

Abstract
Mutations of the chloride channel cystic fibrosis transmembrane conductance regulator (CFTR) that impair its apical localization and function cause cystic fibrosis. A previous report has shown that filamin A (FLNa), an actin-cross-linking and -scaffolding protein, interacts directly with the cytoplasmic N terminus of CFTR and that this interaction is necessary for stability and confinement of the channel to apical membranes. Here, we report that the CFTR N terminus has sequence similarity to known FLNa-binding partner-binding sites. FLNa has 24 Ig (IgFLNa) repeats, and a CFTR peptide pulled down repeats 9, 12, 17, 19, 21, and 23, which share sequence similarity yet differ from the other FLNa Ig domains. Using known structures of IgFLNa·partner complexes as templates, we generated in silico models of IgFLNa·CFTR peptide complexes. Point and deletion mutants of IgFLNa and CFTR informed by the models, including disease-causing mutations L15P and W19C, disrupted the binding interaction. The model predicted that a P5L CFTR mutation should not affect binding, but a synthetic P5L mutant peptide had reduced solubility, suggesting a different disease-causing mechanism. Taken together with the fact that FLNa dimers are elongated (∼160 nm) strands, whereas CFTR is compact (6∼8 nm), we propose that a single FLNa molecule can scaffold multiple CFTR partners. Unlike previously defined dimeric FLNa·partner complexes, the FLNa-monomeric CFTR interaction is relatively weak, presumptively facilitating dynamic clustering of CFTR at cell membranes. Finally, we show that deletion of all CFTR interacting domains from FLNa suppresses the surface expression of CFTR on baby hamster kidney cells.
Keywords: Cell/Mutant, Cell/Surface, Channels/Chloride, Cytoskeleton, Cytoskeleton/Actin, Protein/Structure, CFTR, Filamin
Introduction
A balance of biosynthesis, subcellular trafficking, stability, and metabolism of the cystic fibrosis transmembrane conductance regulator (CFTR)3 determines the density of this molecule at the plasma membrane. Impairment of this balance can result in cystic fibrosis (CF), one of the most common lethal inherited autosomal recessive disorders affecting Caucasians (1–3). The CFTR gene encodes a chloride channel glycoprotein composed of 1,480 amino acid residues. The cytoplasmic N-terminal 80 residues are followed by membrane-spanning domain (MSD)-1, comprising six membrane-spanning helices, cytoplasmic nucleotide-binding domain (NBD)-1, one central regulatory R domain, MSD-2, and NBD-2. Some CFTR mutations that perturb the functions of these domains also result in CF. Although purified monomeric CFTR protein alone is sufficient to function as a chloride channel and an ATPase, it is not clear whether CFTR forms a multimer of two or more CFTR proteins and whether CFTR clustering affects channel activity. To date, there is evidence for monomeric as well as for dimeric CFTR (1, 4–7).
Filamin A and B (FLNa and b, respectively) are ubiquitously expressed nonmuscle isoforms of a family of actin-cross-linking proteins, and FLNc is expressed predominantly in adult striated muscle (8). All three filamins consist of an N-terminal spectrin-related actin-binding domain followed by two dozen Ig-like repeats (IgFLN) with two intervening calpain-sensitive hinges separating repeats 15 and 16 and repeats 23 and 24, and dimerize at the C-terminal repeat 24. FLNa, a dominant and the best characterized filamin isoform, cross-links F-actin to form orthogonal networks and attaches them to membrane receptors and numerous intracellular signaling molecules (8–10).
FLNa and b have recently been identified as proteins that directly associate with the N-terminal 25 residues of CFTR (CFTR1–25) (11). The CF-causing S13F mutation that disrupts this interaction in vivo leads to a reduced pool of CFTR at apical membrane sites and is prematurely delivered to lysosomes and degraded (11). These accumulated data indicate that a filamin-mediated connection of CFTR to the actin cytoskeleton is necessary for proper expression of CFTR at the membrane, consistent with a previous finding that inhibition of actin assembly decreases the cell surface density of CFTR (12).
All characterized atomic structures of FLNa·partner complexes demonstrate that the partners bind in a groove formed between the C and D β strands of FLNa Ig repeat (IgFLNa) and share a conserved motif for FLNa binding (13–16). This motif is also found in CFTR1–25. This finding and the fact that FLNa molecules consist of two subunits, which potentially tether two or more CFTR molecules, led us to reinvestigate the quaternary structure of the FLNa·CFTR complex in the plasma membrane as well as atomic structure of the binding interfaces.
In this study, we have mapped CFTR-binding sites on FLNa using CFTR1–25 peptide-coated beads and found that CFTR interacts with multiple repeats of FLNa. In silico modeling revealed detailed interface between C and D β strands of the repeats and CFTR, which is consistent with mutagenesis.
EXPERIMENTAL PROCEDURES
Protein Constructs
Human full-length FLNa was expressed using a baculovirus expression system (Invitrogen) in Sf9 insect cells and purified as described previously (10). The GST-, maltose-binding protein-His-, or His-tagged constructs were made by PCR using pGEX4T1, pMALc-HT(a or b), or pET-23a(+)-HT(a or b) plasmids (10), respectively, expressed in Escherichia coli, and purified using glutathione, amylose, or nickel-nitrilotriacetic acid affinity columns as described previously (10). The His-EGFP-tagged constructs were made using pFASTBAC-HT(a or b)-EGFP plasmids, which were modified from pFASTBAC-HT(a or b) by inserting EGFP cDNA using NcoI sites and expressed in Sf9 cells. All point or deletion mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene). The cDNA of the full-length HA-CFTR was amplified by PCR using oligonucleotides (5′-ACGCGTCGACATGCAGAGGTCGCCTCTGG, 3′-GTTTAGCGGCCGCTAAAGCCTTGTATCTTGCAC) containing the SalI and NotI sites, respectively, as primers. The PCR product was introduced into the pFASTBAC vector through the SalI and NotI sites to make pFASTBAC-HA-CFTR vector. Using this vector, HA-CFTR was expressed alone or co-expressed with FLAG-FLNa in insect cells. Protein concentration was measured by absorption at 280 nm using parameter calculated by ProtParam tool.
Peptides and Antibodies
The CFTR peptides were purchased from Tufts University core facility (Boston, MA) or Peptide 2.0, Inc. (Chantilly, VA). Anti-FLNa (clone 1–7, 3–14, 1–6, and 1–2) mouse mAbs were generated as described previously. Anti-HA rat mAb and was purchased from Roche Diagnostics Corporation (clone 3F10). Anti-HA mouse monoclonal (clone HA-11) was purchased from Covance. Anti-polyHis (clone HIS-1) mouse mAb conjugated with peroxidase and anti-GST mouse mAb were purchased from Sigma. CFTR mouse mAbs 570, 596, and 217 were kindly provided by J. Riordan (University of North Carolina). CFTR mAb clones 24-1 and 13-1 were purchased from R&D Systems. Anti-FLAG M2 mouse mAb immobilized on agarose beads was purchase from Sigma. Anti-Myc mAb (clone 9E10) was purchased from Abcam. Anti-Myc polyclonal antibody was purchased from GenScript.
Plasmids and Transfection
Deletion mutants of Myc-tagged FLNa were generated using the QuikChange site-directed mutagenesis kit. Wild type and mutant FLNa constructs were transfected into BHK cells alone or into CFTR-expressing BHK cells using Lipofectamine Plus reagent following the manufacturer's instructions (Invitrogen). BHK-CFTR cells were generated and maintained as described previously (17).
Structure-based Sequence Alignment and in Silico Modeling
All available IgFLNa domain structures (supplemental Table S1) were retrieved from the Protein Data Bank (PDB) (18). The sequence alignment between IgFLNa domains was made as follows. (i) Structures of all available IgFLNa domains were superimposed with VERTAA in Bodil (19). (ii) The formed structure-based sequence alignment was frozen, and all 24 Ig-like domains of FLNa were aligned against it. Protein structure-based matrix (20) was used with a gap penalty of 40. Similarly, the available information for FLN-binding peptides was used to build a structure-based sequence alignment, and then the CFTR peptide was aligned against it. CFTR peptide-binding IgFLNa domains were modeled into peptide-binding conformation by using the structure of IgFLNa 21 with bound integrin β7 peptide (PDB code 2brq) (14) by using JACKAL (available at the Columbia University web site). The CFTR·peptide model was made with HOMODGE in Bodil (19). The models of IgFLNa domains with bound CFTR peptide were prepared by using the complex structure of IgFLNa21 with β7 integrin peptide as a template.
CFTR Peptide Pulldown Assay
Various concentration of FLNa constructs were incubated with increasing concentrations of wild-type or S13F mutant biotin·CFTR1–25 peptides immobilized on 20 μl of streptavidin-agarose (50% v/v slurry; Sigma) in 400 μl of TTBS buffer (50 mm Tris-HCl, 150 mm NaCl, 0.1% (w/v) Triton X-100, 0.1 mm β-mercaptoethanol, 0.1 mm EGTA, pH 7.4) containing 2% bovine serum albumin for 1 h at 25 °C. Alternatively, CFTR8–25 peptide (Cys-KASVVSKLFFSWTRPILR) immobilized on Sulfo-Link coupling beads (1 mg of peptide/ml of gel; Pierce) was used. The beads were sedimented and washed three times with binding buffer. Proteins bound to the beads were solubilized in SDS sample buffer and separated by 9.5% or 12.5% (w/v) SDS-PAGE followed by immunoblotting using rabbit polyclonal antibodies against His (Sigma), GST (Sigma), or FLNa. To estimate apparent dissociation constants, the binding of increasing amounts of purified His-EGFP-IgFLNa23 to CFTR8–25 peptide was quantified by densitometry and plotted versus input concentration. The data were fitted to a one-site binding model using GraphPad Prism version 5 for Macintosh (GraphPad Software).
To test the interaction of CFTR N-terminal peptides with expressed Myc-FLNa protein in BHK cells, 1 μg of peptide conjugated to 10 μl of streptavidin-agarose beads was tumbled for 2 h at 4 °C with 100 μg of cell lysate. The beads were washed five times for 5 min/wash with high salt Chaps buffer (50 mm Tris, pH 7.6, 500 mm NaCl, 0.2% Chaps (J. T. Baker, Inc.), 10 mm EDTA), solubilized in SDS sample buffer, and separated by a 3–8% Tris-acetate gel (Invitrogen). Bound proteins were identified by immunoblotting using an anti-Myc mAb used at 1:1,000 dilution followed by analysis using the Odyssey imaging system (LiCor).
CFTR Immunoprecipitation
BHKcells stably expressing Extope-CFTR (BHK-CFTR) were solubilized in Cell Lytic lysis buffer (Sigma) containing protease inhibitors (HaltTM, Pierce). 1 mg protein lysate was incubated with 2 μg each of monoclonal CFTR antibody clones 24-1 and 13-1 conjugated to protein G and incubated for 2 h at 4 °C. The beads were sedimented and washed with lysis buffer. Bound proteins were solubilized in SDS sample buffer and separated by a 3–8% Tris-acetate gel (Invitrogen). Immunoblotting was performed using an anti-Myc antibody at 1:1,000 dilution to detect FLNa proteins and a combination of HA, 596 and 217 antibodies each at 1:10,000 dilution to detect CFTR.
Fluorescence Microscopy
Transfected BHK cells were fixed in 2% formaldehyde prepared in phosphate-buffered saline (PBS) for 15 min, followed by permeabilization with PBS containing 0.25% Triton X-100 for 5 min. The specimens were blocked in PBS containing 2% bovine serum albumin for 1 h at 37 °C and then stained in tandem with polyclonal Myc and monoclonal HA antibodies (1:1,000 and 1:500, respectively) by incubation overnight at 4 °C. Primary antibodies were visualized by incubation with Alexa Fluor 488 rabbit- and 594 mouse- (Invitrogen) conjugated antibodies (for 1 h at 37 °C).
Statistics
Quantitive data were analyzed by one-way analysis of variance using GraphPad Prism (Mac version 5). Multiple comparisons were made using Dunnett's test with p < 0.05 considered significant.
RESULTS
Mapping of the CFTR Interaction Domains of FLNa
His-tagged FLNa fragments covering the entire FLNa molecule were generated and used for CFTR peptide pulldown assays (Fig. 1A). The N-terminal CFTR1–25 peptide pulled down dimerized constructs containing repeats 8–15 and 16–24, but not FLN ABD or a construct composed of repeats 1–8. The results suggested that CFTR interacts with FLNa domains between repeats 9 and 23 (Fig. 1B). Although dimerized FLNa fragments were used for the experiments to synchronize the valence of repeats 1–8 and 8–15 with that of the 16–24 dimer, monomeric forms of the FLNa fragments composed of 8–15 and 16–23, but not repeats 1–8 (data not shown), also bound the CFTR peptide. FLNa fragments did not bind the CF-causing S13F mutant CFTR peptide (Fig. 1B), consistent with previous results (11).
FIGURE 1.

Mapping of CFTR-binding sites on FLNa. A, diagram of human FLNa and its recombinant fragments fused to His6 tag. B, association of the CFTR1–25 peptide with the His-tagged FLNa fragments. Biotinylated wild-type or S13F mutant CFTR peptides (1.0 μm) were incubated with the His-tagged FLNa fragments (0.1 μm). The peptides were pulled with streptavidin-coated beads, and bound protein was detected by immunoblotting (IB) using anti-His mAb conjugated with peroxidase.
To identify a CFTR-binding domain on repeats 8–15, individual FLNa repeats were generated as GST fusion proteins and incubated with the CFTR peptide immobilized on beads. Wild-type CFTR peptide pulled down only IgFLNa9 (Fig. 2A), and deletion of the repeat 9 from His-8–15 + 24 significantly diminished the binding interaction to the CFTR peptide (Fig. 2B). By the same method, CFTR-binding domains were mapped on IgFLNa 17, 19, 21, and 23 within the rod 2 segments (Fig. 3A). When these repeats were fused to maltose-binding protein His, IgFLNa 23 and 21 were found to be a major and minor CFTR-binding domain, respectively, within the rod 2. Repeats 17–20 barely bound CFTR peptide (data not shown). However, deletion of repeat 21 or 23 (supplemental Fig. S1A) or both 21 and 23 (supplemental Fig. S1B) from full-length FLNa was not sufficient to diminish the binding. Further deletions of all odd-numbered repeats in the rod 2 abolished CFTR binding (Fig. 3B).
FIGURE 2.

Localization of CFTR-binding site on IgFLNa9–16. A, GST immunoblot of CFTR1–25 peptide pulldowns from purified GST-tagged FLNa repeats. Biotinylated wild-type or S13F mutant CFTR peptides (1.0 μm) were incubated with the GST-tagged FLNa fragments (0.1 μm). The peptides were pulled with streptavidin-coated beads, and bound protein was detected by immunoblotting (IB) using anti-GST mAb. B, His-8–15 + 24 and deletion mutant (Δ9, deletion of IgFLNa9) were pulled down with increasing amounts of the CFTR1–25 peptide. Bound protein was detected by immunoblotting using anti-His mAb conjugated with peroxidase. WT, wild type.
FIGURE 3.

Localization of CFTR-binding site on the rod 2 segment, IgFLNa15–24. A, GST immunoblot (IB) of CFTR1–25 peptide pulldowns from purified GST-tagged FLNa repeats. B, His-16–24 and deletion mutant (Δ17,19,21,23 deletion of IgFLNa17, 19, 21, and 23) were pulled down with increasing amounts of the wild-type (WT) or S13F mutant CFTR1–25 peptide. Bound protein was detected by immunoblotting using anti-His mAb conjugated with peroxidase.
Identification of a FLNa-binding Site on CFTR
Sequence alignment of the N terminus of CFTR with known FLN-binding motifs on other proteins suggested two potential sites within the first 25 residues of CFTR (Fig. 4A). To test these possibilities, CFTR N-terminal peptides encompassing residues 1–17 and 8–25 were synthesized and used to assess FLNa binding in vitro. Fig. 4C shows that CFTR peptide 8–25, but not 1–17, binds FLNa, indicating that CFTR alignment contains a FLNa-binding site. Amino acid changes reflecting known CF mutations within this peptide (S13F, L15P, or W19C) prevented FLNa binding (Fig. 5C). P5L did not affect the binding interaction of the peptide with FLNa.
FIGURE 4.

Location of FLNa-binding site of CFTR. A, amino acid sequence alignment of the CFTR1–25 (human) and FLNa-binding sites of human glycoprotein Ibα (GPIbα), β integrins, FilGAP, and migfilin. Amino acids indicated with asterisks face a glove generated between the C and D strands of the IgFLNa domain which are mainly involved in binding interaction. Solid and dotted underlines indicate residues 1–17 and 8–25 of CFTR peptides used for binding assay in C. Red and blue amino acids indicate residues mutated in CF patients (P5L, S13F, L15P, and W19C). B, comparison of the amino acid sequence similarity predicted from the full-length nucleotide sequences of the CFTR of monkey (UniProt accession number, Q7JII7), bovine (P35071), sheep (Q00555), horse (Q2QLA3), pig (Q6PQZ2), dog (Q5U820), rabbit (Q00554), rat (P34158), mouse (P26361), and chicken (A0M8U4). Amino acids indicated with asterisks are mainly involved in binding interaction. Residues differ from human CFTR (green). C, CFTR peptide pulldown assay with purified FLNa. Bound FLNa was detected by immunoblotting (IB) with anti-FLNa mAb.
FIGURE 5.

Structure of FLNa-CFTR binding interface. A, model of CFTR (blue, stick model is indicated with gray color) interaction with IgFLNa23 (gray; CD strands, green). B, point mutation in IgFLNa23 (M2474E) abolishes the binding to CFTR. Increasing amounts of wild-type (WT) or M2474E mutant His-EGFP-IgFLNa23 were incubated with CFTR peptide immobilized on Sepharose beads. Bound protein was detected by immunoblotting (IB) using anti-His mAb conjugated with peroxidase. C, effect of CF-causing point mutations of CFTR on FLNa binding. One ml of PBS was added to 1 mg of biotinylated CFTR1–20 peptides (wild-type, P5L, L15P, and W19C), and the solution was centrifuged at 15,000 × g for 10 min at room temperature. All of the peptides were soluble except for P5L mutant peptide (asterisk). The supernatant (2 μl, 2 μm at final) was incubated with 10 nm FLNa in 400 μl of binding buffer, and the peptide was pulled down with streptavidin-agarose. Bound FLNa was detected by immunoblotting (IB) with anti-FLNa mAb. D, structure-based alignment of IgFLNa. Arrows indicate β strands. Green letters indicate amino acid residues of IgFLNa23 responsible for specific interaction with CFTR and corresponding residues of other IgFLNa. The blue box indicates a conserved GPS/C sequence at the turn between C and D strands. Mutation of IgFLNa23 Met2474 (red) to Glu disrupts the CFTR interaction as shown in B. IgFLNa4 Phe-611 (blue) is a bulky amino acid and unique to IgFLNa4. Note that CFTR peptide has higher affinity to top 5 IgFLNa than IgFLNa12, whereas the peptide does not interact with IgFLNa4 and 8 in dose-dependent manner.
In Silico Model of the IgFLNa·CFTR Complex
The CD faces of IgFLNa domains are common binding sites for all FLNa·partners for which atomic structures have been resolved: e.g. IgFLNa17 with glycoprotein Ibα (PDB code 2bp3) (13); IgFLNa21 with integrins β2 (PDB code 2jf1(21), β7 (PDB code 2brq) (14), and migfilin (PDB code 2w0p) (15); and IgFLNa23 with FilGAP (16). Because the amino acid sequence of CFTR could be aligned to the FLNa-binding motif on other proteins (Fig. 4A), we modeled the IgFLNa complexed with CFTR 10–19 peptide based on these known structures (Fig. 5A). The model showed that the CFTR peptide binds to the CD face of the IgFLNa domain by forming 10 main-chain interactions and four side-chain interactions of the CFTR-peptide (IgFLNa23 is shown as a representative in Fig. 5A): (i) the hydroxyl-group of Ser-13 hydrogen bonds to the main-chain carbonyl oxygen atom of Val-2472; (ii) aliphatic residue Leu-15 binds into hydrophobic groove formed by Val-2464 and Met-2474 in IgFLNa23 and Phe-17 in CFTR; (3) Phe-17 binds in between the IgFLNa23 residues Leu-2462, Cys-2476, and Tyr-2483 as well as Leu-15 and Trp-19 of CFTR; (iv) Trp-19 packs against the bottom of the groove that is in between the beginning of the C strand and end of the D strand.
In this model, the S13F mutation of CFTR is incapable of forming hydrogen-bonding interactions with the main-chain carbonyl oxygen atom of Val-2472 (Fig. 5A), consistent with in vitro binding experiments (Figs. 1–4). To validate further the binding mode suggested by the modeling, the hydrophobic Met-2474 residue was mutated to a negatively charged glutamate in IgFLNa23. As predicted, the point mutation M2474E abolished the interaction of IgFLNa23 with CFTR peptide (Fig. 5B). The model also predicted that CF-causing L15P and W19C but not P5L mutants of CFTR perturb the interaction with FLNa. Indeed, L15P and W19C synthetic CFTR1–20 peptides did not or barely bound FLNa (Fig. 5C). The result also eliminated one of the alignments of CFTR (the second from the bottom in Fig. 4A), as W19C should not affect the binding interaction of this alignment. The model predicted that P5L mutation should not have an effect on the binding. Although synthetic P5L CFTR1–20 peptide was barely soluble in PBS at 1 mg/ml, a soluble portion of the peptide bound FLNa (Fig. 5C). The wild-type, L15P, and W19C peptides were soluble at least at concentration of 1 mg/ml in PBS.
Both structural comparison of available IgFLN domains (available for domains 10–24; supplemental Table S1) and sequence comparison of all 24 IgFLNa domains indicate that the sequence length and amino acid composition at the turn between C and D strands have an important role in the definition of whether this CD face can accept peptides to bind as an additional β strand. Seven of 24 IgFLNa domains (4, 9, 12, 17, 19, 21, 23) have very similar characteristics at the turn between C and D strands and contain a conserved GPS/C sequence (Fig. 5D). In addition, the IgFLNa8 contains a similar sequence at the equivalent position, but has additional amino acids at this turn (Fig. 5D). These characters are consistent with the pulldown data demonstrating that IgFLNa 9, 17, 19, 21, and 23 interact with CFTR peptide except for IgFLNa 4 and 12. Therefore, we expressed these IgFLNa domains fused to His-EGFP in insect cells and compared the binding strength of the each repeat to CFTR peptide (supplemental Fig. S2). IgFLNa9, 12, 17, 19, 21 and 23 dose-dependently bound CFTR with different apparent affinities, whereas IgFLNa8 lacked binding to CFTR peptide (supplemental Fig. S2), which was consistent with previous observations (Figs. 2B and 3B). Although IgFLNa4 fused to His-EGFP nonspecifically bound CFTR (data not shown), repeats 1–8 did not bind CFTR (Fig. 1B), indicating that IgFLNa4 does not form a tight complex with CFTR. We expressed FLAG-FLNa deleted for all major CFTR-interacting repeats (9, 17, 19, 21, and 23) in insect cells and purified as a single band on SDS-PAGE (data not shown). The deletion of these repeats significantly diminished interaction with a CFTR peptide immobilized on Sepharose beads (Fig. 6A). This residual binding interaction was lost upon deletion of all structurally similar repeats (Fig. 6B).
FIGURE 6.

Anti-FLAG immunoblotting (IB) of CFTR peptide pulldowns from purified FLAG-FLNa, FLAG-FLNa lacking repeats 9, 17, 19, 21, and 23 (1 nm each) (A) or repeats 4, 9, 12, 17, 19, 21, and 23 (1 nm each) (B). WT, wild type.
Based on these results, we also generated in silico models of IgFLNa 9, 17, 19, and 21 complexed with CFTR peptide (supplemental Fig. S3) to understand the detailed differences in the interactions between each of these IgFLNa domains with bound CFTR peptide. We questioned whether full-length FLNa mutants in cells could interact with CFTR N-terminal peptides. To this end, we performed CFTR peptide pulldown experiments using lysates from BHK-CFTR cells expressing Myc-FLNa fusion constructs (Fig. 7A). As anticipated, Myc-FLNa wild-type protein bound to wild-type, but not the S13F mutant CFTR peptide. Mutant Myc-FLNa lacking repeats 4, 9, 17, 19, 21, and 23 also bound wild-type CFTR peptide, although the binding interaction was somewhat diminished (Fig. 7A). All interactions with wild-type CFTR peptide were abolished in cells expressing Myc-FLNa lacking repeats 9, 12, 17, 19, 21, and 23 (Fig. 7A).
FIGURE 7.
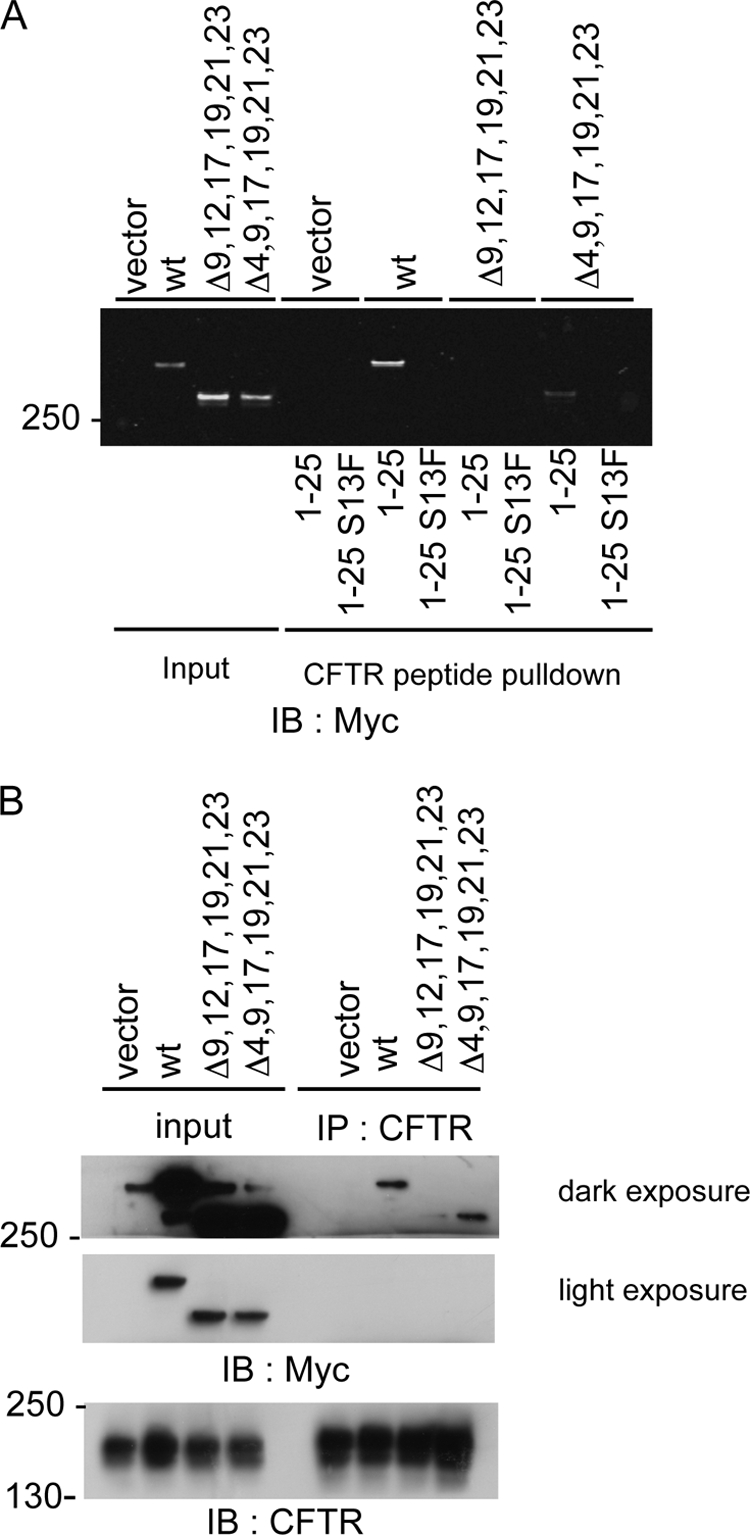
Interaction of FLNa with CFTR in vivo. A, indicated Myc-tagged proteins were expressed in BHK cells. Lysates from these cells were incubated with N-terminal wild type (wt) (1–25) or mutant (1–25 S13F) CFTR peptides. Bound proteins were identified by Western blotting (IB) for Myc. B, indicated Myc-tagged proteins were expressed in BHK cells stably expressing CFTR. CFTR was immunoprecipitated, and samples were analyzed by Western blotting for CFTR or Myc.
To assess the importance of FLNa repeats 9, 12, 17, 19, 21, and 23 in a physiological context, full-length Myc-tagged FLNa constructs were expressed in BHK-CFTR cells, allowing the FLNa and CFTR interaction to be assessed by co-immunoprecipitation experiments (Fig. 7B). As reported above, interactions between CFTR and wild-type FLNa were found (Fig. 7B). Unexpectedly, FLNa Δ(4,9,17,19,21,23) also co-immunoprecipitated with CFTR. This residual interaction was lost when repeat 12 was deleted instead of repeat 4 (Fig. 7B). Taken together, these results, therefore, indicate that FLNa repeat 12 can interact with CFTR.
Exposure of the Cryptic Integrin-binding Site in Repeat 21 of FLNa Does Not Enhance CFTR Binding
A deletion of 41 amino acids between repeats 19 and 20 of FLNa has been shown to expose the CD face of repeat 21 for integrin β chain binding (22). This FLNa variant did not affect CFTR, e.g. it bound the CFTR peptide with the same affinity as wild-type FLNa (supplemental Fig. S4).
FLNa and CFTR Expressed in Sf9 Cells Do Not Form Tight Complex
We have attempted to isolate FLAG-FLNa-HA·CFTR complex co-expressed in Sf9 insect cells and to reconstitute the complex using independently purified proteins for further analysis such as electron microscopy and determination of binding stoichiometry. However, FLAG-FLNa and HA-CFTR did not pull each other down (supplemental Fig. S5). Purified FLAG-FLNa also did not pull down HA-CFTR in cell lysates from HA-CFTR-expressing insect cells (data not shown).
FLNa Repeats 9, 12, 17, 19, 21, and 23 Are Necessary for Optimal Surface Expression of CFTR
We previously reported that an interaction with FLNa is important for the expression of CFTR on the cell surface; the surface expression of the CFTR mutant S13F, which is defective in FLNa binding, is dramatically reduced. Hence, we examined here whether FLNa mutants defective in CFTR binding still influence CFTR expression on the cell surface. CFTR with an extracellular HA tag was transiently co-expressed along with Myc-tagged FLNa in BHK cells. Cells expressing FLNa protein or CFTR were identified by immunofluorescence microscopy using monoclonal HA and polyclonal Myc antibodies and appropriate second antibodies (Fig. 8A). In the absence of exogenous FLNa, ∼50% of the BHK cells exhibited detectable CFTR staining on their plasma membrane (Fig. 8B). Co-expression of FLNa with CFTR increased this number to ∼70% of the BHK cells. FLNa and CFTR also co-localized at membranous sites in these cells. In contrast, FLNa Δ(9,12,17,19,21,23) expression dramatically reduced the number of BHK cells having surface CFTR (28%), a statistically significant decrease from the control cells (Dunnett's test, p < 0.05). Furthermore, the spatial co-localization of the FLNa mutant and exogenous CFTR was lost. We also observed the loss of co-localization and decreased surface CFTR levels in cells co-expressing FLNa Δ(4,9,17,19,21,23). However, this decrease was not statistically significant compared with vector control cells (Fig. 8B). As expected, co-expression of wild-type FLNa had no effect on the surface localization of the FLNa binding-defective CFTR mutant S13F. In conclusion, these data support the hypothesis that multiple FLNa repeats are involved in CFTR function.
FIGURE 8.

Defects in the CFTR-filamin interaction lower the surface expression of wild-type CFTR. A, BHK cells were co-transfected with the indicated myc-tagged constructs and either HA-tagged wild type or S13Fmutant CFTR. Cell surface CFTR was assessed by labeling with a monoclonal HA antibody followed by Alexa Fluor 594-labeled secondary antibodies. Transfected cells were identified by staining with a myc polyclonal antibody. B, myc-positive cells also containing overexpressed CFTR were assessed for CFTR surface expression with at least 50 transfected cells counted/individual experiment *, p < 0.05 versus vector, n = 3. Error bars, S.D.
DISCUSSION
A previous report has shown that the N-terminal 25 residues of CFTR directly interact with FLNa and this interaction regulates the plasma membrane expression and metabolic stability of CFTR (11). Here, we have mapped the FLNa·CFTR-binding sites and generated in silico three-dimensional models that help to explain how CF-causing mutations disrupt FLNa binding. We have also demonstrated that CFTR interacts with multiple Ig repeats of FLNa in vitro, and modeling suggests multiple fits for the CFTR peptide on FLNa. Moreover, we have shown that deletion of the CFTR-interacting domains of FLNa diminish the surface expression of CFTR in vivo. We discuss the implications of these findings within the context of its effect on the quaternary structure of the FLNa·CFTR complex at cell membrane.
Structure of the FLNa·CFTR-binding Interface
Our present data strongly suggest that residues 11–19 of CFTR interact with the CD face of FLNa Ig repeats 9, 12, 17, 19, 21, and 23. These interaction sequences are similar to those of FLNa-binding sites of all FLNa·partners for which atomic structures have been resolved (13–16, 21). In the in silico models (Fig. 5A and supplemental Fig. S3), the amino acids indicated with asterisks in Fig. 4A (Val-11, Ser-13, Leu-15, Phe-17, and Trp-19) face the groove formed between the C and D strands of the IgFLNa repeats and are highly conserved in all vertebrate species compiled in Fig. 4B. Val-11, Ser-13, and Leu-15 are identical to the corresponding amino acids of migfilin and glycoprotein Ibα, which make hydrophobic contacts with FLNa (13, 15). Phe-11 of rat and mouse CFTR is also identical to the corresponding amino acids of glycoprotein Ibα, integrin β2, and FilGAP. Val-11 can be substituted with isoleucine, as seen in pig and chicken. Although Phe-17 and Trp-19 make important contacts with the CD strands of IgFLNa (Fig. 5A), these amino acids are not seen in the corresponding location of other FLNa-binding partners shown in Fig. 4A, suggesting that these amino acids can be also used for identifying potential FLNa-binding partners.
Structural Constraints for CFTR Binding
Comparisons of FLNa Ig repeats that do or do not bind CFTR reveal features in the individual IgFLNas which explain differences in affinity and specificity (Fig. 5D). All nonbinding repeats, with the sole exception of repeat 4, have small sequence insertions at the beginning of strand D that alter the shape of the groove between strands C and D. The C strand in IgFLNa 4 has Phe at position (611), whereas repeats capable of binding CFTR have either Leu or Val at the equivalent position. This Phe residue extends toward the CD face and, due to its large size, can hinder or block binding of CFTR peptide. Otherwise, IgFLNa 4 CD face seems to be very similar to the other studied domains.
Affinity for CFTR is positively modulated by a change from cysteine to a tyrosine at the beginning of the E strand. This tyrosine stacks with Phe-17 and Trp-19 of the CFTR peptide (Fig. 5B), which the smaller cysteine cannot. Thus, the binding affinity of IgFLNa23 for the CFTR peptide is the highest of the repeats. A different change in IgFLNa21 partially compensates for this lack of Tyr with Phe-2476 in the D strand (IgFLNa23 numbering), which can stack with Phe-17 of CFTR. This sequence position is Cys in the other studied domains except for IgFLNa19, where it is a Thr. Again, neither Cys nor Thr can stack with Phe-17 of CFTR.
Structural Basis for Mutations That Impair the FLNa-CFTR Interaction
Three missense mutations in the FLNa-binding site of CFTR have been reported: S13F, L15P, and W19C. The S13F mutation eliminates hydrogen bonding between the main-chain carbonyl oxygen atom of Val or Ala (Val-2472:IgFLNa23 numbering) in IgFLNa repeats 9, 12, 17, 19, 21, and 23, and the bulky Phe residue cannot stack with Val or Ala. The model also predicts that CF-causing L15P and W19C mutations in CFTR perturb the interaction with FLNa. Mutation of Leu-15 to Pro dramatically changes the structure of the CFTR peptide. Because Trp-19 contributes the highest hydrophobic contact surface area in the interface (e.g. glycoprotein Ibα Val-571, 13.67%), loss of this contact by mutation to Cys is disruptive. However, unlike the S13F mutant of CFTR, which is still expressed, albeit reduced ∼50% compared with wild type, on the cell surface in a mature glycosylated form, the W19C mutant of CFTR remains immature and is retained in the endoplasmic reticulum (ER) (11), suggesting that it causes the disease without disrupting the interaction with FLNa. Our modeling further predicted that P5L mutation should not affect FLNa binding. Unfortunately, the insolubility of the synthetic P5L CFTR1–20 peptide made testing this prediction unfeasible. It is possible that the P5L mutation induces aggregation of CFTR molecules in the ER, preventing transfer to Golgi (11).
Assembly State of FLNa·CFTR at the Cell Membrane
Knowledge of the assembly state of CFTR at the cell membrane is essential to understand the physiology and pathophysiology of these molecular interactions (4–7). Our findings suggest that individual FLNa molecules could spatially tether multiple CFTR molecules in the plasma membrane because CFTR peptides interact with six FLNa Ig repeats and because FLNa is an extended filamentous molecule (10). The compact size of the CFTR molecule (23, 24) would facilitate such interactions. However, previous reports have failed to detect stable complexes of CFTR molecules (1). Single molecule fluorescent imaging experiments of CFTR on living cells recently concluded that CFTR exists as a highly motile monomer on the cell surface (7, 11), indicating that FLNa does not simultaneously cluster multiple CFTR molecules. Although FLNa and CFTR expressed in mammalian cells can co-immunoprecipitate (11), our attempt to reconstitute FLNa·CFTR complexes expressed in Sf9 cells was unsuccessful. This failure suggests that the FLNa-binding site of CFTR expressed in insect cells is not properly exposed for FLNa binding presumably due to lack of a CFTR maturation process in Sf9 cells (25).
We have also been unable to perturb endogenous FLNa-CFTR interaction in BHK cells stably expressing CFTR by overexpressing GFP-tagged IgFLNa repeats. Hence, we were unable to deplete CFTR from membranes following expression of these tagged repeats. This could be attributed to mislocalization of the small tandem repeats in cells, capture by other filamin-binding partners, or an intrinsic weak affinity to CFTR due to a relatively high degree of diffusional freedom compared with full-length FLNa.
β-Integrin binding to the CD face of IgFLNa21 is intramolecularly limited (cryptic) by the binding of strand A of repeat 20 in this CD groove. This cryptic binding site is believed to become exposed by mechanical displacements of the repeats in the region of FLNa or by deletion of 41 amino acids in this region that encodes the A strand of repeat 20 (22). However, the FLNa truncate lacking these 41 amino acids interacted equally well with CFTR as wild-type FLNa in vitro, suggesting that mechanical forces do not greatly affect FLNa-CFTR interaction. Although expression level of CFTR varies depending on location and has not been quantitatively determined, relatively high expression of FLNa (low CFTR:FLNa molar ratio) could affect on assembly state of CFTR. In any case, the restricted and dynamic FLNa·CFTR clustering may control exquisite and regulated expression of CFTR at cell membrane, and FLNa may dynamically compartmentalize CFTR with other relevant FLNa-binding partners such as receptors and signaling molecules (26, 27).
A previous report demonstrated that FLNa does not interact with ER-retained ΔF508 CFTR, the most common CF-causing mutant (∼90% of CF patients) (28), presumably because FLNa does not interact with CFTR in the ER. Although small-molecule correctors allow ΔF508 CFTR to escape the ER and the rescued ΔF508 CFTR can reach the cell surface and function as chloride channel (29), the mutant CFTR is unstable and readily degraded at the surface (30). Therefore, great attention has been given to identify a novel compound that enhances CFTR stability at cell membrane. The structural basis of the FLNa-CFTR interaction demonstrated in this study will be useful for developing such therapeutic drugs.
The accompanying article by Smith et al. (32) comes to similar conclusions regarding the multiple binding sites on FLNa for the CFTR. However, the binding affinities reported in the Smith et al. paper for the different FLNa repeats for CFTR are considerably weaker than those determined in this work. We estimate affinities using pulldown assays, whereas Smith et al. used NMR titration. The weak binding affinities determined by the Smith group could explain their inability to demonstrate the critical nature of the W19C mutation in repeat 21. In the NMR titration, the two interacting molecules have free movement, whereas in the pulldown assay, CFTR peptide is immobilized on beads at relatively high density. Hence, it is possible that the concentration and restriction of dynamic motility could increase the apparent binding affinity. A similar affinity difference has been reported with integrin β7-IgFLNa21 interaction (31). These measured affinities in vitro may be more applicable to the physiological situation in vivo where CFTR and FLNa are concentrated at or in the plasma membrane and dynamically restricted. Binding in vivo will also be influenced by the presence or absence of other FLNa partners that could compete for the same sites.
Supplementary Material
Acknowledgments
We thank our collaborators for sharing reagents, Theresa Collins and Lindsey Buckingham for technical assistance.
This work was supported by National Institutes of Health Grants HL-19429 (to T. P. S.) and HL-56252 (to J. H. H.). This work was also supported by the HUSEC Seed Fund for Interdisciplinary Science (T. P. S. and F. N.), the Sigrid Jusélius Foundation (O. T. P.), the University of Jyväskylä (E. N.), and the Finnish IT Center for Science Computational Grant for Project jyy2516 (to O. T. P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and Table S1.
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CF
- cystic fibrosis
- FLNa
- filamin A
- IgFLNa
- filamin A Ig-like domain
- GST
- glutathione S-transferase
- EGFP
- enhanced green fluorescent protein
- HA
- hemagglutinin
- BHK
- baby hamster kidney
- PDB
- Protein Data Bank
- Chaps
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- PBS
- phosphate-buffered saline
- mAb
- monoclonal antibody.
REFERENCES
- 1.Riordan J. R. (2005) Annu. Rev. Physiol. 67, 701–718 [DOI] [PubMed] [Google Scholar]
- 2.Okiyoneda T., Lukacs G. L. (2007) Biochim. Biophys. Acta 1773, 476–479 [DOI] [PubMed] [Google Scholar]
- 3.Riordan J. R. (2008) Annu. Rev. Biochem. 77, 701–726 [DOI] [PubMed] [Google Scholar]
- 4.Chen J. H., Chang X. B., Aleksandrov A. A., Riordan J. R. (2002) J. Membr. Biol. 188, 55–71 [DOI] [PubMed] [Google Scholar]
- 5.Ramjeesingh M., Ugwu F., Li C., Dhani S., Huan L. J., Wang Y., Bear C. E. (2003) Biochem. J. 375, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li C., Roy K., Dandridge K., Naren A. P. (2004) J. Biol. Chem. 279, 24673–24684 [DOI] [PubMed] [Google Scholar]
- 7.Haggie P. M., Verkman A. S. (2008) J. Biol. Chem. 283, 23510–23513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stossel T. P., Condeelis J., Cooley L., Hartwig J. H., Noegel A., Schleicher M., Shapiro S. S. (2001) Nat. Rev. Mol. Cell Biol. 2, 138–145 [DOI] [PubMed] [Google Scholar]
- 9.Popowicz G. M., Schleicher M., Noegel A. A., Holak T. A. (2006) Trends Biochem. Sci. 31, 411–419 [DOI] [PubMed] [Google Scholar]
- 10.Nakamura F., Osborn T. M., Hartemink C. A., Hartwig J. H., Stossel T. P. (2007) J. Cell Biol. 179, 1011–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thelin W. R., Chen Y., Gentzsch M., Kreda S. M., Sallee J. L., Scarlett C. O., Borchers C. H., Jacobson K., Stutts M. J., Milgram S. L. (2007) J. Clin. Invest. 117, 364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantiello H. F. (1996) Exp. Physiol. 81, 505–514 [DOI] [PubMed] [Google Scholar]
- 13.Nakamura F., Pudas R., Heikkinen O., Permi P., Kilpeläinen I., Munday A. D., Hartwig J. H., Stossel T. P., Ylänne J. (2006) Blood 107, 1925–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiema T., Lad Y., Jiang P., Oxley C. L., Baldassarre M., Wegener K. L., Campbell I. D., Ylänne J., Calderwood D. A. (2006) Mol. Cell 21, 337–347 [DOI] [PubMed] [Google Scholar]
- 15.Lad Y., Jiang P., Ruskamo S., Harburger D. S., Ylänne J., Campbell I. D., Calderwood D. A. (2008) J. Biol. Chem. 283, 35154–35163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura F., Heikkinen O., Pentikäinen O. T., Osborn T. M., Kasza K. E., Weitz D. A., Kupiainen O., Permi P., Kilpeläinen I., Ylänne J., Hartwig J. H., Stossel T. P. (2009) PLoS ONE 4, e4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gentzsch M., Chang X. B., Cui L., Wu Y., Ozols V. V., Choudhury A., Pagano R. E., Riordan J. R. (2004) Mol. Biol. Cell 15, 2684–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berman H. M., Battistuz T., Bhat T. N., Bluhm W. F., Bourne P. E., Burkhardt K., Feng Z., Gilliland G. L., Iype L., Jain S., Fagan P., Marvin J., Padilla D., Ravichandran V., Schneider B., Thanki N., Weissig H., Westbrook J. D., Zardecki C. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 899–907 [DOI] [PubMed] [Google Scholar]
- 19.Lehtonen J. V., Still D. J., Rantanen V. V., Ekholm J., Björklund D., Iftikhar Z., Huhtala M., Repo S., Jussila A., Jaakkola J., Pentikäinen O., Nyrönen T., Salminen T., Gyllenberg M., Johnson M. S. (2004) J. Comput. Aided Mol. Des. 18, 401–419 [DOI] [PubMed] [Google Scholar]
- 20.Johnson M. S., Overington J. P. (1993) J. Mol. Biol. 233, 716–738 [DOI] [PubMed] [Google Scholar]
- 21.Takala H., Nurminen E., Nurmi S. M., Aatonen M., Strandin T., Takatalo M., Kiema T., Gahmberg C. G., Ylänne J., Fagerholm S. C. (2008) Blood 112, 1853–1862 [DOI] [PubMed] [Google Scholar]
- 22.Lad Y., Kiema T., Jiang P., Pentikäinen O. T., Coles C. H., Campbell I. D., Calderwood D. A., Ylänne J. (2007) EMBO J. 26, 3993–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosenberg M. F., Kamis A. B., Aleksandrov L. A., Ford R. C., Riordan J. R. (2004) J. Biol. Chem. 279, 39051–39057 [DOI] [PubMed] [Google Scholar]
- 24.Awayn N. H., Rosenberg M. F., Kamis A. B., Aleksandrov L. A., Riordan J. R., Ford R. C. (2005) Biochem. Soc. Trans. 33, 996–999 [DOI] [PubMed] [Google Scholar]
- 25.Kartner N., Hanrahan J. W., Jensen T. J., Naismith A. L., Sun S. Z., Ackerley C. A., Reyes E. F., Tsui L. C., Rommens J. M., Bear C. E., Riordan J. R. (1991) Cell 64, 681–691 [DOI] [PubMed] [Google Scholar]
- 26.Prat A. G., Cunningham C. C., Jackson G. R., Jr., Borkan S. C., Wang Y., Ausiello D. A., Cantiello H. F. (1999) Am. J. Physiol. Cell Physiol. 277, C1160–C1169 [DOI] [PubMed] [Google Scholar]
- 27.Head B. P., Patel H. H., Roth D. M., Murray F., Swaney J. S., Niesman I. R., Farquhar M. G., Insel P. A. (2006) J. Biol. Chem. 281, 26391–26399 [DOI] [PubMed] [Google Scholar]
- 28.Verkman A. S., Galietta L. J. (2009) Nat. Rev. Drug Discov. 8, 153–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pedemonte N., Lukacs G. L., Du K., Caci E., Zegarra-Moran O., Galietta L. J., Verkman A. S. (2005) J. Clin. Invest. 115, 2564–2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jurkuvenaite A., Chen L., Bartoszewski R., Goldstein R., Bebok Z., Matalon S., Collawn J. F. (2010) Am. J. Respir. Cell Mol. Biol. 42, 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ithychanda S. S., Hsu D., Li H., Yan L., Liu D., Das M., Plow E. F., Qin J. (2009) J. Biol. Chem. 284, 35113–35121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith L., Page R. C., Xu Z., Kohli E., Ltman P., Nix J. C., Ithychanda S. S., Liu J., Qin J., Misra S., Liedtke C. M. (2010) J. Biol. Chem. 285, 17166–17176 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.