

Abstract
Voltage-gated sodium channels (VGSCs) are responsible for the initiation and propagation of transient depolarizing currents and play a critical role in the electrical signaling between neurons. A null mutation in the VGSC gene SCN8A, which encodes the transmembrane protein Nav1.6, was identified previously in a human family. Heterozygous mutation carriers displayed a range of phenotypes, including ataxia, cognitive deficits, and emotional instability. A possible role for SCN8A was also proposed in studies examining the genetic basis of attempted suicide and bipolar disorder. In addition, mice with a Scn8a loss-of-function mutation (Scn8amed-Tg/+) show altered anxiety and depression-like phenotypes. Because psychiatric abnormalities are often associated with altered sleep and hormonal patterns, we evaluated heterozygous Scn8amed-jo/+ mutants for alterations in sleep-wake architecture, diurnal corticosterone levels, and behavior. Compared with their wild-type littermates, Scn8amed-jo/+ mutants experience more non-rapid eye movement (non-REM) sleep, a chronic impairment of REM sleep generation and quantity, and a lowered and flattened diurnal rhythm of corticosterone levels. No robust differences were observed between mutants and wild-type littermates in locomotor activity or in behavioral paradigms that evaluate anxiety or depression-like phenotypes; however, Scn8amed-jo/+ mutants did show enhanced spatial memory. This study extends the spectrum of phenotypes associated with mutations in Scn8a and suggests a novel role for altered sodium channel function in human sleep disorders.
Keywords: Ion Channels, Mouse Genetics, Neurobiology, Neurological Diseases, Sodium Channels, HPA Axis, SCN8A, Sleep, Behavior
Introduction
The mammalian genome contains four voltage-gated sodium channel (VGSC)4 α-subunit genes that are highly expressed in the central nervous system (CNS), SCN1A, SCN2A, SCN3A, and SCN8A, which encode the transmembrane proteins Nav1.1, Nav1.2, Nav1.3, and Nav1.6, respectively. VGSCs play a critical role in the initiation and propagation of transient depolarizing currents and electrical signaling between cells. Nav1.6, the most abundantly expressed VGSC in the CNS, is localized in somata and dendrites of projection neurons, nodes of Ranvier of both peripheral and CNS neurons, and axonal initial segments (1–3). Nav1.6 channels are expressed in a wide variety of cells, including Purkinje cells, motor neurons in the brain stem and spinal cord, pyramidal and granule neurons in the hippocampus and cortex, and glial cells and Schwann cells (4). Nav1.6 channels are responsible for a large portion of the persistent sodium current and display several unique biophysical properties that contribute to a propensity for sustained repetitive firing (4).
A broad effect of Scn8a dysfunction on neuronal excitability has been observed in mice with different Scn8a mutations. The Scn8amed-jo mutation, a threonine-to-alanine substitution in the S4–S5 linker of domain III, causes a 10-mV positive shift in the voltage dependence of activation in Purkinje cells (5) and also reduces the resurgent sodium current (6), diminishing the repetitive firing of the Purkinje cells. Reduced transient and persistent sodium current was seen in pyramidal cells of the prefrontal cortex, a site of executive function, in heterozygous Scn8a null mutants (7). Lower sustained and instantaneous firing rates in the initial segment of retinal ganglion cells were also found in heterozygous null mutants (8).
Mice with Scn8a mutations display a variety of recessive movement phenotypes. Homozygous Scn8amed mice that carry a loss-of-function mutation exhibit muscle weakness, progressive paralysis, and death by 3–4 weeks after birth (9). Homozygous Scn8amed-jo/+ mutants have a less severe phenotype (10); by 3 weeks of age, these mutants show an unsteady, wide-based gait and rhythmic tremor of the head and neck during movement (11). However, unlike homozygous null mutants, the Scn8amed-jo homozygous mutants often have a normal life span (11).
Closer examination of heterozygous Scn8a mutants has begun to reveal a number of dominant phenotypes. For instance, heterozygous Scn8amed-Tg mice with a loss-of-function mutation display greater avoidance of the center zone during open field exploration, an increased tendency to remain on the dark side of a light/dark box, and increased immobility in the forced swim test compared with wild-type (WT) littermates (12). These observations suggest that reduced Scn8a expression may be associated with increased anxiety and pronounced stress-induced coping strategies (12). We have found that heterozygous Scn8amed and Scn8amed-jo mutants are more resistant to seizures induced by the chemical convulsants flurothyl and kainic acid compared with WT littermates (13). In contrast, we also observed that heterozygous Scn8amed, Scn8amed-jo, and Scn8a8J mutants display spontaneous spike wave discharges, the hallmark of absence seizures (14). Heterozygous Scn8amed-Tg mutants are also more resistant to electrical kindling (15).
A null mutation in the human SCN8A gene has been reported in a small pedigree in which heterozygous mutation carriers exhibited a range of phenotypes, including ataxia, cognitive deficits, and emotional instability (16). A possible role for SCN8A was also reported in studies examining the genetic basis of attempted suicide (17) as well as bipolar disorder (18). These findings suggest that SCN8A dysfunction may contribute to a variety of neurological and neuropsychiatric disorders.
Neurobiological investigations of psychiatric disorders, particularly those involving alterations in anxiety and depression, often take the sleep-wake cycle into consideration. Moreover, disrupted sleep is a key feature of depression, schizophrenia, mental retardation, dementia, and post-traumatic stress disorder (19–22). To better understand the full range of phenotypes associated with altered Scn8a function, we characterized the sleep-wake architecture and behavioral responses of Scn8amed-jo/+ mutants. Because the dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis plays a major role in psychiatric diseases, we also evaluated Scn8amed-jo/+ mutants for alterations in the circadian variation of corticosterone secretion. We found a robust impairment in the generation and maintenance of rapid eye movement (REM) sleep, an increase in non-REM (NREM) sleep, a lowered and flattened diurnal rhythm of corticosterone secretion indicating hypofunctioning of the HPA axis, and a modest improvement in spatial memory.
EXPERIMENTAL PROCEDURES
Mice
Male Scn8amed-jo/+ mutants were purchased from The Jackson Laboratories (Bar Harbor, ME) and maintained on the C57BL/6J background. The mice were housed in ventilated cages under uniform conditions in a pathogen-free mouse facility with a 12-h light/dark cycle. Food and water were available ad libitum. All experiments were approved by the Emory University Institutional Animal Care and Use Committee.
Genotyping
Genotyping of Scn8amed-jo/+ mutants was performed using primer pair 8aF (5′-ATGCCACAGAAGTGTCATTCC) and 8aR (5′-GGTATTTCCCAGCAAACAGGT) (10). PCR amplification was performed with one cycle at 94 °C for 2 min and 40 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min. The 213-bp PCR product was digested with MslI to produce fragments of 129, 75, and 9 bp from the wild-type allele and 102, 75, 27, and 9 bp from the mutant allele.
Eletrocorticogram (ECoG) Acquisition
Under isoflurane anesthesia, 10 male Scn8amed-jo/+ mutants and 9 male WT littermates (3–4 months old) were surgically implanted with ECoG and electromyography (EMG) electrodes for polysomnographic recordings. The implant consisted of four sterile screw electrodes (Vintage Machine Supplies, Medina, OH) that were placed subdurally. Two screws were placed on the right hemisphere (2 mm anterior-posterior (AP) and 1.2 mm lateral to the midline (LM); 1.5 mm AP and 1.2 mm LM). On the left hemisphere, the two other screw placements were 0.5 mm AP and 2.2 LM and 3.5 mm AP and 2.2 mm LM. Fine wire electrodes were inserted into the left and right neck muscles for EMG acquisition. Postsurgical pain management was achieved with the administration of ibuprofen (0.1 mg/kg) in the drinking water for 3 days. After the surgery, the animals were housed in Plexiglas boxes (20 × 20 × 30 cm) and allowed to recover for 14 days. The animals were then connected to the ECoG cable and, following 48 h of habituation, 48 h of continuous ECoG/EMG data were collected. Amplified ECoG and EMG signals were digitally acquired, collected, and processed by the Somnologica sleep-recording software package (Embla Medical, Reykjavik, Iceland).
Sleep Scoring and Data Analysis
The amount of time spent in each state of vigilance was obtained from 48 h of continuous ECoG base-line sleep recordings and manually scored in 10-s intervals. The time spent in each state of vigilance during the 12-h light and 12-h dark phases from day 1 and day 2 were averaged. To further examine the circadian variation, the average time spent in each state of vigilance during the light and dark phases was also examined in 2-h intervals. The waveforms were classified for wakefulness, NREM sleep, and REM sleep. A low voltage, high frequency ECoG with elevated and variable EMG was defined as wakefulness. NREM sleep was characterized by an ECoG signal that increased in amplitude and decreased in frequency, with the clear presence of high amplitude delta waves (0.5–4 Hz) and an EMG signal that displayed low regular muscular tone. REM sleep was identified by the presence of regular theta waves (4.5–8 Hz) with a lack of muscle tone and phasic bursts of varying duration and amplitude. For power spectral analysis, all 10-s intervals of NREM sleep were subjected to fast Fourier transformation. The power density in the delta frequency (0.5–4 Hz) was determined by the average for each group.
Sleep Deprivation
Following the 48 h of base-line ECoG/EMG recordings, Scn8amed-jo/+ mutants and WT littermates were sleep-deprived during the first 6 h of the light phase (7 a.m. to 1 p.m.). Sleep deprivation (SD) was carried out by the introduction of objects into the cage and tapping the cages whenever the animals appeared drowsy. The animals were not touched during SD and were never disturbed during feeding or drinking. SD was immediately followed by 18 h of continuous ECoG recordings during the recovery period (6-h light phase, 12-h dark phase).
Diurnal Corticosterone Profile
Blood was collected from the facial vein of 10 male Scn8amed-jo/+ mutants and 10 male WT littermates into Microvette CB 300 EDTA tubes (Starstedt) within 5 min of disturbing the cage at either 7 a.m. or 7 p.m. in order to coincide with the expected nadir and peak of circulating corticosterone levels. The 7 a.m. and 7 p.m. samples from each mouse were collected on consecutive days. Plasma was separated by centrifugation at 7000 rpm for 10 min at 4 °C and stored at −20 °C. Plasma corticosterone levels were assayed using a commercial RIA kit (MP Biomedicals) according to the manufacturer's instructions.
Behavioral Tests
Ten male Scn8amed-jo/+ mutants and 10 male WT littermates (3–6 months old) were group-housed and maintained on a 12-h light/dark cycle (lights on at 7 a.m.). All testing was performed during the light phase between 10 a.m. and 2 p.m., and a period of 1 week was allowed between tests. The same group of animals was subjected to the following sequence of behavioral tests: locomotor activity, light/dark box, open field, elevated plus maze, forced swim, and object recognition. An experimenter who was blind to the animal's genotype scored videotaped recordings of each test.
Locomotor Activity
The animals were individually placed in unfamiliar transparent Plexiglas chambers (40 × 20 × 20 cm). Each chamber was placed on a rack containing seven infrared photobeams spaced 5 cm apart, with each beam 5 cm from the cage wall (San Diego Instruments Inc., La Jolla, CA). Ambulatory activity (consecutive beam brakes) was continuously recorded for 50 h (first 2 h, exploratory activity in response to the novel environment; remaining 48 h, endogenous circadian rhythm).
Light/Dark Box Test
The light/dark box test was performed in a rectangular box that was divided to form a light and a dark compartment. The walls of the light compartment (20 × 14 × 14.5 cm) were covered with white paper. The walls of the dark compartment (10 × 14 × 14.5 cm) were covered with black paper and had a roof constructed of black paper. A removable cardboard partition containing a small square opening at floor level (5 × 5 cm) was used to divide the box into light and dark sides. Each animal was placed into the light side of the box, facing away from the dark side and allowed to explore the apparatus for 5 min. The measurements scored were as follows: time spent in the light and dark sides, latency to enter the dark side, and the number of transitions between each side.
Open Field Test
The open field apparatus consisted of a circular arena (96.5-cm diameter) with opaque Plexiglas walls (28 cm high). A marker was used to inscribe a smaller circle 18 cm from the walls. Each mouse was placed in the inner circle of the apparatus and allowed to explore for 5 min. The times spent in the inner and outer circles were scored.
Elevated Plus Maze
The elevated plus maze consisted of two open and two closed arms elevated 76 cm above the floor, with each arm projecting 30.5 cm from the center (5 × 5 cm). Each mouse was individually placed in the central area and allowed to explore for 5 min. The measurements scored were as follows: time spent in closed arms, time spent in open arms, and number of entries into open and closed arms.
Forced Swim Test
The mice were individually placed into a Plexiglas cylinder (10-cm internal diameter, 50 cm high) filled with water (10-cm deep, 25–26 °C) for 6 min. During the last 4 min of the test, the time spent floating was scored. Floating was defined as immobility or minimal movements necessary to maintain the head above the water.
Object Recognition Test
Testing was conducted in an open field box (60 × 60 × 47 cm) constructed of Plexiglas and painted white. In total, four colored ornamental objects were used for testing: a bear, an elephant, a closed bottle, and a dog. The objects were similar in size (∼10 × 6 × 6 cm) but differed in texture. Prior to the test session, the mice were each placed in the empty open field for 5 min on 5 consecutive days (habituation period).
The test session consisted of three successive trials, each 5 min in duration, separated by a 20-min intertrial interval. After completing each trial, the animal was returned to the home cage, and the apparatus and objects were wiped with 70% alcohol. In trial 1, the mouse was placed in the empty open field and allowed to freely explore the testing arena. For trial 2, three objects were introduced into the open field, each placed in a separate corner ∼5 cm from the walls. The selection of the objects and their placement within the open field was randomized for each pair of mice. The mice were paired so that one mutant and one WT littermate were examined using the same layout of the objects. Each mouse was placed in the center of the box, facing the unoccupied corner, and the time spent exploring each object was recorded. For trial 3, one object was maintained in the same position as in trial 2 (repeated object), one object from trial 2 was replaced with a novel object in the same location (novel object), and one object from trial 2 was relocated to the previously unoccupied corner of the open field (relocated object). The time spent exploring each object during the 5-min trial was recorded. A mouse was considered to be exploring an object when it was facing the object at a close distance (≤5 mm) or when the mouse's nose or front paws were in contact with the object.
Statistics
The data were reported as means ± S.E. Homogeneity of variance was assessed by the Levene test and normal distribution of the data by the Shapiro-Wilk test. For sleep analysis, two-way repeated measures analysis of variance (rANOVA) was used to detect differences between genotypes (mutant versus WT) when the light and dark phases were examined in 12-h periods as well as for each 2-h interval. rANOVA was also used to detect differences within genotypes (base line versus recovery following sleep deprivation) during light and dark phases. Post hoc analysis was performed by Tukey's test. For all the behavior tests except the object recognition test, two-tailed t-tests were used to detect differences between genotypes with significance set at p < 0.05. For the object recognition test, a one-tailed t test was used to analyze differences between genotypes. A value of 50% represented chance performance. Therefore, the one-tailed t test showed whether the performance of each genotype was above 50%.
RESULTS
Scn8amed-jo/+ Mice Exhibit Increased NREM Sleep and a Robust Impairment of REM Sleep
Base-line sleep and circadian rhythm dynamics in Scn8amed-jo/+ mutants were compared with WT littermates. Diurnal rhythms for each state of vigilance (wakefulness, NREM sleep, and REM sleep) were present in both genotypes, with expected overall increases in sleep amount during the light phase and increases in wake amount and locomotor activity during the dark phase (Fig. 1). However, sleep and wake amounts and distribution in the mutants differed from WT littermates.
FIGURE 1.

The Scn8amed-jo/+ mutation reduces wakefulness, enhances NREM sleep, and reduces REM sleep amounts. Percentage of time spent in wakefulness (A and B), NREM sleep (C and D), and REM sleep (E and F) during the base-line recording period. Data were averaged over light and dark phases (A, C, and E). The circadian variation of the sleep-wake cycle was obtained by dividing the light and dark periods into 2-h intervals (B, D, and F). Black bars and squares, Scn8amed-jo/+; white bars and squares, WT. Values are presented as mean ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.0001; rANOVA followed by Tukey's post hoc test.
The amount of wakefulness observed in mutants and WT littermates was comparable during the light phase. However, the mutants experienced 12% less wakefulness during the dark phase (Fig. 1A). rANOVA detected effects of group (F(1,17) = 9.48, p < 0.01) and time (F(1,17) = 369.9, p < 0.0001). Post hoc analysis indicated that the decreased amount of wakefulness during the dark phase was statistically significant (p < 0.01, Tukey's test; Fig. 1A).
Although wake amounts during the light phase were not statistically different between genotypes, the mutants did exhibit 14% more NREM sleep during the light phase and 35% more NREM sleep during the dark phase (Fig. 1C). rANOVA detected effects of group (F(1,17) = 23.8, p < 0.0001) and time (F(1,17) = 377.6, p < 0.0001). Post hoc analysis revealed that the amounts of NREM sleep observed in the mutants during the light and dark phases were statistically different from corresponding periods in the WT littermates (light, p < 0.05; dark, p < 0.01, Tukey's test).
In contrast to the increased amount of NREM sleep, the mutants experienced 55% less REM sleep during the light phase when compared with the WT littermates (Fig. 1E). Surprisingly, during the dark phase, the percentage of time spent in REM sleep was similar between genotypes. rANOVA detected effects of group (F(1,17) = 59.8, p < 0.0001), time (F(1,17) = 205.2, p < 0.0001), and an interaction between group and time (F(1,17) = 47.3, p < 0.0001). The impairment of REM sleep amount in the mutants during the light phase was statistically significant (p < 0.0001, Tukey's test).
To perform a more detailed sleep-wake analysis, the different states of vigilance were also analyzed in 2-h intervals. rANOVA followed by post hoc analysis did not reveal any statistically significant differences between genotypes in amounts of wake (Fig. 1B) and NREM sleep (Fig. 1D) during the light and dark phases (p > 0.05 for all comparisons, Tukey's test). In contrast, the mutants spent less time in REM sleep during the last 8 h (11 a.m. to 7 p.m.) of the light period (Fig. 1F). rANOVA detected effects of group (F(1,17) = 90.4, p < 0.001) and time (F(5,85) = 5.8, p < 0.001). The amount of REM sleep was statistically different between genotypes during this period (p < 0.01, Tukey's test). However, differences in amount of REM sleep during the dark phase were not statistically significant (p > 0.05, Tukey's test; Fig. 1F). Because no compensatory increase in REM sleep was observed during the dark phase, the mutants appear to be chronically REM sleep-deprived. Representative examples of ECoG waveforms for each state of vigilance are shown in supplemental Fig. 1.
Scn8amed-jo/+ Mutants Show Decreased REM Sleep Generation and Duration
To determine whether the mutants have altered sleep-wake cycling, we analyzed the number of transitions (stage shifts) between the different states of vigilance and the mean duration of REM sleep bouts (Table 1). For the stage shifts during the light period, the mutants had fewer transitions from NREM sleep to REM sleep and from REM sleep to wakefulness when compared with WT littermates. rANOVA detected effects of group (F(1,17) = 20.2, p < 0.001), shifts (F(4,68) = 255.07, p < 0.0001), and interaction between group and shifts (F(4,68) = 17.1, p < 0.0001). These differences in the number transitions were statistically significant (p < 0.01 for both comparisons, Tukey's test). No statistically significant differences in stage shifts were observed during the dark phase. The diminished number of transitions between NREM and REM sleep in the mutants contributed to the reduced amount of REM sleep during the light phase.
TABLE 1.
Number of arousals and sleep-wake transitions in Scn8amed-jo/+ mutants and WT littermates during the 24-h base-line recording
Shown is a comparison of the total number of arousals and number of stage shifts from wake to NREM sleep, from NREM sleep to wake, from NREM to REM sleep, from REM sleep to NREM sleep, and from REM sleep to wake in Scn8amed-jo/+ mutants and WT littermates. The average REM sleep duration per episode in Scn8amed-jo/+ and WT littermates during the light and dark phases was also calculated. The values are presented as mean ± S.E.
| 12-h light phase |
12-h dark phase |
|||
|---|---|---|---|---|
| Scn8amed-jo/+ | WT | Scn8amed-jo/+ | WT | |
| Total arousals | 105.9 ± 11.3 | 132.7 ± 7.6 | 63.2 ± 5.2 | 64.5 ± 7.2 |
| Wake to NREM | 79.6 ± 4.5 | 97.4 ± 3.8 | 48.5 ± 4.2 | 42.9 ± 4.9 |
| NREM to wake | 53.5 ± 3.7 | 44.7 ± 4 | 37.4 ± 3.6 | 30.6 ± 4.4 |
| NREM to REM | 33.8 ± 2.3a | 59.3 ± 3.3 | 12.7 ± 1.1 | 12.9 ± 1.7 |
| REM to NREM | 7.7 ± 1.7 | 6.8 ± 2.2 | 2.2 ± 0.5 | 0.9 ± 0.2 |
| REM to wake | 26.1 ± 1.9a | 52.6 ± 1.8 | 10.5 ± 1.2 | 12 ± 1.6 |
| REM duration (min) | 0.93 ± 0.13a | 1.17 ± 0.18 | 1.01 ± 0.22 | 1.12 ± 0.15 |
a p < 0.01, rANOVA followed by Tukey's post hoc test for the stage shift and unpaired Student's t test for REM duration.
The average length of each episode of REM sleep was obtained by dividing the total duration of REM sleep by the total number of REM sleep episodes. When compared with WT littermates, REM sleep duration was statistically lower in the mutants during the light phase (Scn8amed-jo/+, 0.93 ± 0.13 min; WT, 1.17 ± 0.18 min; t(17) = 3.11, p < 0.01). However, REM sleep duration was comparable during the dark phase (Scn8amed-jo/+, 1.01 ± 0.2 min; WT, 1.12 ± 0.15 min; t(17) = 1.18, p > 0.05). Therefore, the decrease in overall REM sleep observed during base-line recording is due to a reduction in both the number and duration of REM sleep episodes in the mutants during the light phase.
Scn8amed-jo/+ Mutants Show No Alteration in Delta Power Activity during Basal NREM Sleep
NREM sleep is a restorative phase of sleep that is characterized by slow wave delta oscillations (0.5–4 Hz). The strength of these oscillations, measured as delta power, is positively associated with the duration of prior wakefulness. Consequently, delta power is a quantitative measure of sleep intensity during NREM sleep. Because we observed that Scn8amed-jo/+ mutants are chronically REM sleep-deprived and experience excessive NREM sleep, we sought to determine whether sleep intensity was also altered. Because absolute values of delta power vary substantially between subjects, we performed a within-subject normalization of delta power to allow comparisons between mutants and WT littermates. We accomplished this by averaging the absolute delta power into 2-h intervals for each subject and dividing each 2-h average by the 24-h average (supplemental Fig. 2). rANOVA followed by post hoc analysis did not reveal any statistically significant differences between mutants and WT littermates (p > 0.05, Tukey's test).
Scn8amedjo/+ Mutants Show Impaired REM Sleep Recovery after Sleep Deprivation
We also compared sleep-wake amounts between mutants and WT littermates following a period of SD. After 6 h of SD, continuous ECoG recordings were obtained for 18 h of the subsequent recovery period (6-h light phase, 12-h dark phase). Because the base-line sleep distribution was different between the two genotypes, analyses were performed within each genotype. The amount of NREM and REM sleep during the recovery period was compared with the equivalent time period during base-line recording.
Although 6 h of SD is considered to be mild, increased levels of NREM and REM sleep were expected during the recovery period. The percentage increase in NREM sleep in mutants and WT littermates was comparable during the light phase (Scn8amed-jo/+, 16% increase; WT, 17% increase) (Fig. 2A). rANOVA detected effects of group (F(1,14) = 44.1, p < 0.001) and time (F(1,14) = 58.6, p < 0.001). Both increases in NREM during the light phase were statistically significant (WT, p < 0.001; Scn8amed-jo/+, p < 0.001; Tukey's test). During the dark phase, the amount of NREM sleep increased by 18% in the mutants and by 10% in WT littermates (Fig. 2B). rANOVA detected effects of group (F(1,14) = 19.4, p < 0.001) and time (F(1,14) = 5.6, p < 0.05). Post hoc analysis indicated that only the increase in NREM sleep during the dark period in the mutants was statistically significant (Scn8amed-jo/+, p < 0.05, Tukey's test). We also evaluated the effect of SD on REM sleep. Interestingly, during the 6-h light phase following SD, the increase in REM sleep was ∼2-fold higher in the mutants compared with WT littermates (Scn8amed-jo/+, 65% increase; WT, 34% increase) (Fig. 2C). Nevertheless, the total percentage of REM sleep in the mutants during this period was still less than that of WT littermates during base-line ECoG recording. rANOVA detected effects of group (F(1,14) = 52.2, p < 0.001) and time (F(1,14) = 72.2, p < 0.001), and the increase in REM sleep in both groups was statistically significant (Scn8amed-jo/+, p < 0.001; WT, p < 0.001; Tukey's test). However, the lack of interaction between group and time (F(1,14) = 2.3, p > 0.05) indicated that the difference in the percentage increase of REM sleep during the light period between mutants and WT littermates (65% versus 34%) was not statistically significant. During the 12-h dark period following SD, the increase in REM sleep was higher in WT littermates (Scn8amed-jo/+, 61% increase; WT, 84% increase; Fig. 2D). rANOVA detected effects of group (F(1,14) = 33.6, p < 0.001) and time (F(1,14) = 79.6, p < 0.001); however, only the increase in the WT littermates was statistically significant (p < 0.001, Tukey's test). The interaction between group and time (F(1,14) = 38.0, p < 0.001) indicated that the difference in the percentage increase in REM sleep during the dark period between mutants and WT littermates (61% versus 84%) was statistically significant.
FIGURE 2.

Effect of 6 h of total SD in Scn8amed-jo/+ and WT littermates. Comparison of the amount of time spent in NREM sleep (A and B) and REM sleep (C and D), during 18 h of the recovery period with the equivalent time period during base-line recording. The recovery period comprised 6 h of the light period (1 p.m. to 7 p.m.) (A and C) and 12 h of the dark period (B and D). White bars, base-line period; black bars, recovery period. Values are presented as mean ± S.E. *, p < 0.05; **, p < 0.001; rANOVA followed by Tukey's post hoc test.
These results showed that, during the light period, both genotypes exhibited the expected increase in NREM and REM sleep following SD. However, during the dark period, the increase in NREM in the mutants continued to be significantly higher than the corresponding period during base-line recordings. This excess NREM observed in the mutants was probably achieved at the expense of REM sleep, suggesting that it was a compensatory mechanism for the alteration in the REM sleep structure exhibited by the mutants during the base line and that persisted during recovery from SD.
Scn8amed-jo/+ Mutants Show No Robust Alteration in Delta Power Activity during the Recovery Period
Since NREM sleep intensity is positively related to the duration of prior wakefulness, we analyzed the delta power of NREM sleep during the 18-h period after SD. This analysis provides a measure of the homeostatic sleep response to extended wakefulness. The delta power for each subject was normalized by dividing each 2-h interval of recovery delta power by the corresponding 24-h base-line average of delta power (supplemental Fig. 3). rANOVA detected effects of group (F(1,11) = 9.8, p < 0.01), time (F(2,22) = 6.47, p < 0.01), and interaction between group and time (F(2,22) = 9.7, p < 0.001). Post hoc analysis revealed that the higher percentage of delta power observed in WT littermates during the first 2 h after SD was statistically different from the corresponding 2-h period during base-line recording (p < 0.01, Tukey's test). The analysis of sleep delta power in the mutants revealed no statistically significant differences between base-line and rebound periods.
Scn8amed-jo/+ Mutants Show a Lower and Flattened Diurnal Corticosterone Rhythm
A comorbidity of altered diurnal HPA axis hormone secretion and sleep disturbances has been found in patients with mood disorders (23). To examine the basal activity of the HPA axis, we measured plasma corticosterone (CORT) levels at the beginning of the light (7 a.m.) and dark (7 p.m.) phases (Fig. 3). Although the circadian hormonal variation was maintained in the mutants and WT littermates, the mutant mice showed a flattened diurnal CORT rhythm. rANOVA indicated a significant interaction between genotype and time of day (F(1,25) = 9.357, p < 0.01), and post hoc Tukey's test showed that the mutants had statistically significant lower CORT levels at 7 p.m. (p < 0.01). A smaller difference between the CORT levels at the peak and nadir of the diurnal rhythm was observed in the Scn8amed-jo/+ mutants. Analysis using an independent t test showed that this difference was statistically significant (t(25) = −3.059, p < 0.01) (Fig. 3).
FIGURE 3.

Altered diurnal CORT rhythm in Scn8amed-jo/+ mutants. CORT levels are shown at the beginning of the light phase (7 a.m.) and at the beginning of the dark phase (7 p.m.). The difference in the magnitude of CORT levels between the 7 a.m. and 7 p.m. samples is also shown. White bars, WT; black bars, Scn8amed-jo/+. Error bars, mean ± S.E. *, p < 0.01, rANOVA followed by Tukey's post hoc test, for 7 p.m. CORT levels. *, p < 0.01, t test, for difference between 7 p.m. and 7 a.m. CORT levels.
Scn8amed-jo/+ Mutants Show Enhanced Spatial Memory
Sleep disturbances and hormonal alterations are often correlated with behavioral abnormalities; therefore, we assessed the mutants for alterations in locomotor activity, anxiety-like and depression-like behaviors, and learning and memory.
The analysis of the novelty period (first 2 h of locomotor activity recording) and the following 48 h of normal activity showed no statistically significant differences between genotypes (Table 2). The performances of the mutants and WT littermates were compared in three paradigms that measure anxiety: the elevated plus maze, the light/dark box, and the open field test (Table 2). In the elevated plus maze test, the mutants showed fewer open arm entries than the WT littermates (Scn8amed-jo/+, 2.6 ± 0.5; WT, 5.1 ± 0.9; t(19) = −2.29, p < 0.05), indicating a reluctance to transition from the closed to the open arm. However, there were no statistically significant genotype-specific differences in the total time spent in the closed and open arms or the center. We also did not observe any statistically significant differences between mutants and WT littermates in the parameters analyzed in the light/dark box or open field test (Table 2). In the forced swim test, a measure of depressive-like behavior, there were no statistically significant differences in the amount of time spent immobile between mutants and WT littermates (Table 2).
TABLE 2.
Summary of the behavioral tests
Shown is a comparison of different behavioral tests between Scn8amed-jo/+ mutants and WT littermates. Values are presented as mean ± S.E.
| Scn8amed-jo/+ | WT | |
|---|---|---|
| Locomotor activity | ||
| Novelty (first 2-h beam brakes) | 685 ± 44.8 | 757.4 ± 40 |
| Total ambulance (48-h beam brakes) | 189.8 ± 40 | 189.1 ± 40 |
| Elevated plus maze | ||
| Time spent in closed arm (s) | 178.4 ± 10.5 | 172.2 ± 14.9 |
| Time spent in open arm (s) | 49.3 ± 13.7 | 58.09 ± 9.7 |
| Time spent in center (s) | 72.3 ± 7.17 | 71.8 ± 7.03 |
| No. of open arm entries | 2.6 ± 0.5a | 5.1 ± 0.9 |
| Light/dark box | ||
| Time spent in light side (s) | 116.1 ± 21.2 | 102.5 ± 11.4 |
| Latency to enter dark side (s) | 41 ± 17.3 | 37.2 ± 13.1 |
| Latency to re-enter light side (s) | 58.9 ± 14.7 | 88.2 ± 21.3 |
| No. of transitions | 13.9 ± 2.7 | 17.7 ± 3 |
| Open field | ||
| Time spent in center (%) | 14.1 ± 2.34 | 19.3 ± 3.16 |
| Total distance traveled (m) | 25.3 ± 2.5 | 31.4 ± 2.6 |
| Total distance traveled in center (m) | 4.1 ± 0.7 | 5.9 ± 1.1 |
| Forced swim test | ||
| Time spent immobile (s) | 100.1 ± 8.1 | 89.6 ± 11.2 |
a p < 0.05; unpaired Student's t test.
An object recognition test was used to compare learning and memory between mutants and WT littermates (Fig. 4). This test consisted of three successive trials. In the first trial, each mouse was allowed to explore the empty open arena for 5 min before being returned to the home cage. In the second trial, three objects were introduced into the open field, and the mouse was returned and allowed to explore for 5 min. To ensure that the animals did not have an aversive or preferential response to any of the objects, we analyzed the total time that each object was explored by mutants and WT littermates during the first exposure to the objects (second trial). rANOVA detected an effect of group (F(1,8) = 8.82, p < 0.02) but did not detect an effect of object (F(3,24) = 1.11, p > 0.05) or interaction between group and object (F(3,24) = 0.71, p > 0.05). Post hoc analysis showed that there were no differences in time that each object was explored between genotypes (p > 0.05, Tukey's test). For the evaluation of spatial memory, the first score calculated was the percentage of time in trial 3 spent exploring the novel object compared with the time spent exploring both the novel and the repeated object (i.e. novel/(novel + repeated) × 100). This score provides a measurement of the subject's memory for the repeated object, insofar as preference for the novel object indicates that the presence of the repeated object is remembered. The second score calculated was the percentage of time in trial 3 spent exploring the relocated object compared with the time spent exploring both the relocated and repeated object (i.e. relocated/(relocated + repeated) × 100). This score provides a measurement of the subject's memory for the prior locations of the repeated and relocated objects, insofar as preference for the relocated object could be guided only by spatial memory (the identity of both objects should have been equally familiar). For both scores, a value of 50% represents chance performance, and scores above 50% represent learning and memory.
FIGURE 4.
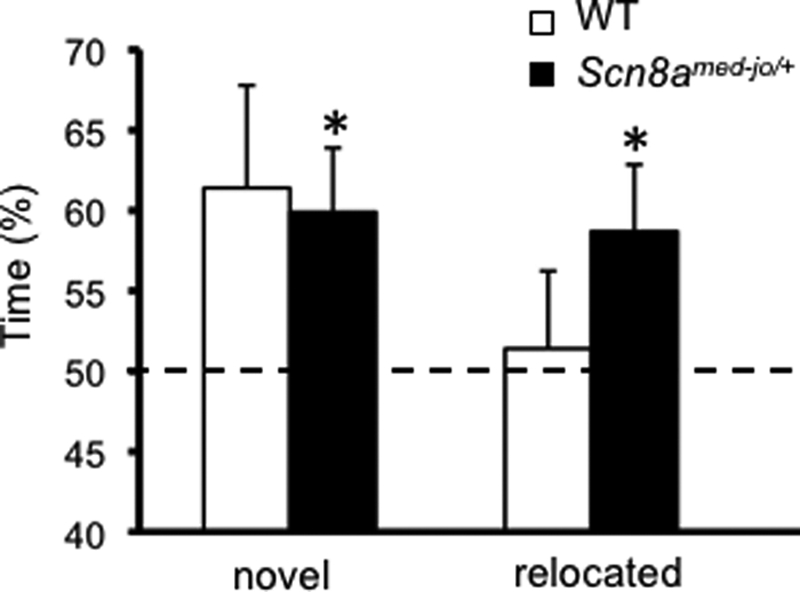
Scn8amed-jo/+ mutants show modest enhancement in spatial memory. Shown is a comparison of the percentage of time spent exploring the novel and relocated objects. A value of 50% represents chance performance. White bars, WT; black bars, Scn8amed-jo/+. Error bars, mean ± S.E. *, p < 0.05, one-tailed t test.
After the first exposure to the three objects in trial 2 and based on the mouse's innate preference for novelty, the expectation was that the mice would spend more time exploring the novel object at the expense of the repeated object in trial 3. Indeed, both mutants and the WT littermates spent more time exploring the novel object compared with the repeated object (Scn8amed-jo/+, 60.0 ± 4.1%; WT, 61.4 ± 6.4%) (Fig. 4). Although the time spent exploring was similar, the percentage was statistically different for the mutants (one-sample t test versus 50% chance: t(9) = 2.45, p < 0.05) and just missed statistical significance for the WT littermates (one-sample t test versus 50% chance: t(8) = 1.79, p = 0.06). Similarly, in trial 3, the expectation was that a mouse would spend more time exploring the repeated object in a new location (the relocated object) compared with a repeated object in the same location. Interestingly, the mutants spent more time than WT littermates exploring the relocated object compared with the repeated object (Scn8amed-jo/+, 58.7 ± 4.2%; WT, 51.4 ± 4.8%) (Fig. 4). The percentage was statistically different for the mutants (one-sample t test versus 50% chance: t(9) = 2.45, p < 0.05) but not the WT littermates (one-sample t test versus 50% chance: t(8) = 0.3, p > 0.1). Thus, although both groups appeared to remember the objects' identities equally well, only the mutants displayed spatial learning and memory in this task.
DISCUSSION
Pharmacological manipulations of different regions of the CNS with tetrodotoxin (TTX), a nonspecific sodium channel blocker (24), yielded the first indirect evidence for the involvement of VGSCs in the regulation of sleep architecture. Independent of the time of day administered, TTX injection into the rat central nucleus of the amygdala resulted in a decrease in the amount of REM sleep and diminished sleep latency (24, 25). When TTX was injected during the dark phase, an increase in the amount of time spent in NREM sleep was also seen (24). In hamsters, injection of TTX into the suprachiasmatic nucleus, the mammalian primary oscillator of different physiological and behavioral functions including circadian rhythms of hormone secretion and the sleep-wake cycle, inhibited the ability of light to increase the expression of Per1 and Per2, the molecular oscillators in the suprachiasmatic nucleus (26). TTX also suppressed pineal levels of melatonin, an important hormone in the regulation of the circadian rhythms of several biological functions (26). Taken together, these results suggested that the inactivation of one or more sodium channel proteins might play a role in generating and maintaining arousal and the different stages of sleep, as well as regulating circadian rhythms of hormone secretion.
The results from this study show that Nav1.6 VGSCs are involved in the generation and maintenance of REM sleep and also in the modulation of NREM sleep. During the total period of recording, an increase in NREM sleep was noted at the expense of REM sleep and wakefulness in Scn8amed-jo/+ mutants. Contributing to the impairment of REM sleep was a reduction in the number of REM sleep episodes as well as a reduction in the mean duration of each REM sleep episode. Overall, this suggests a chronic deprivation of REM sleep in the Scn8amed-jo/+ mutants. We recently identified spike and wave discharges, the hallmark of absence seizures, in Scn8amed-jo/+ mutants and two additional heterozygous Scn8a mutants (14). Thalamocortical oscillations are known to play a critical role in absence seizure generation, suggesting that the normal functioning of these circuits is affected by altered Scn8a function. The transition from wakefulness to sleep involves slow oscillations in cortical neuron membrane potentials that are synchronized by thalamocortical circuits and reflected in the ECoG (27). These slow oscillations play a role in organizing sleep rhythms as slow wave activity and sleep spindles (28). The oscillation itself is composed of a hyperpolarized phase, during which the neurons are deeply silent, followed by a depolarization that is initiated by persistent sodium currents (27). Mutations in Scn8a are responsible for diminishing the persistent sodium current in pyramidal cells of the prefrontal cortex (7). Therefore, we hypothesize that Scn8a dysfunction alters thalamocortical slow oscillations by delaying the depolarization phase, which then contributes to the decrease in wakefulness and the augmented proportion of NREM sleep observed in Scn8amed-jo/+ mutants.
In humans, altered SCN8A function has been implicated in cognitive impairment (16) and psychiatric disorders (17, 18). Endogenous biomarkers, such as the circadian sleep-wake rhythm and the HPA axis, are useful tools that can provide complementary information about various psychiatric disorders in humans and rodent models. For example, the regulation of both sleep and the HPA axis is altered in depressed patients (23) as well as in rodent models of depression (29). In addition to an altered sleep-wake pattern, we also identified a lowered and flattened corticosterone diurnal rhythm in Scn8amed-jo/+ mutants. In humans, decreased cortisol levels are accompanied by a reduction in the amount of REM sleep, and during the last half of the sleep period, higher levels of cortisol are associated with augmented REM sleep duration (30). Similarly, mice with high corticosterone levels experienced more REM sleep, and mice with low corticosterone levels had a significantly higher proportion of NREM sleep (29). Moreover, overexpression of corticotropin-releasing hormone in the entire CNS as well as in specific limbic regions was capable of consistently increasing the amount of REM sleep (31). Therefore, it is possible that the decreased amount of REM sleep in the mutants results from a reduction in the level of corticosterone.
Because altered sleep architecture and altered HPA axis activity are associated with emotional disorders, we also evaluated Scn8amed-jo/+ mutants for anxiety- and depressive-like phenotypes. Although the mutants had a significant decrease in the number of entries into the open arm during the elevated plus maze test, we saw no meaningful alterations in the other anxiety-related parameters evaluated. Likewise, there were no differences between the mutants and WT littermates in the forced swim test. In rats, an increase in anxiety-like behavior was observed following 96 h of total sleep deprivation (32). Surprisingly, the chronic REM sleep reduction seen in the mutants was not capable of triggering a robust anxiety-like phenotype. We hypothesize that the increase in NREM sleep observed during the light and dark phases of base-line sleep-wake ECoG recording compensated for the behavioral consequences of REM sleep deprivation. The fairly normal behavioral responses of Scn8amed-jo/+ mutants differ from the abnormalities observed in heterozygous Scn8a null animals (12). This suggests that some Scn8a mutations may have a greater effect on the neuronal circuitry that influences anxiety- and depression-like phenotypes.
In the object recognition test, we observed that Scn8amed-jo/+ mutants spent more time investigating the relocated object, suggesting that the mutants have a modest enhancement in spatial memory. In healthy individuals, REM sleep deprivation results in memory impairment (33); however, more recently NREM sleep has been linked to memory consolidation (34). In humans with schizophrenia, a direct link is seen between decreased amounts of NREM sleep and impaired visuospatial memory (35). This suggests that the stages of sleep and their electrophysiological characteristics influence neuronal plasticity. The increase in NREM sleep observed during the total ECoG recording in our study might underlie the enhanced memory shown by the mutants.
The identification of altered sleep-wake architecture and HPA axis circadian rhythm in Scn8amed-jo/+ mutants now adds to the range of phenotypes associated with mutations in the CNS VGSCs. Mutations in SCN1A lead to reduced seizure thresholds and spontaneous seizures in mice (36–39) and are involved in a number of human epilepsy disorders, including generalized epilepsy with febrile seizures plus (40) and severe myoclonic epilepsy of infancy (41). SCN1A mutations have also been found in patients with familial hemiplegic migraine (42). Furthermore, SCN2A mutations have been identified in benign familial neonatal infantile seizures (43, 44), and a mutation in SCN3A was recently reported in a patient with pediatric partial epilepsy (45).
Altered sleep-wake architecture has also been observed in other ion channel mutants. Mice lacking expression of the Kcna2 gene, which codes for the voltage-dependent potassium channel Kv1.2, have more waking episodes and less NREM sleep; however, their REM sleep is unaltered (46). These alterations were not seen in the heterozygous Kcna2 null animals. Mice with reduced levels of another potassium channel, Kv3.2, exhibited a normal sleep-wake cycle; however, their EEG power frequency was lower than that of control animals (47). Calcium channels also play an important role in thalamic projections to the cortex during sleep, and deletion of Cav3.1 in the thalamus caused prolonged arousal leading to impaired sleep duration (48). Nonetheless, in a review of the literature, we did not find any other genetic mutant that displayed the chronic REM reduction and increased NREM sleep seen in Scn8amed-jo/+ mice.
The findings of this study extend the range of phenotypes associated with Scn8a dysfunction and suggest a role for VGSCs in human sleep disorders. Additional studies will be required to determine whether altered sleep architecture is a common consequence of VGSC dysfunction or whether the abnormalities observed in Scn8amed-jo/+ mutants reflect a unique role for Scn8a in sleep-wake regulation.
Supplementary Material
Acknowledgments
We thank Dr. Allan Levey, Dr. David Rye, Dr. Michael Decker, Dr. Glenda Keating, and Dr. Gillian Hue for support in sharing space and equipment for ECoG surgeries and data acquisition conducted in the Woodruff Memorial Research Building, Emory University. We thank Dr. David Weinshenker and Dr. Jason Schroeder for assistance in the behavior tests.
This work was supported, in whole or in part, by National Institutes of Health Grants NS046484 and NS065187 (to A. E.) and NS060659 (to K. N. P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–3.
- VGSC
- voltage-gated sodium channel
- CNS
- central nervous system
- WT
- wild type
- HPA
- hypothalamic-pituitary-adrenal
- REM
- rapid eye movement
- NREM
- non-REM
- ECoG
- eletrocorticogram
- EMG
- electromyography
- SD
- sleep deprivation
- rANOVA
- repeated measures analysis of variance
- CORT
- corticosterone
- TTX
- tetrodotoxin.
REFERENCES
- 1.Caldwell J. H., Schaller K. L., Lasher R. S., Peles E., Levinson S. R. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5616–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boiko T., Rasband M. N., Levinson S. R., Caldwell J. H., Mandel G., Trimmer J. S., Matthews G. (2001) Neuron 30, 91–104 [DOI] [PubMed] [Google Scholar]
- 3.Boiko T., Van Wart A., Caldwell J. H., Levinson S. R., Trimmer J. S., Matthews G. (2003) J. Neurosci. 23, 2306–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y., Yu F. H., Sharp E. M., Beacham D., Scheuer T., Catterall W. A. (2008) Mol. Cell Neurosci. 38, 607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith M. R., Goldin A. L. (1999) Neuroreport 10, 3027–3031 [DOI] [PubMed] [Google Scholar]
- 6.Raman I. M., Sprunger L. K., Meisler M. H., Bean B. P. (1997) Neuron 19, 881–891 [DOI] [PubMed] [Google Scholar]
- 7.Maurice N., Tkatch T., Meisler M., Sprunger L. K., Surmeier D. J. (2001) J. Neurosci. 21, 2268–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Wart A., Matthews G. (2006) J. Neurosci. 26, 7172–7180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgess D. L., Kohrman D. C., Galt J., Plummer N. W., Jones J. M., Spear B., Meisler M. H. (1995) Nat. Genet. 10, 461–465 [DOI] [PubMed] [Google Scholar]
- 10.Kohrman D. C., Smith M. R., Goldin A. L., Harris J., Meisler M. H. (1996) J. Neurosci. 16, 5993–5999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meisler M. H., Kearney J., Escayg A., MacDonald B. T., Sprunger L. K. (2001) Neuroscientist 7, 136–145 [DOI] [PubMed] [Google Scholar]
- 12.McKinney B. C., Chow C. Y., Meisler M. H., Murphy G. G. (2008) Genes Brain Behav. 7, 629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin M. S., Tang B., Papale L. A., Yu F. H., Catterall W. A., Escayg A. (2007) Hum. Mol. Genet. 16, 2892–2899 [DOI] [PubMed] [Google Scholar]
- 14.Papale L. A., Beyer B., Jones J. M., Sharkey L. M., Tufik S., Epstein M., Letts V. A., Meisler M. H., Frankel W. N., Escayg A. (2009) Hum. Mol. Genet. 18, 1633–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blumenfeld H., Lampert A., Klein J. P., Mission J., Chen M. C., Rivera M., Dib-Hajj S., Brennan A. R., Hains B. C., Waxman S. G. (2009) Epilepsia 50, 44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trudeau M. M., Dalton J. C., Day J. W., Ranum L. P., Meisler M. H. (2006) J. Med. Genet. 43, 527–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wasserman D., Geijer T., Rozanov V., Wasserman J. (2005) Am. J. Med. Genet. B. Neuropsychiatr. Genet. 133B, 116–119 [DOI] [PubMed] [Google Scholar]
- 18.Wang Y., Zhang J., Li X., Ji J., Yang F., Wan C., Feng G., Wan P., He L., He G. (2008) Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 1902–1904 [DOI] [PubMed] [Google Scholar]
- 19.Bourgeois J. A., Coffey S. M., Rivera S. M., Hessl D., Gane L. W., Tassone F., Greco C., Finucane B., Nelson L., Berry-Kravis E., Grigsby J., Hagerman P. J., Hagerman R. J. (2009) J. Clin. Psychiatry 70, 852–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young A. H. (2004) Stress 7, 205–208 [DOI] [PubMed] [Google Scholar]
- 21.de Kloet C. S., Vermetten E., Geuze E., Kavelaars A., Heijnen C. J., Westenberg H. G. (2006) J. Psychiatr. Res. 40, 550–567 [DOI] [PubMed] [Google Scholar]
- 22.Walker E., Mittal V., Tessner K. (2008) Annu. Rev. Clin. Psychol. 4, 189–216 [DOI] [PubMed] [Google Scholar]
- 23.Friess E., Schmid D., Modell S., Brunner H., Lauer C. J., Holsboer F., Ising M. (2008) J. Psychiatr. Res. 42, 1154–1162 [DOI] [PubMed] [Google Scholar]
- 24.Sanford L. D., Yang L., Liu X., Tang X. (2006) Brain Res. 1084, 80–88 [DOI] [PubMed] [Google Scholar]
- 25.Tang X., Yang L., Liu X., Sanford L. D. (2005) Sleep 28, 923–930 [DOI] [PubMed] [Google Scholar]
- 26.Paul K. N., Gamble K. L., Fukuhara C., Novak C. M., Tosini G., Albers H. E. (2004) Eur. J. Neurosci. 19, 2808–2814 [DOI] [PubMed] [Google Scholar]
- 27.Hill S., Tononi G. (2005) J. Neurophysiol. 93, 1671–1698 [DOI] [PubMed] [Google Scholar]
- 28.Steriade M., Timofeev I. (2003) Neuron 37, 563–576 [DOI] [PubMed] [Google Scholar]
- 29.Touma C., Fenzl T., Ruschel J., Palme R., Holsboer F., Kimura M., Landgraf R. (2009) PLoS ONE 4, e4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fehm H. L., Spath-Schwalbe E., Pietrowsky R., Kern W., Born J. (1993) Exp. Clin. Endocrinol. 101, 267–276 [DOI] [PubMed] [Google Scholar]
- 31.Kimura M., Muller-Preuss P., Lu A., Wiesner E., Flachskamm C., Wurst W., Holsboer F., Deussing J. M. (2010) Mol. Psychiatry 15, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andersen M. L., Martins P. J., D'Almeida V., Bignotto M., Tufik S. (2005) J. Sleep Res. 14, 83–90 [DOI] [PubMed] [Google Scholar]
- 33.Rauchs G., Bertran F., Guillery-Girard B., Desgranges B., Kerrouche N., Denise P., Foret J., Eustache F. (2004) Sleep 27, 395–401 [DOI] [PubMed] [Google Scholar]
- 34.Stickgold R., James L., Hobson J. A. (2000) Nat. Neurosci. 3, 1237–1238 [DOI] [PubMed] [Google Scholar]
- 35.Göder R., Boigs M., Braun S., Friege L., Fritzer G., Aldenhoff J. B., Hinze-Selch D. (2004) J. Psychiatr. Res. 38, 591–599 [DOI] [PubMed] [Google Scholar]
- 36.Yu F. H., Mantegazza M., Westenbroek R. E., Robbins C. A., Kalume F., Burton K. A., Spain W. J., McKnight G. S., Scheuer T., Catterall W. A. (2006) Nat. Neurosci. 9, 1142–1149 [DOI] [PubMed] [Google Scholar]
- 37.Martin M. S., Dutt K., Papale L. A., Dubé C. M., Dutton S. B., de Haan G., Shankar A., Tufik S., Meisler M. H., Baram T. Z., Goldin A. L., Escayg A. (2010) J. Biol. Chem. 285, 9823–9834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogiwara I., Miyamoto H., Morita N., Atapour N., Mazaki E., Inoue I., Takeuchi T., Itohara S., Yanagawa Y., Obata K., Furuichi T., Hensch T. K., Yamakawa K. (2007) J. Neurosci. 27, 5903–5914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang B., Dutt K., Papale L., Rusconi R., Shankar A., Hunter J., Tufik S., Yu F. H., Catterall W. A., Mantegazza M., Goldin A. L., Escayg A. (2009) Neurobiol. Dis. 35, 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Escayg A., MacDonald B. T., Meisler M. H., Baulac S., Huberfeld G., An-Gourfinkel I., Brice A., LeGuern E., Moulard B., Chaigne D., Buresi C., Malafosse A. (2000) Nat. Genet. 24, 343–345 [DOI] [PubMed] [Google Scholar]
- 41.Claes L., Del-Favero J., Ceulemans B., Lagae L., Van Broeckhoven C., De Jonghe P. (2001) Am. J. Hum. Genet. 68, 1327–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dichgans M., Freilinger T., Eckstein G., Babini E., Lorenz-Depiereux B., Biskup S., Ferrari M. D., Herzog J., van den Maagdenberg A. M., Pusch M., Strom T. M. (2005) Lancet 366, 371–377 [DOI] [PubMed] [Google Scholar]
- 43.Herlenius E., Heron S. E., Grinton B. E., Keay D., Scheffer I. E., Mulley J. C., Berkovic S. F. (2007) Epilepsia 48, 1138–1142 [DOI] [PubMed] [Google Scholar]
- 44.Striano P., Bordo L., Lispi M. L., Specchio N., Minetti C., Vigevano F., Zara F. (2006) Epilepsia 47, 218–220 [DOI] [PubMed] [Google Scholar]
- 45.Holland K. D., Kearney J. A., Glauser T. A., Buck G., Keddache M., Blankston J. R., Glaaser I. W., Kass R. S., Meisler M. H. (2008) Neurosci. Lett. 433, 65–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Douglas C. L., Vyazovskiy V., Southard T., Chiu S. Y., Messing A., Tononi G., Cirelli C. (2007) BMC Biol. 5, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vyazovskiy V. V., Deboer T., Rudy B., Lau D., Borbély A. A., Tobler I. (2002) Brain Res. 947, 204–211 [DOI] [PubMed] [Google Scholar]
- 48.Anderson M. P., Mochizuki T., Xie J., Fischler W., Manger J. P., Talley E. M., Scammell T. E., Tonegawa S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 1743–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.