

Abstract
Elucidating the mechanism of human immunodeficiency virus, type 1 (HIV-1) provirus transcriptional silencing in latently infected cells is crucial for understanding the pathophysiological process of HIV-1 infection. It is well established that hypoacetylation of histone proteins by histone deacetylases is involved in the maintenance of HIV-1 latency by repressing viral transcription. Although histone methylation is involved in the organization of chromatin domains and plays a central epigenetic role in gene expression, the role of histone methylation in the maintenance of HIV-1 latency has not been clarified. Here we present evidence that histone H3 Lys9 (H3K9) methyltransferase G9a is responsible for transcriptional repression of HIV-1 by promoting repressive dimethylation at H3K9 and for the maintenance of viral latency. We observed that G9a significantly inhibited basal, as well as, the induced HIV-1 gene expression by tumor necrosis factor-α or Tat. Mutant G9a, however, lacking the SET domain responsible for the catalytic activity of histone methyltransferase, did not show such an effect. When G9a expression was knocked down by small interfering RNA, HIV-1 replication was augmented from cells transiently transfected with a full-length HIV-1 clone. Moreover, a specific inhibitor of G9a, BIX01294, could reactivate expression of HIV-1 from latently infected cells such as ACH-2 and OM10.1. Furthermore, chromatin immunoprecipitation assays revealed the presence of G9a and H3K9 dimethylation on nucleosome histones in the vicinity of the HIV-1 long terminal repeat promoter. These results suggest that G9a is responsible for the transcriptional quiescence of latent HIV-1 provirus and provide a molecular basis for understanding the mechanism by which HIV-1 latency is maintained.
Keywords: Chromatin Immunoprecipitation (ChiP), Chromatin Histone Modification, Epigenetics, HIV, Transcription
Introduction
In eukaryotic cells, transcriptional activity of each gene is largely regulated at the epigenetic level, in which biochemical modifications of chromatin-associated proteins such as histones play critical roles (1, 2). The DNA-associated histones conform an octamer configuration containing two copies of each core histone protein including H2A, H2B, H3, and H4. The N-terminal region of these histone proteins is unstructured and, thus, considered to be in a highly dynamic structure (3). Histone tails protrude out from the globular center of the nucleosome where they may interact with other nuclear factors (2, 4, 5). The N-terminal tails are subjective to a variety of post-translational modifications, such as phosphorylation, acetylation, methylation, and ubiquitination (4–7). These modifications affect the affinity of other nuclear proteins in binding to the histone tail and, thus, regulate the nature of histone-protein complexes associated with each chromatin region. The ability of nuclear proteins to specifically associate with certain histone modifications is the basis of the histone code postulate (2, 5). According to this idea, nuclear proteins appear to function in activating or inhibiting transcription or, similarly, serve to maintain a specific chromatin structure.
Human immunodeficiency virus, type 1 (HIV-1)2 gene expression is the major determinant of viral replication leading to disease progression of acquired immunodeficiency syndrome (AIDS). After HIV-1 infection, integrated HIV-1 proviral DNA is incorporated into nucleosomes and the transcriptional activity of its long terminal repeat (LTR) is under the control of local nucleosomal structure (8–11). It has been suggested that epigenetic modifications of the nucleosomal structure (called “Nuc-1”) near the viral mRNA start site may play regulatory roles in induction of LTR-driven transcription and viral expression (10, 11). The compaction of HIV-1 proviral DNA and its permissiveness for viral transcription are directly dependent on histone post-translational modifications, such as acetylation and methylation (8, 12–14). These distinct modifications serve to recruit various regulatory protein complexes toward HIV-1 LTR and eventually up-regulate or down-regulate HIV-1 gene expression and viral replication. Activation of HIV-1 gene expression by cytokines and virally encoded transactivator Tat is also accompanied by histone acetylation, leading to loss or rearrangement of Nuc1 (8–12). In contrast to productively infected cells, latently infected cells harbor the proviral HIV-1 genome integrated into the silent chromatin allowing persistence of transcriptionally inactive proviruses (8, 9). Although evidence has been accumulated to elucidate the molecular mechanisms involved in viral promoter activation, as described above, the identity of cellular chromatin modifiers and the biochemical mechanism involved in the repression of the HIV-1 promoter is still unclear.
Previous studies have shown that the presence of histone deacetylases (HDACs) at the vicinity of the HIV LTR is correlated with transcriptional repression leading to viral latency (15–18). HDAC1 mediates chromatin remodeling resulting in both LTR promoter activity and viral production repression. Negative transcription factors such as Ying Yang protein 1 (15), nuclear factor κB (NF-κB), p50 homodimer (16), and C-promoter binding factor (17) have been shown to mediate HDAC1 recruitment to the LTR and, consequently, inhibit transcription from the viral promoter. We also reported that activator protein 4 acts as a transcriptional repressor by recruiting HDAC molecules and is involved in the maintenance of viral latency (18). Others observed that drugs that inhibit HDAC activity such as trichostatin A, butyric acid, and valproic acid, could effectively induce HIV transcription in latently infected cells (12, 13, 19, 20). These studies suggest that epigenetic silencing is involved in the maintenance of HIV-1 transcriptional latency.
In addition to histone acetylation, histone Lys methylation also plays an epigenetic role in the organization of chromatin domains and the regulation of gene expression (6, 21, 22). Methylation of histones at Lys and Arg residues play both positive and negative roles in transcriptional regulation. For example, methylation of at Lys4 and Arg17 of histone H3 is generally associated with active genes, whereas methylation of H3 at Lys9 (H3K9me) and Lys27 (H3K27me) has been associated with inactive genes (21, 22). It was also noted that H3K9me exhibited more definitive transcriptional repression over H3K27me (21, 22). The repressive methylation of H3K9 has been detected at the promoter regions of many silenced genes, together with increased DNA methylation and reduced histone acetylation (23–27). Two recent reports (28, 29) demonstrated that the histone methyltransferase (HMT) Suppressor of variegation 3–9 Homolog (Suv39H) 1, which is primarily involved in Lys9 trimethylation of histone H3 (H3K9me3), is responsible for HIV-1 transcriptional silencing. Although H3K9 methylation is known to play a crucial role in chromatin-mediated transcriptional silencing, the molecular mechanism of H3K9 methylation and another HMT on HIV-1 gene repression has yet to be clarified.
In addition to Suv39H1, there are at least four other mammalian H3K9 HMTs, including Suv39H2, a close relative of Suv39H1, G9a, G9a-like protein (GLP)/EuHMTase1, and SETDB1/ERG-associated protein with SET domain (ESET) (21, 30–34). These proteins commonly contain a SET (Suv39, enhancer of zeste, trithorax) domain that is responsible for catalytic action (6, 21, 22). Among these HMTs, G9a is a key enzyme responsible for H3K9 dimethylation (H3K9me2) in mammals as disruption of the G9a gene resulted in a drastic decrease in H3K9 methylation primarily in the silenced region within euchromatin (30, 31, 35, 36). Thus, G9a has been implicated in silencing the gene expression (25–27). Interestingly, in vitro experiments revealed that G9a exhibited a 10–20-fold stronger HMT activity toward H3K9 compared with Suv39H1 (30).
In this study we investigated the role of G9a in HIV-1 gene expression. We demonstrate that G9a is responsible for the maintenance of chromatin-mediated HIV-1 silencing through histone modification of H3K9me2. Biological and therapeutic implications are discussed.
EXPERIMENTAL PROCEDURES
Reagents (Chemicals and Immunoreagents)
5-Aza-2′-deoxycytidine (5-aza-CdR), suberoylanilide hydroxamic acid (SAHA), and anti-G9a antibody were obtained from Sigma and BIX01294, a (1H-1,4-diazepin-1-yl)quinazolin-4-y-yl amine derivative, was purchased from ALEXIS (San Diego, CA). The antibodies against H3K9me2, H3K9me3, and H3K27me2 were purchased from Abcam (Cambridge, MA) and anti-H3 antibody from Upstate Biotechnology. The anti-RNA polymerase II, and anti-α-tubulin antibodies were obtained from Santa Cruz Biotechnology.
Cell Culture
ACH-2 and OM10.1 cells were obtained from the AIDS Research and Reference Reagent Program (NIAID, National Institutes of Health, Bethesda, MD). These cells were maintained at 37 °C in RPMI 1640 (Sigma) with 10% fetal bovine serum (Sigma). To maintain the latency of HIV-1 in ACH-2 and OM10.1, 20 μm azidothymidine (Sigma) was added in the culture medium and excluded prior to the experiments. Human embryonic kidney 293T and HeLa cells were purchased form ATCC (Manassas, VA) and were grown at 37 °C in Dulbecco's modified Eagle's medium (Sigma) with 10% heat-inactivated fetal bovine serum.
Plasmids
Construction of the HIV-1 LTR-based luciferase expression plasmids: HIV-1 LTR-luc (containing the HIV-1 LTR U3 and R), pCMV-Tat, full-length HIV-1 molecular clone (pNL4-3), human T cell lymphotropic virus type 1 (HTLV-1)-luc, cytomegalovirus (CMV)-luc (purchased from Promega), and murine mammary tumor virus (MMTV)-luc were described previously (18, 20, 37, 38). The G9a expression vector and its mutant (G9aΔSET) (39) were generous gifts from Dr. Martin J. Walsh (Mount Sinai School of Medicine of New York University).
RNA Interference
The short interference (si)RNAs against G9a and its control gfp were synthesized by Takara (Ohtsu, Japan). The target sequences are: G9a (5′-CCA UGC UGU CAA CUA CCA UGG TT-3′) and gfp (5′-GGC UAC GUC CAG GAG CGC ACC-3′). For siRNA studies, 293 cells cultured in 12-well plates were transfected with 0.02 μg of HIV-1 LTR-luc or 0.1 μg of pNL4-3, together with 100 nm siRNAs using Lipofectamine 2000 reagent (Invitrogen) as previously described (18, 37, 40). The transfected cells were incubated in culture flasks with a complete medium for 36 h. Then, cells were incubated for an additional 24 h in the presence or absence of tumor necrosis factor (TNF)-α (3 ng/ml). To ensure the knockdown of G9a protein production, Western blotting was performed with anti-G9a antibody. The transfected cells were harvested and subjected to luciferase assay or ELISA for detecting p24.
Immunoblot Assay
The experimental procedures for immunoprecipitation and immunoblotting were performed as described (18, 20). Briefly, cells were harvested with lysis buffer (25 mm HEPES-NaOH, pH 7.9, 150 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.3% Nonidet P-40, 1 mm dithiothreitol, 0.5 mm phenylmethylsulfonyl fluoride), the proteins were separated by SDS-PAGE and transferred to a nitrocellulose membrane (Hybond-C; Amersham Biosciences). The membrane was probed with the respective antibodies, and immunoreactive proteins were visualized by enhanced chemiluminescence (SuperSignal, Pierce). To detect HIV-1 proteins, the cell lysates were subjected to immunoblotting using collected sera from AIDS patients. To evaluate the levels of histone methylation, samples were prepared from cultured cells by acid extraction as described previously (41).
Transfection and Luciferase Assay
CEM T cells were transfected by NucleofectorTM kit V for Jurkat cells (Amaxa Biosystems) according to the manufacturer's protocol (18, 40). Briefly, 3 × 106 cells were mixed with 0.5 μg of HIV-1 LTR-luc either with or without 1.0 μg of pCMV-Tat and the indicated amounts of G9a in 100 μl of Nucleofector solution V. These samples were transferred into a transfection cuvette and subjected to electroporation using program T-14. The transfected cells were incubated in culture flasks with a complete medium for 24 h. Cells were then incubated for an additional 24 h in either the presence or absence of TNF-α. The transfected cells were harvested and the extracts subjected to luciferase assay using the Luciferase Assay SystemTM (Promega). All experiments were carried out in triplicates and the data presented as the fold-increase in luciferase activities (mean ± S.D.) relative to the control for three independent transfections.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assays were performed according to the provider's protocol (Upstate Biotechnology) with some modifications as previously described (18, 20, 40, 42). In brief, chromatin from cross-linked cells was sheared by sonication 13 times for 10 s at one-third of the maximum power of a Microson XL sonicator (Wakenyaku, Co., Ltd., Kyoto, Japan) with 20 s cooling on ice between each pulse and incubated with specific antibody followed by incubation with protein A-agarose beads saturated with salmon sperm DNA. The precipitated DNA was analyzed by PCR (31–33 cycles) with Taq Mastermix (Qiagen) and primers for the HIV-1 LTR (−176 to +61; forward, 5′-CGA GAC CTG CAT CCG GAG TA-3′ and reverse, 5′-AGT TTT ATT GAG GCT TAA GC-3′). For each reaction, 10% of the recovered DNA was used as an input control.
Measurement of Viral p24 Antigen
The p24 antigen level in the cell culture supernatant was measured by p24 antigen capture ELISA using a RETRO-TEK HIV-1 p24 Antigen ELISA kit (Zepto Metrix Corp., Buffalo, NY) as described previously (18, 20, 42). All experiments were performed in triplicates and the data presented as mean ± S.D.
RESULTS
Induction of Latent HIV-1 Replication by 5-Aza-CdR through Down-regulation of G9a
DNA methylation is a critical epigenetic process that helps control chromatin structure and gene regulation (8, 21). It is known that aberrant DNA methylation and H3K9 hypermethylation are associated with the epigenetic silencing (23–27, 41). To examine the effect of histone methylation, we treated T-cell-derived ACH-2 and macrophage-derived OM10.1 cells that are latently infected with replication competent HIV-1 (43–45) with 5-aza-CdR, which is a nucleoside antimetabolite originally synthesized over 40 years ago and is a potent inhibitor of DNA methyltransferase (DNMT) activity through irreversible binding to DNMTs (46). It was previously reported that 5-aza-CdR reactivated epigenetically silenced genes independently from its effects on DNA methylation and was associated with down-regulation of G9a that is known to be a H3K9 methyltransferase causing dimethylation of H3K9 (H3K9me2) (41). Upon treatment with 5-aza-CdR, G9a expression was down-regulated (41).
In Fig. 1, we examined the effects of 5-aza-CdR on the level of HIV-1 production in latently infected ACH-2 and OM10.1 cells. Upon treatment with 5-aza-CdR, the levels of viral protein expression in the cytoplasm were induced (Fig. 1A). In addition, when purified histone fractions were examined, the level of H3K9me2 was correlated with the G9a level although no significant change in the H3K9me3 level was detected (Fig. 1, A and B). These results suggested that H3K9me2 and, thus, G9a could be involved in expression of HIV-1 from latently infected cells.
FIGURE 1.

Activation of HIV-1 by 5-aza-CdR. A, reactivation of the silenced HIV-1 gene in the latently infected cells by 5-aza-CdR. Latent HIV-1-infected ACH-2 and OM10.1 cells were incubated with 5-aza-CdR (0, 1, 5, or 15 μm) for 48 h. The cell lysates were analyzed for viral proteins by immunoblotting with the collected AIDS patients sera or G9a antibody. Positions of HIV-1 proteins are indicated on the right. The α-tubulin was used as an internal control. B, down-regulation of H3K9me2 by 5-aza-CdR. The experiments were similarly performed as in A except that the histone protein fraction was partially purified from these cells. The cell lysates were analyzed for the dimethylated histone H3 at Lys9 (H3K9me2) or trimethylated histone H3 at Lys9 (H3K9me3) by immunoblotting using specific antibodies. The unmodified H3 protein was used as a control.
Repression of HIV-1 LTR Gene Expression by G9a
Considering HIV-1 induction from latent cells was inversely correlated with G9a expression and dimethylation of H3K9 upon treatment with 5-aza-CdR, we examined the effect of G9a overexpression in HIV-1 LTR transcription. As shown in Fig. 2A, the luciferase reporter plasmid under the control of HIV-1 LTR was co-transfected with the G9a expression vector in CEM T cells. As demonstrated, the basal transcription level from HIV-1 LTR was inhibited by G9a in a dose-dependent manner. Upon stimulation of the HIV-1 promoter either by TNF-α, a physiological inducer of NF-κB, or Tat, a viral-encoded transcriptional activator, G9a could similarly exert its negative effect. As shown in the figure, the amounts of H3K9me2, the dimethylated form of H3 at Lys9, were modestly up-regulated by G9a overexpression. Similar results were observed using 293 cells (data not shown). These results demonstrated, for the first time, that G9a exhibits repressive action on HIV-1 gene expression in cultured cells in vivo.
FIGURE 2.

Repression of HIV-1 LTR gene expression by G9a overexpression. A, effects of G9a on gene expression from transiently transfected HIV-1 LTR. The expression plasmid pcDNA-G9a was co-transfected with the HIV-1 LTR-luc reporter construct (20) into CEM T cells. Amounts of G9a plasmid co-transfected were 2 and 8 μg/transfection. Extents of HIV-1 gene expression and the effects of G9a are shown in CEM cells at the basal level (left panel), upon TNF-α stimulation (middle panel), or upon co-transfecting the Tat-expressing plasmid pCMV-Tat at 2 μg/transfection (right panel). Upon TNF-α stimulation, cells were incubated with 3 ng/ml of TNF-α after 24 h of transfection and incubated for an additional 24 h. B, effects of G9a SET domain deletion on HIV-1 gene expression. CEM cells were transfected with HIV-1 LTR-luc together with the SET-defective G9a deletion mutant (G9aΔSET). The cells were harvested and luciferase activity was measured. Amounts of G9a, tagged with EGFP, and the dimethylated form of histone 3 at the Lys9 position were analyzed by Western blotting using specific antibodies. Each value shown is the fold-increase in the luciferase activity (mean ± S.D.) relative to the control transfection of three independent experiments.
The SET Domain of G9a Is Essential for the Repression of the HIV-1 Gene Expression
G9a contains SET domain in a C-terminal and a previous study (30, 39) showed that the SET domain is essential for the G9a-mediated H3K9 methylation, thus, we examined the effect of deleting the SET domain, responsible for the catalytic action of G9a, from G9a (G9aΔSET) in down-regulating the HIV-1 gene expression. As shown in Fig. 2B, overexpression of G9aΔSET abolished the repressive action of G9a on both basal and TNF-α- or Tat-stimulated HIV-1 expression. The amount of H3K9me2 was not changed by overexpression of G9aΔSET. These findings indicate that the HMT activity is involved in the G9a-mediated repression of HIV-1 gene expression.
The Effect of G9a Knockdown
To examine the effect of endogenous G9a, we adopted a siRNA technique to knockdown G9a expression and examined HIV-1 production when the endogenous G9a was depleted. Transduction of G9a siRNA caused the depletion of G9a protein and thus down-regulation of H3K9me2 (Fig. 3A), which resulted in increased basal transcription from HIV-1 LTR, as shown in Fig. 3B (2.3-fold as compared with control siRNA). Moreover, TNF-α-stimulated LTR gene expression was further augmented by G9a depletion (3.6-fold). To assess the physiological relevance of the repressive action of G9a, we examined the effect of depleting endogenous G9a on HIV-1 replication using siRNA on G9a (Fig. 3C). 293 cells were transfected with a replication-competent full-length HIV-1 clone (pNL4–3) together with G9a siRNA, and viral production was evaluated by measuring HIV-1 p24 antigen levels in the culture supernatant. We found that G9a depletion resulted in an increase in basal HIV-1 production (1.9-fold as compared with control siRNA). In the right column of Fig. 3C, we observed a synergism between TNF-α stimulation and G9a knockdown upon HIV-1 production (2.2-fold with TNF-α-stimulation and further 2.7-fold by G9a knockdown). These results indicate that endogenous G9a acts as a negative regulator of HIV-1 gene expression and replication.
FIGURE 3.

Effects of G9a knockdown. A, confirmation of the siRNA-mediated G9a knockdown. 293 cells were transfected with 100 nm siRNAs directed against either G9a or gfp (control) mRNAs. After 36 h of transfection, cells were lysed and levels of G9a and H3K9me2 were assessed by immunoblotting using specific antibodies. Afterward, the immunoblotted membrane was stripped and reprobed with anti-α-tubulin antibody. B, augmentation of HIV-1 gene expression by G9a depletion. 293 cells were transfected with HIV-1 LTR-luc concurrently with either G9a siRNA or its control. After 24 h of transfection, cells were either untreated or treated with 3 ng/ml of TNF-α and incubated for an additional 24 h. Cells were harvested and luciferase activity was measured. Each value is the fold increase in the luciferase activity (means ± S.D.) relative to the control transfection of three independent experiments. C, effects of G9a knockdown on HIV-1 viral replication. 293 cells were transfected with pNL4-3, a replication competent full-length HIV-1 clone, and either G9a siRNA or control siRNA (GFP). After 24 h of transfection, cells were either untreated (left panel) or treated (right panel) with 3 ng/ml of TNF-α and incubated for an additional 24 h. The culture supernatants were collected and subjected to HIV-1 p24 antigen level determination by ELISA. Each value shown is the fold increase in the HIV p24 level (means ± S.D.) relative to the control transfection of three independent experiments.
Effect of a Specific G9a Inhibitor BIX01294
To confirm the negative effect of G9a on HIV-1 gene expression, we examined the effect of BIX01294, a specific inhibitor of G9a, on the level of viral replication from the latently infected cells with HIV-1. BIX01294 was initially identified as a specific inhibitor against HMT by high throughput in vitro screening of a chemical library comprising ∼125,000 preselected compounds using recombinant G9a (47). BIX01294 selectively inhibited G9a HMT activity and the generation of H3K9me2 without affecting the cofactor S-adenosylmethionine and other HMTs such as Suv39H1, protein arginine methyltransferase 1, SET7/9, and ESET (47).
As shown in Fig. 4A (right), HIV-1 LTR gene expression in CEM T cells was augmented in a dose-dependent manner for BIX01294 concentration. In addition, BIX01294 could restore the G9a-induced repression of HIV-1 gene expression (Fig. 4A, left). In Fig. 4B, effects of BIX01294 were examined for various promoters including human T cell lymphotropic virus type 1 (HTLV-1), CMV, and MMTV. Although the transcriptional activity of HTLV-1 was augmented in a dose-dependent manner for BIX01294, no such effect was observed with the CMV and MMTV promoters. Fig. 4C shows that upon treatment of ACH-2 and OM10.1 cells with BIX01294, viral production was dramatically up-regulated either in the culture supernatant or within the cytoplasm. When we purified the histone fraction and examined the levels of histone H3 methylation, we found that methylation of H3K9me2, but not H3K27me2, was down-regulated by BIX01294. Similar effects were observed with another cell line, U1 (48), a macrophage-monocyte cell line latently infected with HIV-1 (data not shown). These results clearly show that the induction of dimethylation at H3K9 by G9a is involved in repression of HIV-1 replication.
FIGURE 4.

Effects of G9a inhibitor BIX01294 on HIV-1 replication. A, activation of HIV-1 gene expression by BIX01294 (left) and restoration of the G9a-mediated repression of HIV-1 gene expression by BIX01294 (right). The HIV-1 LTR-luc plasmid was transfected into CEM cells, incubated for 24 h, treated with BIX01294 (0, 1.5, 5, or 10 μm) for another 24 h, and luciferase activity was measured. In the right panel, the HIV-1 LTR-luc and G9a plasmids were transfected into CEM cells, incubated for 24 h, treated with BIX01294 (0, 5 or 10 μm) for another 24 h, and the luciferase activity was measured. B, effects of BIX01294 on gene expression from HTLV-1, CMV, and MMTV promoters. As positive controls, each promoter was treated with known inducers. PMA, phorbol 12-myristate 12-acetate; Dex, dexamethasone. Experiments with MMTV-luc were performed with HeLa cells where the glucocorticoid receptor is expressed. For the data in A and B, each data point represents the mean ± S.D. of three independent experiments. C, activation of HIV-1 replication by BIX01294. Latently infected cell lines, ACH-2 and OM10.1, were incubated in the presence of various concentrations of BIX01294 for 48 h and both the culture supernatant and the cell lysate were measured for viral p24 antigen levels: upper panels, viral p24 proteins in the culture supernatant; middle panels, major viral proteins in the cytoplasm; and lower panels, the dimethylated histones, H3K9me2 and H3K27me2, in the nuclear histone fractions through immunoblotting using specific antibodies. Each value shown is the HIV p24 level (means ± S.D.) of three independent experiments.
BIX01294 Facilitates HIV-1 Reactivation via Chromatin Remodeling
As demonstrated above, we found that BIX01294 could down-regulate H3K9me2 by inhibiting G9a and re-activate latent HIV-1 at the transcriptional level. In Fig. 5, we performed ChIP assays to further examine the effects of BIX01294 in ACH-2 cells using antibodies against G9a, H3K9me2 and H3K27me2, H3, and RNA polymerase II. We observed that G9a, H3K9me2, and H3K27me2, and only traceable amounts of RNA polymerase II bound to the core promoter region (from −176 to +61) within HIV-1 LTR when these cells were maintained in the latent state (without any stimulation). However, when ACH-2 cells were treated with either BIX01294 or TNF-α to induce HIV-1 replication, G9a and H3K9me2 were readily dissociated from the HIV-1 promoter and RNA polymerase II became detectable in the HIV-1 promoter (Fig. 5). The amount of H3K27me2 was not changed by BIX01294 treatment.
FIGURE 5.
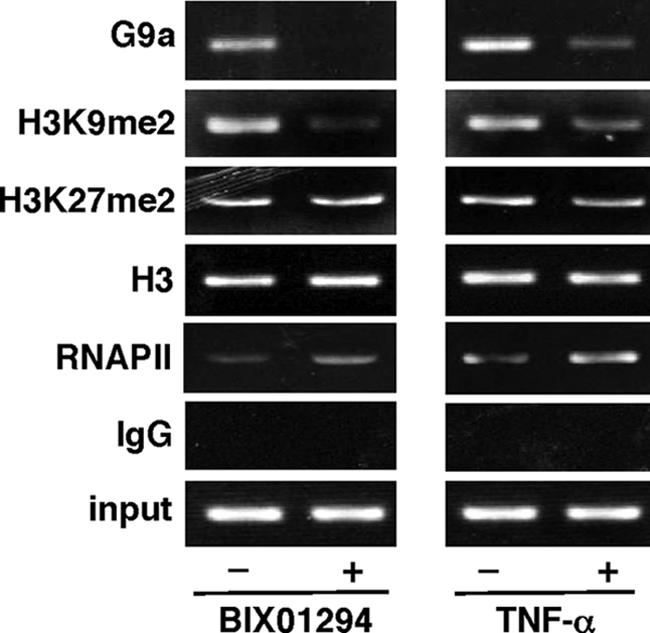
Presence of G9a and H3K9me2 in the proviral HIV-1 LTR revealed by ChIP assay. ChIP assays were carried out to detect the dimethylated histone H3 in the vicinity of HIV-1 proviral DNA in ACH-2 cells. ACH-2 cells were either treated or untreated with 10 μm BIX01294 (left panel) or 3 ng/ml of TNF-α (right panel) for 90 min and subjected to ChIP assays as described under “Experimental Procedures.” The final PCR was carried out to amplify the viral sequence from −176 to +61 within the viral LTR. The antibodies used in the ChIP assays are indicated on the left. Input DNA (input) represents 10% of total input chromatin DNA, whereas immunoprecipitation with non-immune IgG (“IgG”) served as a negative control. RNAP II, RNA polymerase II.
Synergistic Activation of HIV-1 Replication by BIX01294 and HDAC Inhibitor SAHA or DNMT Inhibitor 5-Aza-CdR
Because we found that the G9a HMT inhibitor BIX01294, HDAC inhibitors SAHA (49, 50) or butyric acid (13, 19, 20), and DNMT inhibitor 5-aza-CdR could independently induce HIV-1 production from latently infected cells, we examined whether the effect of BIX01294 could synergize with either SAHA or 5-aza-CdR in inducing HIV-1 production. As shown in Fig. 6A, where ACH-2 cells were treated with BIX01294 alone or in combination with SAHA, we observed a clear synergism between BIX01294 and SAHA. Whereas SAHA alone or BIX01294 alone (10 μm) could induce HIV-1 production by 13.5- and 4.9-fold, respectively, treatment with both BIX01294 and SAHA augmented HIV-1 production by 47.1-fold. When ACH-2 cells were treated with both BIX01294 and 5-aza-CdR, a similar synergism was observed (Fig. 6B). Synergism was also observed with BIX01294 and butyric acid (data not shown). These findings collectively suggest that DNA methylation, acetylation, and methylation of histones are independently involved in gene expression and viral replication.
FIGURE 6.

Synergistic activation of HIV-1 replication by BIX01294 and SAHA or 5-aza-CdR. To investigate the mode of actions of these chemical compounds with distinct biochemical actions, the HIV-1 latently infected ACH-2 cells were treated with various combinations of SAHA, 5-aza-CdR, and BIX01294 for 48 h. The culture supernatants and the cell lysates were analyzed for p24 antigen levels by ELISA (upper panels) and detection of virus proteins by immunoblotting (lower panels), respectively. Each value shown is the HIV p24 level (means ± S.D.) of three independent experiments.
DISCUSSION
The ability of HIV-1 to establish latent infection is considered momentous for AIDS progression (8, 9). Elucidating the transcriptional silencing mechanism of HIV-1 provirus in latently infected cells is crucial in understanding the pathophysiological process of HIV-1 infection and to further develop novel therapies. Viral latency involves certain chromatin modifications, in particular those of histone proteins, leading to proviral quiescence. It is well established (15–18) that proviral quiescence is crossly associated with HDAC recruitment to HIV-1 LTR. In addition, although H3K9 trimethylation is triggered by Suv39H1 and is known to be associated with gene silencing (6, 21, 22), the role of histone methylation by G9a in the maintenance of HIV-1 latency, however, has not yet been elucidated. Because the extent of histone methylation by another HMT G9a is much greater that Suv39H1 (30), we explored the effect of H3K9me2 on the transcriptional activity of latent HIV-1 proviral chromatin. We found that G9a is overexpressed, HIV-1 gene expression was greatly suppressed, whereas G9a knockdown mediated by siRNA strikingly augmented HIV-1 transcription, thus, vital replication. Moreover, when cell lines latently infected with HIV-1 were treated with BIX01294, a specific inhibitor of G9a, H3K9 dimethylation was down-regulated and reactivated HIV-1 transcription and viral replication in the latently infected cells. These findings were confirmed by ChIP assays. These results suggest that G9a is responsible for the maintenance of transcriptional quiescence of latent HIV-1 provirus.
Although accumulating evidence suggests that HDACs are critical regulators of HIV-1 latency, du Chéné et al. (29) observed that knockdown of HDAC1 had only a marginal effect on the basal transcriptional activity of latent HIV-1. However, by using a general HDAC inhibitor, trichostatin A, HIV-1 promoter activity was stimulated nearly 40-fold indicating that HDAC-mediated repression of the HIV-1 promoter involves more than one HDAC species (29). Considering the same authors observed that knockdown of heterochromatin protein 1 (HP1) γ, a heterochromatin packaging protein that specifically recognizes tri- or dimethylated histone H3, could stimulate HIV-1 transcription over 70-fold (29), it was suggested that histone methylation plays a major role in controlling the transcriptional activity of the latent HIV-1 provirus.
Regarding the role of histone methylation on HIV-1 provirus transcription, Suv39H1 and its associating factors, such as HP1 proteins, were found to be accumulated in the vicinity of latent HIV-1 proviral DNA and conform the repressive or transcriptionally silent heterochromatin (28, 29). There have been a number of interesting observations regarding the chromatin configurations and the transcriptional status of latent HIV-1 proviral DNA. For example, du Chéné et al. (29) and Marban et al. (28) reported that HP1γ was associated with the latent HIV-1 proviral DNA and when HP1γ was knocked down, latent HIV-1 was reactivated. In contrast, Mateescu et al. (51) reported that H3K9 methylation and HP1β, but not HP1γ, were associated with latent HIV-1 proviral DNA upon ChIP analyses and that knockdown of HP1β reactivated latent HIV-1 gene expression, whereas HP1γ knockdown suppressed HIV-1 transcription. Thus, the effect of HP1 in the maintenance of HIV-1 latency is still controversial.
Another HMT catalyst, G9a, primarily located at the silenced euchromatin (21, 30, 31, 35, 47) is considered to have distinct target genes. It is possible that Suv39H and G9a have distinct roles in chromatin modification by having distinct molecular partners, thus, recognizing different modalities of distinct histone modifications (21, 22, 30–32, 35). In addition, a recent report by Gazzar et al. (52) demonstrated that G9a is involved in endotoxin tolerance and that G9a knock-down was correlated with the loss of Suv39H binding to the TNF-α promoter, and not vice versa, suggesting that Suv39H appears to be located downstream of G9a. Thus, it is suggested that when the gene, such as HIV-1 provirus, is silenced by the recruitment of G9a and, subsequently, with dimethylation of histone H3 (H3K9me2), which was then recognized by a heterochromatin protein complex containing HP1 and HDACs (33, 52–55), followed by recruitment of Suv39H thus converting the silent euchromatin to a heterochromatin. G9a-mediated chromatin silencing might have a critical role in establishment of the latent HIV-1 provirus. In fact, a G9a-specific inhibitor, BIX01294, could efficiently reactivate the latent HIV-1 genes in a wide variety of cells (Fig. 4).
In addition to histone modifications, the spatial distribution of genes within the nucleus might also contribute to the transcriptional control. For example, a recent report demonstrated an interesting correlation between transcriptional repression of the HIV-1 provirus and its spatial interaction with a pericentromeric heterochromatin region located in certain chromosomes, such as chromosome 12 in multiple distinctive Jurkat-derived cell clones where HIV-1 proviral DNAs are latently infected (56). Furthermore, as euchromatins are known to be dispersed within the nuclear core, certain portions of the nucleus, where G9a is predominantly present, could provide an intranuclear environment allowing reversible silencing (8, 56). Nevertheless, further studies are needed to clarify the possible mechanism that links histone and DNA methylation. At present, it is well established that the latent HIV-1 provirus is associated with DNA methylation in its vicinity (57–59). In addition, because G9a is known to associate with DNMT (26, 52, 60) and G9a knockout cells revealed significantly reduced DNA methylation (26, 27, 60, 61), it is possible that G9a indirectly induces DNA methylation. Thus, molecular actions of G9a in regulating the transcriptional activity of the HIV-1 promoter need to be further explored.
Acknowledgments
We thank Marni Cueno for critical reading of the manuscript and expert language editing, as well as Drs. Martin J. Walsh and Alain Israël for the G9a and HTLV-1-luc plasmids, respectively.
This work was supported by grants-in-aid from the Ministry of Health, Labor and Welfare of Japan, the Ministry of Education, Culture, Sports, Science and Technology of Japan, the Japan Human Sciences Foundation, and the Takeda Science Foundation.
- HIV-1
- human immunodeficiency virus type 1
- 5-aza-CdR
- 5-aza-2′-deoxycytidine
- DNMT
- DNA methyltransferase
- HDAC
- histone deacetylase
- HMT
- histone methyltransferase
- HP1
- heterochromatin protein 1
- SAHA
- suberoylanilide hydroxamic acid
- SET
- Su(var)3-9 enhancer-of-zeste and trithorax
- Suv39H
- suppressor of variegation 3-9 homolog
- TNF-α
- tumor necrosis factor-α
- LTR
- long terminal repeat
- siRNA
- short interference RNA
- CMV
- cytomegalovirus
- ChIP
- chromatin immunoprecipitation
- MMTV
- murine mammary tumor virus
- ELISA
- enzyme-linked immunosorbent assay
- GFP
- green fluorescent protein
- HTLV-1
- human T cell lymphotropic virus type 1.
REFERENCES
- 1.Narlikar G. J., Fan H. Y., Kingston R. E. (2002) Cell 108, 475–487 [DOI] [PubMed] [Google Scholar]
- 2.Strahl B. D., Allis C. D. (2000) Nature 403, 41–45 [DOI] [PubMed] [Google Scholar]
- 3.Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 4.Berger S. L. (2007) Nature 447, 407–412 [DOI] [PubMed] [Google Scholar]
- 5.Jenuwein T., Allis C. D. (2001) Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 6.Lachner M., O'Sullivan R. J., Jenuwein T. (2003) J. Cell Sci. 116, 2117–2124 [DOI] [PubMed] [Google Scholar]
- 7.Li B., Carey M., Workman J. L. (2007) Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 8.Colin L., Van Lint C. (2009) Retrovirology 6, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marcello A. (2006) Retrovirology 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verdin E. (1991) J. Virol. 65, 6790–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verdin E., Paras P., Jr., Van Lint C. (1993) EMBO J. 12, 3249–3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Lint C., Emiliani S., Ott M., Verdin E. (1996) EMBO J. 15, 1112–1120 [PMC free article] [PubMed] [Google Scholar]
- 13.Sheridan P. L., Mayall T. P., Verdin E., Jones K. A. (1997) Genes Dev. 11, 3327–3340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lusic M., Marcello A., Cereseto A., Giacca M. (2003) EMBO J. 22, 6550–6561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coull J. J., Romerio F., Sun J. M., Volker J. L., Galvin K. M., Davie J. R., Shi Y., Hansen U., Margolis D. M. (2000) J. Virol. 74, 6790–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams S. A., Chen L. F., Kwon H., Ruiz-Jarabo C. M., Verdin E., Greene W. C. (2006) EMBO J. 25, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyagi M., Karn J. (2007) EMBO J. 26, 4985–4995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imai K., Okamoto T. (2006) J. Biol. Chem. 281, 12495–12505 [DOI] [PubMed] [Google Scholar]
- 19.Lehrman G., Hogue I. B., Palmer S., Jennings C., Spina C. A., Wiegand A., Landay A. L., Coombs R. W., Richman D. D., Mellors J. W., Coffin J. M., Bosch R. J., Margolis D. M. (2005) Lancet 366, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imai K., Ochiai K., Okamoto T. (2009) J. Immunol. 182, 3688–3695 [DOI] [PubMed] [Google Scholar]
- 21.Jenuwein T. (2006) FEBS J. 273, 3121–3135 [DOI] [PubMed] [Google Scholar]
- 22.Lee D. Y., Teyssier C., Strahl B. D., Stallcup M. R. (2005) Endocr. Rev. 26, 147–170 [DOI] [PubMed] [Google Scholar]
- 23.Baylin S. B., Ohm J. E. (2006) Nat. Rev. Cancer. 6, 107–116 [DOI] [PubMed] [Google Scholar]
- 24.Yoo C. B., Jones P. A. (2006) Nat. Rev. Drug Discov. 5, 37–50 [DOI] [PubMed] [Google Scholar]
- 25.Feldman N., Gerson A., Fang J., Li E., Zhang Y., Shinkai Y., Cedar H., Bergman Y. (2006) Nat. Cell Biol. 8, 188–194 [DOI] [PubMed] [Google Scholar]
- 26.Epsztejn-Litman S., Feldman N., Abu-Remaileh M., Shufaro Y., Gerson A., Ueda J., Deplus R., Fuks F., Shinkai Y., Cedar H., Bergman Y. (2008) Nat. Struct. Mol. Biol. 15, 1176–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong K. B., Maksakova I. A., Mohn F., Leung D., Appanah R., Lee S., Yang H. W., Lam L. L., Mager D. L., Schübeler D., Tachibana M., Shinkai Y., Lorincz M. C. (2008) EMBO J. 27, 2691–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marban C., Suzanne S., Dequiedt F., de Walque S., Redel L., Van Lint C., Aunis D., Rohr O. (2007) EMBO J. 26, 412–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.du Chéné I., Basyuk E., Lin Y. L., Triboulet R., Knezevich A., Chable-Bessia C., Mettling C., Baillat V., Reynes J., Corbeau P., Bertrand E., Marcello A., Emiliani S., Kiernan R., Benkirane M. (2007) EMBO J. 26, 424–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tachibana M., Sugimoto K., Fukushima T., Shinkai Y. (2001) J. Biol. Chem. 276, 25309–25317 [DOI] [PubMed] [Google Scholar]
- 31.Tachibana M., Sugimoto K., Nozaki M., Ueda J., Ohta T., Ohki M., Fukuda M., Takeda N., Niida H., Kato H., Shinkai Y. (2002) Genes Dev. 16, 1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rea S., Eisenhaber F., O'Carroll D., Strahl B. D., Sun Z. W., Schmid M., Opravil S., Mechtler K., Ponting C. P., Allis C. D., Jenuwein T. (2000) Nature 406, 593–599 [DOI] [PubMed] [Google Scholar]
- 33.Shi Y., Sawada J., Sui G., el Affar B., Whetstine J. R., Lan F., Ogawa H., Luke M. P., Nakatani Y., Shi Y. (2003) Nature 422, 735–738 [DOI] [PubMed] [Google Scholar]
- 34.Schultz D. C., Ayyanathan K., Negorev D., Maul G. G., Rauscher F. J., 3rd (2002) Genes Dev. 16, 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rice J. C., Briggs S. D., Ueberheide B., Barber C. M., Shabanowitz J., Hunt D. F., Shinkai Y., Allis C. D. (2003) Mol. Cell 12, 1591–1598 [DOI] [PubMed] [Google Scholar]
- 36.Tachibana M., Ueda J., Fukuda M., Takeda N., Ohta T., Iwanari H., Sakihama T., Kodama T., Hamakubo T., Shinkai Y. (2005) Genes Dev. 19, 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imai K., Asamitsu K., Victoriano A. F., Cueno M. E., Fujinaga K., Okamoto T. (2009) FEBS J. 276, 7124–7133 [DOI] [PubMed] [Google Scholar]
- 38.Yamaoka S., Courtois G., Bessia C., Whiteside S. T., Weil R., Agou F., Kirk H. E., Kay R. J., Israël A. (1998) Cell 93, 1231–1240 [DOI] [PubMed] [Google Scholar]
- 39.Nishio H., Walsh M. J. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 11257–11262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Imai K., Nakata K., Kawai K., Hamano T., Mei N., Kasai H., Okamoto T. (2005) J. Biol. Chem. 280, 26701–26713 [DOI] [PubMed] [Google Scholar]
- 41.Wozniak R. J., Klimecki W. T., Lau S. S., Feinstein Y., Futscher B. W. (2007) Oncogene 26, 77–90 [DOI] [PubMed] [Google Scholar]
- 42.Victoriano A. F., Asamitsu K., Hibi Y., Imai K., Barzaga N. G., Okamoto T. (2006) Antimicrob. Agents Chemother. 50, 547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clouse K. A., Powell D., Washington I., Poli G., Strebel K., Farrar W., Barstad P., Kovacs J., Fauci A. S., Folks T. M. (1989) J. Immunol. 142, 431–438 [PubMed] [Google Scholar]
- 44.Folks T. M., Clouse K. A., Justement J., Rabson A., Duh E., Kehrl J. H., Fauci A. S. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 2365–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Butera S. T., Perez V. L., Wu B. Y., Nabel G. J., Folks T. M. (1991) J. Virol. 65, 4645–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creusot F., Acs G., Christman J. K. (1982) J. Biol. Chem. 257, 2041–2048 [PubMed] [Google Scholar]
- 47.Kubicek S., O'Sullivan R. J., August E. M., Hickey E. R., Zhang Q., Teodoro M. L., Rea S., Mechtler K., Kowalski J. A., Homon C. A., Kelly T. A., Jenuwein T. (2007) Mol. Cell 25, 473–481 [DOI] [PubMed] [Google Scholar]
- 48.Folks T. M., Justement J., Kinter A., Schnittman S., Orenstein J., Poli G., Fauci A. S. (1988) J. Immunol. 140, 1117–1122 [PubMed] [Google Scholar]
- 49.Contreras X., Schweneker M., Chen C. S., McCune J. M., Deeks S. G., Martin J., Peterlin B. M. (2009) J. Biol. Chem. 284, 6782–6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Archin N. M., Espeseth A., Parker D., Cheema M., Hazuda D., Margolis D. M. (2009) AIDS Res. Hum. Retroviruses 25, 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mateescu B., Bourachot B., Rachez C., Ogryzko V., Muchardt C. (2008) EMBO Rep. 9, 267–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El Gazzar M., Yoza B. K., Chen X., Hu J., Hawkins G. A., McCall C. E. (2008) J. Biol. Chem. 283, 32198–32208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogawa H., Ishiguro K., Gaubatz S., Livingston D. M., Nakatani Y. (2002) Science 296, 1132–1136 [DOI] [PubMed] [Google Scholar]
- 54.Roopra A., Qazi R., Schoenike B., Daley T. J., Morrison J. F. (2004) Mol. Cell 14, 727–738 [DOI] [PubMed] [Google Scholar]
- 55.Sampath S. C., Marazzi I., Yap K. L., Sampath S. C., Krutchinsky A. N., Mecklenbräuker I., Viale A., Rudensky E., Zhou M. M., Chait B. T., Tarakhovsky A. (2007) Mol. Cell 27, 596–608 [DOI] [PubMed] [Google Scholar]
- 56.Dieudonné M., Maiuri P., Biancotto C., Knezevich A., Kula A., Lusic M., Marcello A. (2009) EMBO J. 28, 2231–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishida T., Hamano A., Koiwa T., Watanabe T. (2006) Retrovirology 3, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kauder S. E., Bosque A., Lindqvist A., Planelles V., Verdin E. (2009) PLoS Pathog. 5, e1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blazkova J., Trejbalova K., Gondois-Rey F., Halfon P., Philibert P., Guiguen A., Verdin E., Olive D., Van Lint C., Hejnar J., Hirsch I. (2009) PLoS Pathog. 5, e1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Estève P. O., Chin H. G., Smallwood A., Feehery G. R., Gangisetty O., Karpf A. R., Carey M. F., Pradhan S. (2006) Genes Dev. 20, 3089–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xin Z., Tachibana M., Guggiari M., Heard E., Shinkai Y., Wagstaff J. (2003) J. Biol. Chem. 278, 14996–15000 [DOI] [PubMed] [Google Scholar]