the corresponding binding partner (32, 34–36), we first examined the ability of FLAG-tagged forms of Timeless and Tipin to form complexes with the endogenous binding partner in human cells. Whereas FLAG-Timeless readily co-immunoprecipitated ∼20–25% of endogenous Tipin in transiently transfected HEK293T cells and led to a slight (2–3-fold) increase in total Tipin protein levels in the cells (Fig. 4A, left panel), very little endogenous Timeless (<1%) co-immunoprecipitated with the ectopically expressed FLAG-Tipin (Fig. 4A, right panel). These results suggested to us that Tipin protein stability or heterodimeric complex formation with Timeless may be dependent upon the presence of sufficient levels of free Timeless protein in the cell, although we cannot rule out the possibility that some other property of the ectopically expressed Tipin interferes with its ability to form complexes with Timeless.
FIGURE 4.

Characterization of Tipin protein expression and nuclear localization in human cells. A, an empty vector (Vec) and vectors encoding FLAG-Timeless (F-Tim; left panel) or FLAG-Tipin (F-Tip; right panel) were transiently transfected into HEK293T cells. The lysates were prepared and then immunoprecipitated (IP) with anti-FLAG resin. Input lanes show 5% of the lysate used for immunoprecipitation. B, HEK293T cells fractionated to yield a hypotonic cytosolic (Cyto) extract and a high salt nuclear (Nuc) fraction were separated by SDS-PAGE and immunoblotted with antibodies against the indicated proteins. C, HEK293T cells were transfected with an empty vector (lanes 1 and 7), vectors expressing FLAG-Tipin and increasing amounts of FLAG-Timeless (lanes 2–5 and 8–11), or a vector expressing FLAG-Timeless alone (lane 6 and 12). The total amount of plasmid DNA used per transfection (15 μg) was identical and was normalized with an empty vector plasmid DNA. The cells were fractionated as in B to yield cytosolic and nuclear fractions. D, HEK293T cells were transfected with empty vector or vectors expressing FLAG-Tipin and/or FLAG-Timeless, as indicated. The cells were fractionated to yield Triton-soluble (soluble cytosolic), soluble nuclear, or chromatin-enriched extracts. E, Flp-InTM T-RExTM-293 cell lines were generated to express FLAG-Tipin-WT or FLAG-Tipin-L195A under control of a tetracycline-inducible promoter. Uninduced (− Tetracycline) or cells induced with tetracycline (+ Tetracycline) for 3 days were lysed and immunoprecipitated with anti-FLAG-agarose, and the immunoprecipitates were probed for Timeless and Tipin. Input represents 5% of the lysate used for the immunoprecipitation. F, nuclear extracts from HEK293T or induced Flp-InTM T-RExTM-293-FLAG-Tipin (WT and L195A) were immunoprecipitated with anti-FLAG resin, separated by SDS-PAGE, and immunoblotted with antibodies against the indicated proteins.
We next tested whether the ectopically expressed Tipin properly localized to the nuclear fraction of cells, because it has been reported that co-transfection of vectors encoding both Timeless and Tipin in NIH3T3 cells greatly enhanced the nuclear retention of the ectopically expressed Tipin (31). Furthermore, previous immunofluorescence microscopy studies showed that the majority of endogenous Timeless and Tipin protein are found in nuclei (34–36), but knockdown of either protein results in the residually expressed binding partner localizing to the cytosol instead (34). We therefore sought to confirm these results and monitor the localization of ectopically expressed Timeless and Tipin by using subcellular fractionation procedures. Lysis of HEK293T cells in hypotonic buffers resulted in ∼60% of the endogenous Timeless and Tipin to be released from nuclei and be detected in the cytosolic fraction (Fig. 4B), similar to the distribution of RPA, of which a large fraction is known to be readily extracted from nuclei under hypotonic conditions (56). We then expressed FLAG-Timeless and FLAG-Tipin alone or in combination in HEK293T cells and then examined the protein expression level and localization by subcellular fractionation. Maximal FLAG-Timeless and FLAG-Tipin protein levels and enrichment in a high salt nuclear extract required co-transfection of vectors encoding both proteins (Fig. 4C). Using a different protocol to generate detergent-soluble and -resistant fractions (41), we further found enrichment of both FLAG-Tipin-WT and FLAG-Tipin-L195A in a detergent-resistant, chromatin-enriched fraction in HEK293T cells when FLAG-Timeless was co-expressed (Fig. 4D). Collectively, these results suggested that functional analysis of ectopically expressed Tipin may require Timeless co-expression to enable stable formation and nuclear localization of Timeless-Tipin complexes.
We also generated stable HEK293 lines that express FLAG-tagged forms of Tipin-WT and Tipin-L195A under control of a tetracyline-inducible promoter to study the interaction of Tipin with Timeless and other checkpoint factors. As shown in Fig. 4E, this approach yielded FLAG-Tipin that co-precipitated with a much larger fraction of endogenous Timeless than with transient transfection (Fig. 4A, compare input and immunoprecipitation signals). Interestingly, constitutive expression of FLAG-Tipin-WT and FLAG-Tipin-L195A led to expression of both forms of FLAG-Tipin at levels 2–3-fold higher than endogenous, untagged Tipin (data not shown) but had no observable effect on cell growth rate (data not shown), suggesting that neither the FLAG tag nor the L195A mutation affects Tipin activity in cell proliferation under nonstressed conditions. Because we previously reported that Timeless co-immunoprecipitated with ATR and ATRIP in human cells (30), we tested whether FLAG-Tipin-WT and FLAG-Tipin-L195A were capable of forming complexes with ATR-ATRIP in these cells. Interestingly, ATR and ATRIP both co-immunoprecipitated with FLAG-Tipin-WT but co-immunoprecipitated only weakly with FLAG-Tipin-L195A (Fig. 4F), suggesting that the interaction of Timeless-Tipin with ATR-ATRIP is mediated in large part through RPA. Importantly, equivalent amounts of Claspin were co-immunoprecipitated with both forms of Tipin, consistent with results using purified recombinant proteins (Fig. 3E) and indicating that Tipin-L195A may potentially sequester Claspin from functioning in ATR-Chk1 signaling.
Tipin-L195A Abrogates Chk1 Phosphorylation after Genotoxic Stress
With Timeless-Tipin complex expression and nuclear localization conditions optimized, we transiently transfected HeLa cells with vectors encoding FLAG-Timeless and either FLAG-Tipin-WT or FLAG-Tipin-L195A that were made resistant to siRNA by modification of the Tipin cDNA sequence. Approximately 27 h after co-transfection of these vectors with either nontargeting control siRNA or siRNA targeting endogenous Tipin, the cells were treated with HU for 30 min to induce activation of ATR. Under these conditions we observed a nearly complete abrogation of HU-induced Chk1 phosphorylation in cells expressing FLAG-Tipin-L195A and transfected with siRNA targeting endogenous Tipin, in comparison with cells expressing FLAG-Tipin-WT (Fig. 5A, compare lanes 4 and 8). Although the effect was much more pronounced when endogenous Tipin levels were reduced by siRNA transfection, an approximately 40% reduction in Chk1 phosphorylation was observed in control siRNA-transfected cells expressing FLAG-Tipin-L195A (Fig. 5, A, lanes 3 and 7, and B), suggesting that this form of Tipin acts in a dominant negative manner to interfere with Chk1 phosphorylation during replication stress.
FIGURE 5.

Tipin-L195A does not support maintenance of Chk1 phosphorylation in response to DNA damage and replication stress. A, HeLa cells transiently transfected with vectors encoding FLAG-Timeless and siRNA-resistant FLAG-Tipin (WT or L195A) and either a nontargeting control (Con) siRNA or an siRNA targeting Tipin (Tip) were treated with 1 mm HU for 30 min. B, quantitation of phospho-Chk1 signals from experiments performed as in A. Phospho-Chk1 signals for each HU-treated sample were normalized to cells transfected with control siRNA and FLAG-Tipin-WT. The data show the averages and standard deviation from three independent experiments. C, HeLa cells transiently transfected with Tipin siRNA and vectors encoding FLAG-Timeless and siRNA-resistant FLAG-Tipin (WT or L195A) were treated with 100 ng/ml NCS for 1 h. D, uninduced (− Tet) and induced (+ Tet) Flp-In T-REx-293-FLAG-Tipin-L195A (siRNA-resistant) cells were transfected with either a control siRNA or an siRNA targeting endogenous Tipin and then treated with 1 mm HU for 2 h. E, HEK293T cells transfected with vectors expressing FLAG-Timeless and either FLAG-Tipin-WT or FLAG-Tipin-L195A were treated with 1 mm HU for 6 h or left untreated before fractionation to enrich for chromatin-associated proteins. Lysate from an equivalent number of cells was separated by SDS-PAGE and immunoblotted with antibodies against the indicated proteins. F, Flp-In T-REx-293-FLAG-Tipin-L195A cells induced with tetracycline to express FLAG-Tipin-L195A (siRNA-resistant) were transfected with either control or Tipin siRNA and then treated with 1 mm HU for the indicated lengths of time. The graph shows average Chk1 phosphorylation at each time point, relative to untreated control, from two independent experiments.
Because Tipin contributes to DNA damage checkpoint activation and Chk1 phosphorylation in response to agents that induce double-strand breaks in DNA (35) (Fig. 1B), we examined the Chk1 phosphorylation status in NCS-treated HeLa cells expressing Tipin-WT or Tipin-L195A and transfected with siRNA targeting endogenous Tipin. As shown in Fig. 5C, we observed a 3–4-fold reduction in Chk1 phosphorylation after NCS treatment in FLAG-Tipin-L195A-expressing cells compared with cells expressing FLAG-Tipin-WT. These results indicate that the RPA-Tipin interaction is important for Chk1 phosphorylation in response to multiple genotoxic stressors.
Similar results were obtained with a HEK293 cell line capable of expressing a siRNA-resistant form of FLAG-Tipin-L195A upon induction with tetracycline. As shown in Fig. 5D, expression of FLAG-Tipin-L195A under conditions where endogenous Tipin was reduced via RNA interference caused a significant abrogation of HU-induced Chk1 phosphorylation. Importantly, no significant effect was observed on the phosphorylation of RPA2 at Ser33 under these conditions, demonstrating that Tipin-L195A expression does not interfere with the ability of ATR to phosphorylate other substrates.
Because we showed that Tipin was required for Claspin to stably associate with RPA-coated ssDNA in vitro, we fractionated untreated or HU-treated HEK293T cells expressing FLAG-Timeless along with either FLAG-Tipin-WT or FLAG-Tipin-L195A to enrich for chromatin-associated proteins. As in HeLa cells, we observed a dominant negative effect of the Tipin-L195A mutant on phosphorylation of Chk1 after HU (Fig. 5E). Importantly, in comparison with cells expressing wild type Tipin, we observed a reduction in the level of chromatin-associated Claspin after HU treatment in cells expressing Tipin-L195A, suggesting that the interaction of Tipin with RPA may be required to stabilize the association of Claspin with chromatin at sites of replication stress. We repeatedly observed no difference in chromatin association between Tipin-WT and Tipin-L195A, either in the absence or presence of HU (Fig. 5E), indicating that the Tipin-RPA interaction is not essential to recruit or stabilize the Timeless-Tipin complex on bulk chromatin. This observation is consistent with a recent report showing that Timeless and Tipin associate with undamaged chromatin in Xenopus egg extracts depleted of RPA (33). Whether Tipin stably associates with RPA at sites of DNA damage and replication stress is unclear, because we have not observed co-localization of Tipin with RPA by indirect immunofluorescence microscopy (data not shown). However, neither Claspin nor Chk1 stably associates with RPA-ssDNA compartments by immunofluorescence microscopy either (24, 57), indicating that like Claspin and Chk1, Tipin function at sites of DNA damage may be transient or dynamic to facilitate turnover or release of phosphorylated, active Chk1 kinase.
Although we observed a reduction in Claspin association with chromatin in HU-treated HEK293T cells expressing Tipin-L195A, we noticed that Claspin protein levels were also reduced in the soluble fraction of cells as well. To confirm this reduction and to compare the kinetics of Claspin loss with Chk1 phosphorylation status, we performed time course experiments in HU-treated Flp-In T-REx-293 cells stably expressing siRNA-resistant FLAG-Tipin-L195A and transfected with either control or Tipin siRNA. As shown in Fig. 5F, we observed that the loss of Claspin protein appeared slightly delayed relative to the reduction in Chk1 phosphorylation, indicating that the Claspin loss may be a consequence and not a cause of reduced ATR-Chk1 signaling. This is consistent with the observation that genetic or chemical abrogation of the ATR-Chk1 signaling pathway leads to the proteosomal degradation of Claspin (58).
DISCUSSION
Currently available data indicate that the ATR kinase is activated by multiple mechanisms in response to DNA damage, including its recruitment to the sites of damage by repair proteins and the direct recognition of DNA damage by checkpoint factors (59–65). However, the predominant signal for the ATR-Chk1 signaling pathway appears to be RPA-coated ssDNA that is generated as a common intermediate of replication stress and damage processing by double-strand break and nucleotide excision repair pathways. ATR stably associates with RPA-coated ssDNA through direct interaction of the ATRIP subunit of the ATR-ATRIP heterodimer with the 70-kDa RPA1 subunit of RPA (6, 7). An issue that has not been widely addressed is precisely how the key substrate of ATR, the Chk1 signal transducing kinase, interacts with ATR on DNA. Our data now provide a potential mechanism: through a direct interaction of the Tipin subunit of the Timeless-Tipin complex with the 34-kDa RPA2 subunit of RPA. Further, we show that Timeless-Tipin serves as a platform for ATR-Claspin interaction that is essential for efficient phosphorylation of Chk1 by ATR (Fig. 6). Importantly, because Claspin and Chk1 do not appear to stably associate with sites of DNA damage (24, 57), Tipin may similarly only transiently interact with RPA and ATR to facilitate the release of phosphorylated, active Chk1 from sites of damage so that Chk1 can perform its checkpoint functions throughout the nucleus. Clearly additional work is necessary to better understand the mechanism of ATR-Chk1 signaling, and ultimately this model for Tipin function will need to be tested experimentally in vitro with purified factors. It should be noted that an in vitro system for Claspin-mediated phosphorylation of Chk1 by ATR has been reconstituted with purified human checkpoint proteins (23), and with this system it has been possible to demonstrate, for the first time, that RPA-ssDNA can directly stimulate ATR kinase activity.3 The availability of these defined in vitro systems should eventually enable us to test and refine the model we have proposed for Timeless-Tipin in ATR-Chk1 signaling.
FIGURE 6.

Model for Tipin function in ATR-Chk1 signaling. In response to DNA damage or replication stress, the generation of ssDNA and presence of primer-template junctions leads to the association of RPA and 9-1-1 at sites of DNA damage. Through an interaction of the RPA1 subunit of RPA with ATRIP, the ATR kinase is recruited to these sites. Through the C-terminal domain of Rad9, TopBP1 also stably associates with damage sites, resulting in activation of ATR kinase activity. Through an interaction of the RPA2 subunit of RPA with Tipin, the Timeless-Tipin complex and then Claspin are able to associate with ATR at sites of DNA damage. The presence of Claspin allows binding of Chk1 and then phosphorylation by ATR. The data showing that Timeless-Tipin, Claspin, and Chk1 do not appear to stably associate with sites of damage in human cells indicate that these proteins may be released to allow phosphorylation of additional Chk1 molecules.
It should also be noted that the ATR-Chk1 signaling pathway is a potential target for cancer therapy. Through abrogation of cell cycle checkpoint function, inhibitors of this pathway are expected to sensitize cancer cells to chemotherapeutic agents by forcing cells containing DNA damage and/or unreplicated DNA to undergo catastrophic mitoses. The development of small molecules that inhibit Chk1 kinase activity for use in combination therapies with traditional chemotherapeutics supports this hypothesis (21). Because the Tipin-RPA interaction site is now known, our studies suggest that a peptide inhibitor or chemical compound that prevents the interaction of Tipin with RPA could potentially serve as a useful therapeutic agent to sensitize cancer cells to DNA damage-inducing chemotherapeutic drugs.
Acknowledgments
We thank the University of North Carolina Center for Environmental Health and Susceptibility, which was supported by National Institutes of Health Grant ES10126.
This work was supported, in whole or in part, by National Institutes of Health Grants GM32833 (to A. S.), ES014635 (to W. K. K.), ES015856 (to M. C.-S.), T32-ES07017 (to S. L. S.-R.), and T32-CA09156 (to M. G. K.).
J.-H. Choi and A. Sancar, unpublished data.
- ATM
- ataxia telangiectasia mutated
- ATR
- ATM and Rad3-related
- ATRIP
- ATR-interacting protein
- IR
- ionizing radiation
- RPA
- replication protein A
- aRPA
- alternative RPA
- ssDNA
- single-stranded DNA
- HU
- hydroxyurea
- NCS
- neocarzinostatin
- siRNA
- small interfering RNA
- WT
- wild type
- MCM
- minichromosome maintenance.
REFERENCES
- 1.Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., Linn S. (2004) Annu. Rev. Biochem. 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 2.Kerzendorfer C., O'Driscoll M. (2009) DNA Repair 8, 1139–1152 [DOI] [PubMed] [Google Scholar]
- 3.Lee J. H., Paull T. T. (2007) Oncogene 26, 7741–7748 [DOI] [PubMed] [Google Scholar]
- 4.Palm W., de Lange T. (2008) Annu. Rev. Genet. 42, 301–334 [DOI] [PubMed] [Google Scholar]
- 5.Cimprich K. A., Cortez D. (2008) Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.You Z., Kong L., Newport J. (2002) J. Biol. Chem. 277, 27088–27093 [DOI] [PubMed] [Google Scholar]
- 7.Zou L., Elledge S. J. (2003) Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 8.Fanning E., Klimovich V., Nager A. R. (2006) Nucleic Acids Res. 34, 4126–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wold M. S. (1997) Annu. Rev. Biochem. 66, 61–92 [DOI] [PubMed] [Google Scholar]
- 10.Ball H. L., Myers J. S., Cortez D. (2005) Mol. Biol. Cell 16, 2372–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ball H. L., Ehrhardt M. R., Mordes D. A., Glick G. G., Chazin W. J., Cortez D. (2007) Mol. Cell. Biol. 27, 3367–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cortez D., Guntuku S., Qin J., Elledge S. J. (2001) Science 294, 1713–1716 [DOI] [PubMed] [Google Scholar]
- 13.Kumagai A., Lee J., Yoo H. Y., Dunphy W. G. (2006) Cell 124, 943–955 [DOI] [PubMed] [Google Scholar]
- 14.Delacroix S., Wagner J. M., Kobayashi M., Yamamoto K., Karnitz L. M. (2007) Genes Dev. 21, 1472–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., Elledge S. J. (2007) Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 16.Stokes M. P., Rush J., Macneill J., Ren J. M., Sprott K., Nardone J., Yang V., Beausoleil S. A., Gygi S. P., Livingstone M., Zhang H., Polakiewicz R. D., Comb M. J. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 19855–19860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Q., Guntuku S., Cui X. S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A., Donehower L. A., Elledge S. J.(2000) Genes Dev. 14, 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao H., Piwnica-Worms H. (2001) Mol. Cell. Biol. 21, 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smits V. A., Reaper P. M., Jackson S. P. (2006) Curr. Biol. 16, 150–159 [DOI] [PubMed] [Google Scholar]
- 20.Stracker T. H., Usui T., Petrini J. H. (2009) DNA Repair 8, 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bucher N., Britten C. D. (2008) Br. J. Cancer 98, 523–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chini C. C., Chen J. (2003) J. Biol. Chem. 278, 30057–30062 [DOI] [PubMed] [Google Scholar]
- 23.Lindsey-Boltz L. A., Serçin O., Choi J. H., Sancar A. (2009) J. Biol. Chem. 284, 33107–33114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S., Bekker-Jensen S., Mailand N., Lukas C., Bartek J., Lukas J. (2006) Mol. Cell. Biol. 26, 6056–6064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee J., Kumagai A., Dunphy W. G. (2003) Mol. Cell 11, 329–340 [DOI] [PubMed] [Google Scholar]
- 26.Kumagai A., Dunphy W. G. (2000) Mol. Cell 6, 839–849 [DOI] [PubMed] [Google Scholar]
- 27.Lupardus P. J., Cimprich K. A. (2006) Mol. Biol. Cell 17, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin S. Y., Li K., Stewart G. S., Elledge S. J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 6484–6489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoo H. Y., Jeong S. Y., Dunphy W. G. (2006) Genes Dev. 20, 772–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unsal-Kaçmaz K., Mullen T. E., Kaufmann W. K., Sancar A. (2005) Mol. Cell. Biol. 25, 3109–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gotter A. L. (2003) J. Mol. Biol. 331, 167–176 [DOI] [PubMed] [Google Scholar]
- 32.Unsal-Kaçmaz K., Chastain P. D., Qu P. P., Minoo P., Cordeiro-Stone M., Sancar A., Kaufmann W. K. (2007) Mol. Cell. Biol. 27, 3131–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka H., Kubota Y., Tsujimura T., Kumano M., Masai H., Takisawa H. (2009) Genes Cells 14, 949–963 [DOI] [PubMed] [Google Scholar]
- 34.Yoshizawa-Sugata N., Masai H. (2007) J. Biol. Chem. 282, 2729–2740 [DOI] [PubMed] [Google Scholar]
- 35.Chou D. M., Elledge S. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 18143–18147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gotter A. L., Suppa C., Emanuel B. S. (2007) J. Mol. Biol. 366, 36–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Unsal-Kaçmaz K., Sancar A. (2004) Mol. Cell. Biol. 24, 1292–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henricksen L. A., Umbricht C. B., Wold M. S. (1994) J. Biol. Chem. 269, 11121–11132 [PubMed] [Google Scholar]
- 39.Mason A. C., Haring S. J., Pryor J. M., Staloch C. A., Gan T. F., Wold M. S. (2009) J. Biol. Chem. 284, 5324–5331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sar F., Lindsey-Boltz L. A., Subramanian D., Croteau D. L., Hutsell S. Q., Griffith J. D., Sancar A. (2004) J. Biol. Chem. 279, 39289–39295 [DOI] [PubMed] [Google Scholar]
- 41.Méndez J., Stillman B. (2000) Mol. Cell. Biol. 20, 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Errico A., Costanzo V., Hunt T. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 14929–14934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olson E., Nievera C. J., Klimovich V., Fanning E., Wu X. (2006) J. Biol. Chem. 281, 39517–39533 [DOI] [PubMed] [Google Scholar]
- 45.Vassin V. M., Anantha R. W., Sokolova E., Kanner S., Borowiec J. A. (2009) J. Cell Sci. 122, 4070–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cortez D., Glick G., Elledge S. J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10078–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y. W., Otterness D. M., Chiang G. G., Xie W., Liu Y. C., Mercurio F., Abraham R. T. (2005) Mol. Cell 19, 607–618 [DOI] [PubMed] [Google Scholar]
- 48.Zhang Y. W., Brognard J., Coughlin C., You Z., Dolled-Filhart M., Aslanian A., Manning G., Abraham R. T., Hunter T. (2009) Mol. Cell 35, 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoo H. Y., Shevchenko A., Shevchenko A., Dunphy W. G. (2004) J. Biol. Chem. 279, 53353–53364 [DOI] [PubMed] [Google Scholar]
- 50.Tsao C. C., Geisen C., Abraham R. T. (2004) EMBO J. 23, 4660–4669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith K. D., Fu M. A., Brown E. J. (2009) J. Cell Biol. 187, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keshav K. F., Chen C., Dutta A. (1995) Mol. Cell. Biol. 15, 3119–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haring S. J., Humphreys T. D., Wold M. S. (2010) Nucleic Acids Res. 38, 846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kemp M. G., Mason A. C., Carreira A., Reardon J. T., Haring S. J., Borgstahl G. E., Kowalczykowski S. C., Sancar A., Wold M. S. (2010) J. Biol. Chem. 285, 4788–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mer G., Bochkarev A., Gupta R., Bochkareva E., Frappier L., Ingles C. J., Edwards A. M., Chazin W. J. (2000) Cell 103, 449–456 [DOI] [PubMed] [Google Scholar]
- 56.Fairman M. P., Stillman B. (1988) EMBO J. 7, 1211–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bekker-Jensen S., Lukas C., Kitagawa R., Melander F., Kastan M. B., Bartek J., Lukas J. (2006) J. Cell Biol. 173, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chini C. C., Wood J., Chen J. (2006) Oncogene 25, 4165–4171 [DOI] [PubMed] [Google Scholar]
- 59.Choi J. H., Lindsey-Boltz L. A., Sancar A. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13301–13306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang G., Sancar A. (2006) Mol. Cell. Biol. 26, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi J. H., Lindsey-Boltz L. A., Sancar A. (2009) Nucleic Acids Res. 37, 1501–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Unsal-Kaçmaz K., Makhov A. M., Griffith J. D., Sancar A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 6673–6678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshioka K., Yoshioka Y., Hsieh P. (2006) Mol. Cell 22, 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y., Qin J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 15387–15392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu Y., Fang Y., Shao H., Lindsey-Boltz L., Sancar A., Modrich P. (2010) J. Biol. Chem. 285, 5974–5982 [DOI] [PMC free article] [PubMed] [Google Scholar]