

Abstract
Recent studies have found that smoking is associated with an increased risk of dementia, but the effects of secondhand smoke (SHS) on dementia risk are not known to have previously been studied. The authors used Cox proportional hazards marginal structural models to examine the association between self-reported lifetime household SHS exposure and risk of incident dementia over 6 years among 970 US participants in the Cardiovascular Health Cognition Study (performed from 1991 to 1999) who were never smokers and were free of clinical cardiovascular disease (CVD), dementia, and mild cognitive impairment at baseline. In addition, because prior studies have found that SHS is associated with increased risk of CVD and that CVD is associated with increased risk of dementia, the authors tested for interactions between SHS and measures of clinical and subclinical CVD on dementia risk. Moderate (16–25 years) and high (>25 years) SHS exposure levels were not independently associated with dementia risk; however, subjects with >25 years of SHS exposure and >25% carotid artery stenosis had a 3-fold increase (hazard ratio = 3.00, 95% confidence interval: 1.03, 9.72) in dementia risk compared with subjects with no/low (0–15 years) SHS exposure and ≤25% carotid artery stenosis. High lifetime SHS exposure may increase the risk of dementia in elderly with undiagnosed CVD.
Keywords: aged; dementia; longitudinal studies; models, statistical; tobacco smoke pollution
Tobacco smoke contains hundreds of chemicals known to be toxic or carcinogenic, which can occur in greater concentrations in secondhand smoke (SHS) than in the smoke inhaled by smokers (1, 2). Exposure to SHS is associated with developmental and respiratory problems in children as well as increased risk of lung cancer and coronary heart disease in adults (1, 2). In addition, a recent study found that SHS exposure was associated with greater risk of cognitive impairment in adults (3). However, to our knowledge, the association between SHS and dementia has not previously been studied.
The effects of active smoking on the brain have been somewhat controversial, with some early reports suggesting that active smoking might have beneficial effects (4, 5) or even be associated with a reduced risk of dementia (6, 7). However, more recent evidence clearly suggests that active smoking has neurotoxic effects (8, 9) and is associated with approximately a doubling in dementia risk for older adults (10–12). Therefore, it is plausible that nonsmokers who are exposed to high levels of SHS might also experience increased dementia risk.
SHS also might represent an indirect risk of dementia by exacerbating the risks associated with underlying vascular disease. SHS causes vascular changes, including carotid artery thickening, lesion formation, platelet aggregation, and compromised endothelial function, and may contribute to stroke (1, 2, 13, 14). Vascular disease, in turn, has been associated with an increased risk of developing dementia (15). Additionally, several recent studies have found that measures of subclinical vascular disease (cerebral magnetic resonance imaging (MRI) findings of small/silent infarcts, enlarged ventricles, and white matter disease (16, 17)) and ultrasound evidence of carotid artery thickening (18–20)—and their potential co-occurrence with SHS (13, 21)—are associated with increased dementia risk and evidence of cognitive impairment.
The primary objective of this study was to determine whether SHS exposure is associated with increased dementia risk for older nonsmokers. In addition, given the known deleterious effects of SHS on the vascular system (1, 2) and the growing evidence that vascular disease contributes to the clinical manifestation of dementia (15–19), we hypothesized, a priori, that SHS increases dementia risk in vulnerable subpopulations with underlying clinical or subclinical vascular disease (Figure 1).
Figure 1.

Hypothesized causal pathways by which lifetime exposure to secondhand smoke could increase the risk of dementia—directly (solid arrow) or indirectly (dashed arrows)—by exacerbating the effects of clinical cardiovascular disease (myocardial infarction, angina, stroke/transient ischemic attack) or subclinical cardiovascular or cerebrovascular disease.
MATERIALS AND METHODS
Subject population
The subject population was participants in the Cardiovascular Health Cognition Study (17, 22, 23), which is nested within the larger Cardiovascular Health Study (24). The Cardiovascular Health Study is a prospective, population-based, longitudinal study of risk factors for coronary heart disease and stroke in adults aged 65 years or older. Subjects were recruited from randomized Medicare eligibility lists in 4 US communities: Forsyth County, North Carolina; Washington County, Maryland; Sacramento County, California; and Pittsburgh, Pennsylvania. The Cardiovascular Health Study enrolled 5,201 participants from 1989 to 1990 and an additional 687 African-American participants in 1992–1993.
In 1998–1999, the Cardiovascular Health Cognition Study was initiated to identify subjects who had developed dementia during follow-up (17, 22, 23). Participants included 3,608 subjects from both groups who had a cerebral MRI scan and a Modified Mini-Mental State Examination in 1991–1994. A standardized protocol was administered across the 4 sites to classify subjects as having prevalent dementia at the time of the MRI examination or incident dementia from the time of the MRI to the end of the follow-up period (1998–1999), death, or loss to follow-up.
Of the 3,608 participants, we included only those who had normal cognitive function, were lifelong nonsmokers, and did not have clinical cardiovascular disease (CVD) at baseline so that we could focus on incident disease pathways (i.e., to ensure that SHS exposure clearly preceded both clinical CVD and dementia). Participants from the African-American cohort were excluded from our analyses because it was not possible to differentiate between prevalent and incident CVD at the time of the MRI in this group; however, African-American participants from the original cohort were included. Of the 3,171 potential subjects from the original cohort, we excluded 175 who had prevalent dementia at baseline, 415 with mild cognitive impairment for whom year of onset was unknown and it was not possible to differentiate between prevalent and incident disease, 1,335 current or former smokers, 2 with missing smoking status, 244 with underlying CVD at baseline, 10 with Modified Mini-Mental State Examination scores of less than 70 or missing at baseline, and 20 with missing SHS exposure. Thus, data on 970 subjects remained available for analysis.
Cardiovascular Health Study procedures were approved by institutional review boards at each site, and all participants signed an informed consent at entry and periodically throughout the study. Moreover, the secondary data analyses described here were approved by the Cardiovascular Health Study Steering Committee; the Committee on Human Research at the University of California, San Francisco; the San Francisco Veteran's Administration Medical Center R&D Committee; and the institutional review board at the University of California, Berkeley.
Dementia diagnosis
Dementia was defined as a progressive or static deficit in at least 2 cognitive domains that did not necessarily include memory and was of sufficient severity to affect subjects’ daily activities, combined with a previous history of normal intellectual function (17, 22, 23). This definition differs slightly from the standard Diagnostic and Statistical Manual of Mental Disorders, 4th Edition, definition (25), which requires a memory deficit. Individuals who did not meet the dementia criteria but who exhibited poor cognitive function that reflected a decline from a prior level were classified as having mild cognitive impairment. Standard criteria were used to classify dementia type as probable or possible Alzheimer's disease, probable or possible vascular dementia, mixed dementia, or other (25–28).
Diagnoses were made by an adjudication committee that consisted of neurologists and psychiatrists (one from each site) with expertise in dementia diagnosis (17, 22, 23). Data available for review consisted of information collected annually as part of the main Cardiovascular Health Study and included cognitive test scores, depressive symptoms, level of difficulty with activities of daily living and instrumental activities of daily living, hearing and vision problems, alcohol intake, use of drugs to treat dementia, and recent hospital records. Moreover, detailed neuropsychological, neurologic, and neuropsychiatric data were available for a subgroup of high-risk participants who were examined in 1998–1999. Subjects who died or were lost to follow-up were classified on the basis of their status at the time of death or their last evaluation, respectively.
Secondhand smoke
Subjects were asked whether they had ever lived with anyone who smoked cigarettes regularly and, if so, the total number of years and the time period (childhood, ages 20–50 years, after age 50 years). A summary exposure variable was created to reflect the total number of years of SHS exposure. In preliminary analyses, risk of dementia was calculated for deciles of SHS exposure, and categories were collapsed when age-adjusted risk estimates were roughly equivalent. Final analyses included 3 categories of SHS exposure: none/low (≤15 years), moderate (16–25 years), and high (>25 years). Subjects with no exposure and low exposure were combined into the lowest category because there was no observed difference in risk of dementia between these 2 groups. Workplace and other external sources of SHS were not assessed.
Vascular disease measures
CVD was defined as having a history of myocardial infarction, stroke, transient ischemic attack, angina pectoris, claudication, angioplasty, or bypass surgery. All vascular events were identified at baseline and during follow-up as part of the main Cardiovascular Health Study by using a protocol that required validation by either physician questionnaire or medical record review (24, 29, 30).
Subclinical MRI measures were based on cerebral MRI examinations performed by using a standard protocol (31, 32). Images were interpreted by trained neuroradiologists who were blinded to subjects’ age, sex, race, ethnicity, and other clinical information. Infarcts on MRI were defined as lesions with abnormal signal in a vascular distribution and no mass effect. White matter disease was estimated as the total volume of periventricular and subcortical white matter signal abnormality on spin-density-weighted axial images compared with 8 “reference” images and was classified from grade 0 (none) to grade 9 (extensive). Specific subclinical MRI measures used in this study included small infarcts (<3 mm), large infarcts (≥3 mm), and white matter disease (grade 3 or higher).
Subclinical carotid artery measures were based on duplex ultrasonography performed with 2-dimensional brightness mode imaging to detect thickening of the arterial wall, disruption of normal wall surfaces, and development of focal plaques bilaterally (33). Images were interpreted at the Cardiovascular Health Study Ultrasound Reading Center by trained readers. Specific subclinical carotid artery measures used in this study included internal or common carotid artery thickness above the 80th percentile and stenosis >25% of the internal carotid artery.
Other measures
Time-independent measures included age (categorized as <70, 70–73, 74–79, ≥80 years), race (African American/other, Caucasian), gender, income (categories ranged from <$5,000 to ≥$50,000), education (high school equivalent or greater), apolipoprotein-E genotype (ϵ4 allele present/absent), C-reactive protein (milliliters per liter), and occupation (professional, sales/clerical, farmer/craftsman, housewife, other). Time-dependent measures included self-reported health (excellent, good, fair, poor), hypertension (ever hypertensive/borderline or not hypertensive based on history of hypertension and/or measured blood pressure at visit), diabetes (fasting glucose ≥126 mg/dL and/or oral hypoglycemic/insulin therapy), physical activity (number of blocks walked in the last week), depression (Center for Epidemiologic Studies Depression (34) Scale score of ≥16), weight (kilograms), cholesterol (milligrams per deciliter), and alcohol intake (number of beverages per week). The time-dependent measures were recorded annually over follow-up with the exception of the diabetes measure, which was recorded for 2 of the possible 6 annual visits. These variables were accounted for as potential confounders in the analysis (refer to the Appendix).
Statistical analysis
Bivariate and multiway associations between subclinical and clinical CVD, SHS exposure, and dementia were examined. Associations between potential confounding factors and dementia also were evaluated and incorporated into subsequent multivariable analyses (refer to the Appendix). Stratified Kaplan-Meier plots were used to examine the distribution of incident dementia by subclinical and clinical CVD status.
Cox proportional hazards marginal structural models (MSMs) were used to study different causal pathways between SHS and dementia risk (Figure 1) and to account for incident clinical CVD on the causal pathway. These models can provide unbiased estimates of causal effects in the context of time-dependent confounders and causal intermediates (35–38), whereas standard analytic methods are likely to produce biased risk estimates under these conditions (39–41). These models do not adjust for incident CVD. Rather, they account specifically for incident CVD as a potential causal intermediate. Therefore, the estimates of the observed associations from these models (e.g., SHS) reflect effects that are independent of incident CVD. Details of application of MSMs for this study are provided in the Appendix and in methods described elsewhere (39).
For our analyses, Cox proportional hazards MSMs were fit by using weighted logistic regression estimated with generalized estimating equations, with individual weights derived for each subject (36). Specific models included direct effects of SHS and CVD (model 1), additional direct effects of subclinical MRI measures (model 2) and subclinical carotid artery measures (model 3), and joint effects of SHS and subclinical carotid artery measures (model 4). For some of the models (e.g., joint effects of SHS × CVD), sample sizes were too small to test our hypotheses.
RESULTS
Descriptive and crude analyses
Subjects were a mean age of 74 years (standard deviation, 5), and 74% were women. More than 60% had lived with a smoker for ≤15 years (n = 600), including 470 who had never lived with a smoker; 13% (n = 123) had lived with a smoker for 16–25 years, and 25% (n = 247) had lived with a smoker for >25 years. Subjects were followed for a mean of 5.5 years (range: 0.5–8.4), during which time 15% (n = 148) developed dementia (94 Alzheimer's disease, 41 vascular dementia, 10 mixed dementia, 3 other).
Participants who had lived with a smoker for ≤15 years were older and were less likely to be female compared with those who had lived with a smoker for >15 years (Table 1). No differences were observed between the 3 SHS groups based on education or presence of clinical vascular disease, subclinical MRI measures, or subclinical carotid artery measures.
Table 1.
Baseline Characteristicsa of 970 Nonsmokers by Level of Secondhand Smoke Exposure, Cardiovascular Health Cognition Study, 1991–1994
| Variable | Level of Exposure |
P Valueb | ||
| 0–15 Years (n = 600) | 16–25 Years (n = 123) | >25 Years (n = 247) | ||
| Demographics | ||||
| Age, years | 75.0 (4.9) | 73.9 (4.3) | 73.8 (4.3) | <0.001 |
| Gender: female | 68.7 | 82.1 | 83.0 | <0.001 |
| Education (≥high school diploma) | 76.6 | 84.3 | 78.7 | 0.12 |
| Clinical vascular disease | 11.3 | 9.8 | 9.7 | 0.74 |
| Subclinical MRI measures | ||||
| Large infarcts | 28.8 | 22.8 | 23.1 | 0.14 |
| Small infarcts | 13.6 | 16.3 | 11.7 | 0.48 |
| White matter disease | 32.1 | 24.6 | 32.7 | 0.23 |
| Subclinical carotid artery measures | ||||
| Internal artery thickness >80th percentile | 20.2 | 18.7 | 19.6 | 0.92 |
| Common artery thickness >80th percentile | 21.8 | 13.8 | 15.9 | 0.04 |
| Stenosis >25% | 39.0 | 35.0 | 34.7 | 0.42 |
Abbreviation: MRI, magnetic resonance imaging.
Values are expressed as mean (standard deviation) or %.
Based on the F test for continuous data and the χ2 test for categorical data.
In crude analyses, we found no evidence of an association between SHS exposure and risk of dementia (Table 2). Moreover, incident clinical vascular disease was not significantly associated with dementia risk. In contrast, most of our measures of subclinical vascular disease were associated with increased dementia risk.
Table 2.
Unadjusted Association Between Secondhand Smoke Exposure, Vascular Measures, and Dementia Incidence in the Cardiovascular Health Cognition Study From 1991 to 1999a
| Characteristic | Dementia |
No Dementia |
CIRb | 95% CI | ||
| No.c | % | No. | % | |||
| Secondhand smoke exposure | ||||||
| 0–15 years | 95 | 15.8 | 505 | 84.2 | Ref | |
| 16–25 years | 17 | 13.8 | 106 | 86.2 | 0.87 | 0.54, 1.41 |
| >25 years | 36 | 14.6 | 211 | 85.4 | 0.92 | 0.65, 1.31 |
| Clinical vascular disease | ||||||
| Absent | 138 | 15.9 | 728 | 84.1 | Ref | |
| Present | 10 | 9.6 | 94 | 90.4 | 0.60 | 0.33, 1.11 |
| Subclinical MRI measures | ||||||
| Large infarcts ≥3 mm | ||||||
| Absent | 94 | 13.2 | 616 | 86.8 | Ref | |
| Present | 52 | 20.2 | 205 | 79.8 | 1.53 | 1.12, 2.08 |
| Small infarcts <3 mm | ||||||
| Absent | 117 | 14.0 | 720 | 86.0 | Ref | |
| Present | 29 | 22.3 | 101 | 77.7 | 1.60 | 1.11, 2.29 |
| White matter disease | ||||||
| Absent | 64 | 9.7 | 595 | 90.3 | Ref | |
| Present | 81 | 27.0 | 219 | 73.0 | 2.78 | 2.06, 3.74 |
| Subclinical carotid artery measures | ||||||
| Internal artery thickness >80th percentile | ||||||
| Absent | 103 | 13.4 | 667 | 86.6 | Ref | |
| Present | 42 | 22.0 | 149 | 78.0 | 1.64 | 1.19, 2.27 |
| Common artery thickness >80th percentile | ||||||
| Absent | 105 | 13.5 | 671 | 86.5 | Ref | |
| Present | 40 | 21.6 | 145 | 78.4 | 1.60 | 1.15, 2.22 |
| Stenosis >25% | ||||||
| Absent | 83 | 13.8 | 518 | 86.2 | Ref | |
| Present | 62 | 17.3 | 297 | 82.7 | 1.25 | 0.92, 1.69 |
Abbreviations: CI, confidence interval; CIR, cumulative incidence ratio; MRI, magnetic resonance imaging; Ref, referent.
For various characteristics, 0–10 values are missing.
Unadjusted value based on dementia status at the last follow-up visit.
The 148 dementia cases included 94 with Alzheimer's disease only, 41 with vascular dementia, 10 with mixed vascular dementia/Alzheimer's disease, and 3 with other types of dementia.
MSM analyses
In our MSM analyses (Table 3), incident clinical CVD was the “causal effects” parameter, whereas the other variables in the model were treated as “stratification” variables (refer to the Appendix). Therefore, the hazard ratio estimates for clinical CVD reflect the population-level change in the relative hazard of incident dementia if, contrary to fact, everyone in the population experienced clinical CVD compared with if no one experienced clinical CVD. In contrast, the hazard ratio estimates for the other variables in the models reflect the change in the relative hazard of dementia associated with a particular exposure if, contrary to fact, no one in the population experienced clinical CVD (i.e., independent of any effects of clinical CVD).
Table 3.
Effects of Secondhand Smoke Exposure and Vascular Disease on Dementia Incidence, per Cox Proportional Hazards Marginal Structural Models,a in the Cardiovascular Health Cognition Study From 1991 to 1999
| Variable | Model 1 |
Model 2 |
Model 3 |
Model 4 |
||||
| HR | 95% CI | HR | 95% CI | HR | 95% CI | HR | 95% CI | |
| Clinical vascular diseaseb | 1.65 | 0.62, 3.16 | 1.56 | 0.53, 3.10 | 1.59 | 0.62, 3.21 | 1.60 | 0.60, 3.27 |
| SHS exposurec | ||||||||
| 16–25 years | 1.02 | 0.48, 1.88 | 1.08 | 0.53, 2.02 | 1.02 | 0.48, 1.90 | 1.13 | 0.38, 2.43 |
| >25 years | 1.43 | 0.80, 2.32 | 1.28 | 0.71, 2.14 | 1.46 | 0.82, 2.40 | 0.81 | 0.34, 1.50 |
| Subclinical MRI measuresd | ||||||||
| Infarct ≥3 mm | 0.90 | 0.50, 1.62 | ||||||
| Infarct <3 mm | 1.67 | 0.92, 2.98 | ||||||
| White matter disease | 2.65 | 1.70, 4.34 | ||||||
| Subclinical carotid artery measuresd | ||||||||
| Internal artery thickness >80th percentile | 1.60 | 0.80, 3.07 | 1.52 | 0.77, 2.92 | ||||
| Common artery thickness >80th percentile | 1.07 | 0.66, 1.69 | 1.08 | 0.66, 1.73 | ||||
| Stenosis >25% | 0.99 | 0.59, 1.67 | 0.76 | 0.40, 1.41 | ||||
| Stenosis >25% × SHS exposure 16–25 years | 0.87 | 0.21, 3.35 | ||||||
| Stenosis >25% × SHS exposure >25 years | 3.00 | 1.03, 9.72 | ||||||
Abbreviations: CI, confidence interval; HR, hazard ratio; MRI, magnetic resonance imaging; SHS, secondhand smoke.
All models adjusted for age, gender, and education. Other variables listed in the Materials and Methods section of the text were included in the “treatment” model; therefore, it was not necessary to adjust for them in the marginal structural models. Refer to the Appendix for more details. Results were similar when subclinical MRI measures and subclinical carotid artery measures were included in the same model to determine whether their effects were independent, and when Modified Mini-Mental State Examination score was included as a stratification variable to further control for residual confounding by baseline cognitive function, education, and socioeconomic status.
Marginal (population) relative hazard of clinical cardiovascular disease on dementia at time t for a given stratum of age, SHS exposure, gender, and education.
Associated relative hazard of dementia at time t for various SHS exposure levels compared with that for subjects with 0–15 years of SHS exposure for a given stratum of age, gender, and education, independent of any effects of cardiovascular disease.
Associated relative hazard of dementia at time t for subjects for various subclinical MRI measures (model 2), subclinical carotid artery measures (model 3), or interactions between SHS and subclinical carotid artery measures (model 4) for a given stratum of age, gender, education, and SHS exposure, independent of any effects of cardiovascular disease.
Model 1 indicates that the population-level relative hazard of dementia was estimated to be larger by 65% for those with CVD if, contrary to fact, no one in the population experienced CVD, although this increase was not statistically significant (hazard ratio = 1.65, 95% confidence interval: 0.62, 3.16). Additionally if, contrary to fact, no one experienced clinical CVD in the population, there was no evidence of an associated change in the relative hazard of dementia in those with moderate SHS exposure (hazard ratio = 1.02, 95% confidence interval: 0.48, 1.88) or high SHS exposure (hazard ratio = 1.43, 95% confidence interval: 0.80, 2.32) relative to those with low/no SHS exposure.
When subclinical MRI measures and subclinical carotid artery measures were added, there was little change in the associations between clinical CVD, SHS exposure, and dementia risk. However, if, contrary to fact, no one in the population experienced clinical CVD, then the associated relative hazard of dementia was estimated to increase by two-thirds for those with small MRI infarcts (hazard ratio = 1.67, 95% confidence interval: 0.92, 2.98) and was more than 2.5 times higher for those with evidence of white matter disease (hazard ratio = 2.65, 95% confidence interval: 1.70, 4.34). The associated change in dementia risk was not significantly altered by large MRI infarcts (model 2) or subclinical carotid artery measures (model 3).
When the joint effects of SHS and subclinical vascular measures were examined (model 4), there was evidence of interaction between SHS exposure and internal carotid artery stenosis on dementia risk. If, contrary to fact, no one experienced clinical CVD, neither SHS exposure nor internal carotid artery stenosis alone was associated with dementia risk (Figure 2; Table 3, model 4); however, the relative hazard of dementia for those with both >25% stenosis and >25 years of SHS exposure was 3 times higher (hazard ratio = 3.00, 95% confidence interval: 1.03, 9.72) than for those with neither of these characteristics. We found no suggestion that the relative hazard of dementia associated with other subclinical measures increased with increased SHS exposure (data not shown).
Figure 2.

Association of secondhand smoke and internal carotid artery stenosis with dementia risk in the Cardiovascular Health Cognition Study, performed from 1991 to 1999. Cox proportional hazards marginal structural models were used to calculate hazard ratios for dementia as a function of secondhand smoke exposure and internal carotid artery stenosis, neither of which increased dementia risk when considered alone. However, the risk of dementia was 3 times higher for those with high levels of secondhand smoke exposure (>25 years) and internal carotid artery stenosis (>25%) compared with those with no/low secondhand smoke exposure (<15 years) and ≤25% stenosis.
DISCUSSION
In this study of almost 1,000 lifetime nonsmokers, we found that exposure to high levels of SHS in combination with carotid artery stenosis was associated with an elevated risk of developing dementia over 6 years. The risk of dementia was tripled for participants who had lived with a smoker for >25 years over their lifetimes and also had carotid artery stenosis of >25% at baseline. There was no evidence of a direct effect of SHS exposure on the risk of incident dementia independent of the pathway through carotid artery stenosis.
This is the first study known to examine the potential causal association of lifetime exposure to SHS with dementia in older adults. However, its findings are consistent with studies that have found an association between SHS exposure and worse cognitive function in children (8, 42, 43) and, more recently, adults (3).
Several factors support the biologic plausibility of SHS exposure as a risk factor for dementia. First, SHS is highly toxic and contains at least 250 chemicals known to be harmful or carcinogenic (2); therefore, such exposure could negatively impact the brain and render it more susceptible to dementia. Second, exposure to SHS can cause both immediate and long-term adverse effects in the cardiovascular system that include increased “stickiness” of blood platelets, endothelial dysfunction, decreased coronary flow velocity reserves, and reduced heart rate variability (2, 13). Moreover, endothelial dysfunction may be related to the reduced clearance of beta-amyloid protein, which is considered to be related to the pathogenesis of Alzheimer's disease (44). Third, evidence suggests that nonsmokers exposed to high levels of SHS may develop atherosclerosis of the carotid and large arteries of the brain as well as degeneration of the intracerebral arteries. These changes, in turn, may increase the risk of stroke and dementia (1, 13, 15, 21). Therefore, there are several mechanisms through which SHS could directly and indirectly affect dementia risk.
In this study, dementia risk was increased 3-fold for subjects with >25 years of SHS exposure and >25% stenosis of the internal carotid artery, although the association was imprecisely estimated. We found no evidence of an associated increased risk of dementia by SHS exposure or carotid artery stenosis alone. This finding may reflect a true lack of association, or it may reflect low statistical power to detect a small effect or bias toward the null associated with measurement error. Potentially, the effects of these variables alone were not sufficient to be observed as independent risk factors, but their combined effects may have acted synergistically to induce an association with dementia.
Similar interactions between SHS exposure and other measures of underlying CVD (e.g., MRI measures, intimal wall thickness) were not observed, however. This absence was partly due to insufficient data. However, it may have been due to biologic reasons as well. For example, the absence of interactions between SHS and the MRI measures and associated dementia risk might be explained by the fact that the MRI variables are specific to the brain and may represent pathways separate from those that involve general, underlying CVD. Therefore, the added effects of SHS exposure by way of these pathways might be negligible when compared with the risk of positive levels of different MRI variables for dementia and cerebrovascular disease. We also did not have adequate statistical power to perform analyses stratified by dementia subtype. Most of our subjects with dementia had Alzheimer's disease (64%), and it is possible that SHS has a more direct effect on vascular dementia.
Strengths of the study include its well-characterized study population, detailed measures of underlying CVD, and the use of MSMs, which made investigation of the different causal pathways between SHS and dementia possible. A limitation was that SHS was based on self-report and did not include measured work exposure or other external sources of SHS exposure. However, the study population consisted mainly of older women (∼74%), whose most likely source of SHS was household exposure.
To ensure causal directionality and to identify effects specific to SHS exposure, our study was restricted to nonsmokers who had no preexisting clinical dementia or CVD. Specific types of subclinical CVD were examined in combination with SHS exposure to represent selective biologic pathways by which SHS might differentially affect risk of dementia. However, these restrictions and the specificity of the study reduced the sample size of the analysis and likely decreased the precision of our effect estimates.
Potentially, the significant interaction observed between SHS and carotid artery stenosis was due to type I error. Correction for multiple comparisons using a resampling-based approach widened the confidence interval slightly (95% confidence interval: 0.97, 10.32) (45, 46). Such adjustments are considered controversial, particularly when underlying causal associations are suspected, because of an increased probability of type II error (47). Regardless, given that the estimate was imprecisely measured, the result should be considered preliminary and warrants additional follow-up. The study methodology addresses the fact that clinical CVD reflects, for many people, a causal pathway between SHS exposure and cerebrovascular disease. Moreover, we hypothesized that clinical CVD represents a causal pathway between SHS exposure and incident dementia. Thus, a direct effect of SHS on dementia risk could be examined, as well as the potential contribution of SHS exposure among subjects with subclinical levels of CVD. MSMs provide a method for the control of factors on the causal pathway, which is not possible with standard statistical approaches. The application of MSMs, in this instance, has relevant public health implications because it provides a quantitative basis for assessing the need to target older individuals, whose underlying characteristics put them at greater risk of dementia.
In summary, in this cohort of elder nonsmokers, we found that exposure to high levels of SHS alone did not increase dementia risk. However, those elders who had a history of high SHS exposure combined with a history of carotid artery stenosis experienced a 3-fold increase in the risk of dementia.
Acknowledgments
Author affiliations: Department of Psychiatry, University of California, San Francisco, San Francisco, California (Deborah E. Barnes); Department of Medicine, University of California, San Francisco, San Francisco, California (Kala M. Mehta); Department of Mental Health, San Francisco Veterans Affairs Medical Center, San Francisco, California (Deborah E. Barnes); Department of Geriatrics, San Francisco Veterans Affairs Medical Center, San Francisco, California (Kala M. Mehta); Department of Epidemiology and Biostatistics, University of California, Berkeley, Berkeley, California (Thaddeus J. Haight, Ira B. Tager); Department of Mental Health, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland (Michelle C. Carlson); Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, Maryland (Michelle C. Carlson); and Department of Epidemiology, School of Public Health, University of Pittsburgh, Pittsburgh, Pennsylvania (Lewis H. Kuller).
This work was supported by the National Institutes of Health (contracts N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01-HC-15103, N01-HC-55222, N01-HC-75150, N01-HC-45133 and grant U01 HL080295 from the National Heart, Lung, and Blood Institute, with an additional contribution from the National Institute of Neurological Disorders and Stroke. The Cardiovascular Health Cognition Study was supported by a grant from the National Institute on Aging (R01 AG15928-02). Additional support for this study was provided by the Flight Attendants Medical Research Institute. Dr. Barnes was supported in part by a K01 Career Development Award (K01 AG024069).
A full list of principal Cardiovascular Health Study investigators and institutions can be found at the following website: http://www.chs-nhlbi.org/pi.htm.
Conflict of interest: none declared.
Glossary
Abbreviations
- CVD
cardiovascular disease
- MRI
magnetic resonance imaging
- MSM
marginal structural model
- SHS
secondhand smoke
APPENDIX
Implementation of Cox Proportional Hazards MSMs
Overview.
MSMs are based on the concept of counterfactuals, which represent the set of outcomes subjects might have experienced if they experienced exposures other than the ones actually received (37, 46, 48, 49). Hypothetically, if we knew the outcomes that corresponded to all possible exposures that a given subject could experience (i.e., the outcomes associated with his or her “actual” as well as “counterfactual” experiences), then each subject would serve as his or her own control and we could assess whether differences in the outcome were attributable causally to differences in level of exposure. In practice, we do not observe all possible outcomes. MSMs represent one class of causal statistical models for modeling this hypothetical world, based on observed data, and for examining causal parameters of interest (46). Moreover, MSM analyses enable modeling of time-dependent confounders and informative censoring and therefore can provide unbiased estimates of these causal parameters (37).
Identification of causal effects with MSMs, based on observed data, depends on a set of assumptions and estimation procedures (e.g., inverse probability of treatment weight) (37). In addition to inverse probability of treatment weights, weights are obtained to account for systematically missing covariates and loss to follow-up (inverse probability of censoring weights) (37). A weighted logistic regression estimated with generalized estimating equations, with weights derived for each subject, was used to fit the Cox proportional hazards MSMs in this analysis (36).
Typically, MSMs include both “causal parameters” and “stratification variables.” In our analyses, it was not possible to define SHS as the causal parameter because doing so would have required knowledge of both when SHS exposure occurred and what factors contributed to greater or lesser exposure to SHS (both required to define causal effects). Therefore, we examined SHS as a baseline stratification variable and treated clinical CVD as the causal effect parameter in the analyses. This decision seemed reasonable because SHS exposure has been associated with increased risk of CVD. Additional baseline stratification variables included subclinical measures of carotid artery disease and cerebral MRI status, age, gender, and education.
This approach enabled us to examine the causal contribution of population-level differences in CVD incidence to the change in the hazard of onset dementia at time t during the 6-year study. In addition, we examined the associated change in the hazard of dementia for different baseline stratification variables given that, contrary to fact, no one in the population had clinical CVD. In this manner, we examined the association of the different levels of these stratification variables with respect to the risk of onset dementia independent of these variables’ influence on clinical CVD. Identification and estimation of parameters, used to represent the contributions of these variables to the risk of dementia, are based on a set of assumptions that underlie MSMs. For example, confounders (refer to the “Other measures” paragraph in the Materials and Methods section) of clinical CVD and dementia were examined in a separate “treatment” model to satisfy one of these assumptions, and results from that model were applied toward estimating the Cox proportional hazards MSM parameters. In this manner, only those variables of direct interest are included in the MSM.
In summary, Cox proportional hazards MSMs provide parameters that represent estimates of the causal relative hazard of exposure and disease onset more directly than do the parameters from the usual Cox proportional hazards association model. Moreover, these MSMs provide unbiased estimates in the presence of causal intermediates that cannot occur with standard analytical methods.
Causal model.
Given a time-dependent process (t = 1, …, 6) that includes clinical CVD incidence, recorded covariates (time dependent and time independent), and occurrence of dementia, we define the following Cox proportional hazards MSM to examine the causal contributions of population-level CVD incidence and baseline risk covariates “v” (e.g., age, cumulative lifetime SHS exposure) to the hazard of onset dementia at time t:
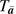 denotes a counterfactual outcome process of time to dementia (years after baseline interview) that corresponds with all possible courses of CVD incidence subjects could have experienced during the 6-year study
denotes a counterfactual outcome process of time to dementia (years after baseline interview) that corresponds with all possible courses of CVD incidence subjects could have experienced during the 6-year study 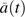 . Once a subject experienced an event, he or she was classified as having CVD for the length of the study. represents the unspecified baseline hazard of dementia at time t. The parameter β1 signifies the causal log relative change to the baseline hazard of dementia at time t if, contrary to fact, everyone in the population experienced clinical CVD through observation time t compared with no one in the population having CVD. These counterfactual hazards were examined for subpopulations defined by v (v includes categories of exposure to SHS). is a parameter vector that signifies the associated log relative hazard change to the baseline hazard at any time t for different subpopulations of subjects as defined by v—for example, category of SHS, age—given that everyone in the population did not experience clinical CVD. It is important to note that the parameters that correspond with the baseline covariates are not causal but association parameters; however, they can be interpreted as having taken into account clinical CVD as a causal intermediate.
. Once a subject experienced an event, he or she was classified as having CVD for the length of the study. represents the unspecified baseline hazard of dementia at time t. The parameter β1 signifies the causal log relative change to the baseline hazard of dementia at time t if, contrary to fact, everyone in the population experienced clinical CVD through observation time t compared with no one in the population having CVD. These counterfactual hazards were examined for subpopulations defined by v (v includes categories of exposure to SHS). is a parameter vector that signifies the associated log relative hazard change to the baseline hazard at any time t for different subpopulations of subjects as defined by v—for example, category of SHS, age—given that everyone in the population did not experience clinical CVD. It is important to note that the parameters that correspond with the baseline covariates are not causal but association parameters; however, they can be interpreted as having taken into account clinical CVD as a causal intermediate.
MSM assumptions.
In general terms, we make the following 4 assumptions to identify causal effects with MSMs: 1) temporal ordering of variables—for example, covariates at t precede clinical CVD status at t and dementia status at t + 1; 2) consistency assumption—observed data are just one realization of the “full” counterfactual data; 3) no unmeasured confounding—the treatment (e.g., clinical CVD) at any given time t, conditional on covariates, is independent of the counterfactual outcome; and 4) experimental treatment assignment—all possible treatments (e.g., occurrence/absence of clinical CVD) are observed for given covariates (37, 46, 48, 49).
Estimation of causal parameters.
Identification of Cox proportional hazards MSM estimates requires satisfaction of the above 4 assumptions. In particular, satisfaction of the no-unmeasured-confounding assumption is based on solving the inverse probability of treatment weight estimating equation (36, 37). Solving this equation is equivalent to performing a weighted Cox proportional hazards regression with subject-specific treatment weights (Sw):
These weights were obtained by using 2 probability models. The first was a pooled logistic regression of incident clinical CVD A(k) on time-dependent covariates L(k) (e.g., hypertension, physical activity at time k) and time-independent covariates (e.g., gender, education, income), described in the “Other measures” paragraph of the Materials and Methods section. The second model was similar to the first but was a fit of incident clinical CVD on subsets of the baseline covariates V specified in the different Cox proportional hazards MSMs. The probabilities from the 2 models were used to create subject-specific stabilized weights at each time point for which a subject's data were observed.
Informally, the denominator of the formulation above represents the probability of observing a subject's history of incident clinical CVD given covariates and no previous history of clinical CVD through time k (36).
In addition to inverse probability of treatment weights, subject-specific weights were obtained to account for systematically missing covariates and loss to follow-up (Swc) using inverse probability of censoring weights (36):
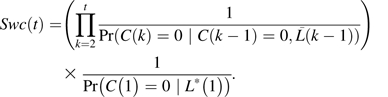 |
Here, a pooled logistic model was fit to evaluate censoring at follow-up C(k) based on previous levels of covariates 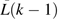 among those present at time k – 1. Past clinical CVD was not considered as a covariate in the model because of limited data. Separate logistic models evaluated systematically missing data in 2 baseline variables (apolipoprotein E, income) by using other baseline covariates L*(1), and the estimates were used to determine the joint probability of no censoring in either of these variables, C(1) = 0. Other data missing at baseline were considered missing at random. It was not necessary to develop a set of stabilized weights for these inverse probability of censoring weights given that they were relatively stable (i.e., no large weights). Informally, these weights represent the inverse probability of observing a subject in the data given his or her past covariate history.
among those present at time k – 1. Past clinical CVD was not considered as a covariate in the model because of limited data. Separate logistic models evaluated systematically missing data in 2 baseline variables (apolipoprotein E, income) by using other baseline covariates L*(1), and the estimates were used to determine the joint probability of no censoring in either of these variables, C(1) = 0. Other data missing at baseline were considered missing at random. It was not necessary to develop a set of stabilized weights for these inverse probability of censoring weights given that they were relatively stable (i.e., no large weights). Informally, these weights represent the inverse probability of observing a subject in the data given his or her past covariate history.
We implemented the Cox proportional hazards MSM with a weighted logistic regression, for example, of clinical dementia Y(t) on clinical CVD 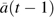 and baseline covariates V among those with no previous clinical dementia,
and baseline covariates V among those with no previous clinical dementia,
with the product of the weights Sw(t) × Swc(t) derived for each subject. Generalized estimating equations were used to account for within-subject correlation induced by use of the weights (36). Given that the standard errors with this approach tend to be too conservative, we obtained empirical estimates of the standard errors with 1,000 bootstrap replications (46).
Experimental treatment assignment assumption.
Experimental treatment assignment violation was assessed informally by comparing the distributions of the conditional probabilities of clinical CVD, given covariates, in subjects with and without CVD. Given the low frequency of clinical CVD in the population, both distributions indicated a low probability of disease, but there was sufficient variability in both distributions (i.e., covariates not deterministic of CVD status) to indicate that a violation of the experimental treatment assignment was unlikely.
References
- 1.California Environmental Protection Agency: Air Resources Board and Office of Environmental Health Hazard Assessment. Sacramento, CA: California Environmental Protection Agency; June 24, 2005. Proposed identification of environmental tobacco smoke as a toxic air contaminant as approved by the Scientific Review Panel on June 24, 2005 (Appendix III) ( ftp://ftp.arb.ca.gov/carbis/regact/ets2006/app3exe.pdf). (Accessed October 7, 2009) [Google Scholar]
- 2.US Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. Rockville, MD: US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; June 27, 2006. ( http://www.surgeongeneral.gov/library/secondhandsmoke/) [Google Scholar]
- 3.Llewellyn DJ, Lang IA, Langa KM, et al. Exposure to secondhand smoke and cognitive impairment in non-smokers: national cross sectional study with cotinine measurement [electronic article] BMJ. 2009;338:b462. doi: 10.1136/bmj.b462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elrod K, Buccafusco JJ, Jackson WJ. Nicotine enhances delayed matching-to-sample performance by primates. Life Sci. 1988;43(3):277–287. doi: 10.1016/0024-3205(88)90318-9. [DOI] [PubMed] [Google Scholar]
- 5.Salomon AR, Marcinowski KJ, Friedland RP, et al. Nicotine inhibits amyloid formation by the beta-peptide. Biochemistry. 1996;35(42):13568–13578. doi: 10.1021/bi9617264. [DOI] [PubMed] [Google Scholar]
- 6.Lee PN. Smoking and Alzheimer's disease: a review of the epidemiological evidence. Neuroepidemiology. 1994;13(4):131–144. doi: 10.1159/000110372. [DOI] [PubMed] [Google Scholar]
- 7.Van Duijn CM, Clayton DG, Chandra V, et al. Interaction between genetic and environmental risk factors for Alzheimer's disease: a reanalysis of case-control studies. EURODEM Risk Factors Research Group. Genet Epidemiol. 1994;11(6):539–551. doi: 10.1002/gepi.1370110609. [DOI] [PubMed] [Google Scholar]
- 8.Swan GE, Lessov-Schlaggar CN. The effects of tobacco smoke and nicotine on cognition and the brain. Neuropsychol Rev. 2007;17(3):259–273. doi: 10.1007/s11065-007-9035-9. [DOI] [PubMed] [Google Scholar]
- 9.Reitz C, Luchsinger J, Tang MX, et al. Effect of smoking and time on cognitive function in the elderly without dementia. Neurology. 2005;65(6):870–875. doi: 10.1212/01.wnl.0000176057.22827.b7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almeida OP, Hulse GK, Lawrence D, et al. Smoking as a risk factor for Alzheimer's disease: contrasting evidence from a systematic review of case-control and cohort studies. Addiction. 2002;97(1):15–28. doi: 10.1046/j.1360-0443.2002.00016.x. [DOI] [PubMed] [Google Scholar]
- 11.Anstey KJ, von Sanden C, Salim A, et al. Smoking as a risk factor for dementia and cognitive decline: a meta-analysis of prospective studies. Am J Epidemiol. 2007;166(4):367–378. doi: 10.1093/aje/kwm116. [DOI] [PubMed] [Google Scholar]
- 12.Hernán MA, Alonso A, Logroscino G. Cigarette smoking and dementia: potential selection bias in the elderly. Epidemiology. 2008;19(3):448–450. doi: 10.1097/EDE.0b013e31816bbe14. [DOI] [PubMed] [Google Scholar]
- 13.Barnoya J, Glantz SA. Cardiovascular effects of secondhand smoke: nearly as large as smoking. Circulation. 2005;111(20):2684–2698. doi: 10.1161/CIRCULATIONAHA.104.492215. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Shu XO, Yang G, et al. Association of passive smoking by husbands with prevalence of stroke among Chinese women nonsmokers. Am J Epidemiol. 2005;161(3):213–218. doi: 10.1093/aje/kwi028. [DOI] [PubMed] [Google Scholar]
- 15.Snowdon DA, Greiner LH, Mortimer JA, et al. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277(10):813–817. [PubMed] [Google Scholar]
- 16.Vermeer SE, Prins ND, den Heijer T, et al. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348(13):1215–1222. doi: 10.1056/NEJMoa022066. [DOI] [PubMed] [Google Scholar]
- 17.Kuller LH, Lopez OL, Newman A, et al. Risk factors for dementia in the Cardiovascular Health Cognition Study. Neuroepidemiology. 2003;22(1):13–22. doi: 10.1159/000067109. [DOI] [PubMed] [Google Scholar]
- 18.Mathiesen EB, Waterloo K, Joakimsen O, et al. Reduced neuropsychological test performance in asymptomatic carotid stenosis: the Tromsø Study. Neurology. 2004;62(5):695–701. doi: 10.1212/01.wnl.0000113759.80877.1f. [DOI] [PubMed] [Google Scholar]
- 19.Kearney-Schwartz A, Rossignol P, Bracard S, et al. Vascular structure and function is correlated to cognitive performance and white matter hyperintensities in older hypertensive patients with subjective memory complaints. Stroke. 2009;40(4):1229–1236. doi: 10.1161/STROKEAHA.108.532853. [DOI] [PubMed] [Google Scholar]
- 20.Newman AB, Fitzpatrick AL, Lopez O, et al. Dementia and Alzheimer's disease incidence in relationship to cardiovascular disease in the Cardiovascular Health Study cohort. J Am Geriatr Soc. 2005;53(7):1101–1107. doi: 10.1111/j.1532-5415.2005.53360.x. [DOI] [PubMed] [Google Scholar]
- 21.Hanon O, Haulon S, Lenoir H, et al. Relationship between arterial stiffness and cognitive function in elderly subjects with complaints of memory loss. Stroke. 2005;36(10):2193–2197. doi: 10.1161/01.STR.0000181771.82518.1c. [DOI] [PubMed] [Google Scholar]
- 22.Fitzpatrick AL, Kuller LH, Ives DG, et al. Incidence and prevalence of dementia in the Cardiovascular Health Study. J Am Geriatr Soc. 2004;52(2):195–204. doi: 10.1111/j.1532-5415.2004.52058.x. [DOI] [PubMed] [Google Scholar]
- 23.Lopez OL, Kuller LH, Fitzpatrick A, et al. Evaluation of dementia in the Cardiovascular Health Cognition Study. Neuroepidemiology. 2003;22(1):1–12. doi: 10.1159/000067110. [DOI] [PubMed] [Google Scholar]
- 24.Fried LP, Borhani NO, Enright P, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1(3):263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 25.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV. 4th ed. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 26.Chui HC, Victoroff JI, Margolin D, et al. Criteria for the diagnosis of ischemic vascular dementia proposed by the State of California Alzheimer's Disease Diagnostic and Treatment Centers. Neurology. 1992;42(3 pt 1):473–480. doi: 10.1212/wnl.42.3.473. [DOI] [PubMed] [Google Scholar]
- 27.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 28.Román GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 1993;43(2):250–260. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- 29.Ives DG, Fitzpatrick AL, Bild DE, et al. Surveillance and ascertainment of cardiovascular events. The Cardiovascular Health Study. Ann Epidemiol. 1995;5(4):278–285. doi: 10.1016/1047-2797(94)00093-9. [DOI] [PubMed] [Google Scholar]
- 30.Psaty BM, Kuller LH, Bild D, et al. Methods of assessing prevalent cardiovascular disease in the Cardiovascular Health Study. Ann Epidemiol. 1995;5(4):270–277. doi: 10.1016/1047-2797(94)00092-8. [DOI] [PubMed] [Google Scholar]
- 31.Bryan RN, Manolio TA, Schertz LD, et al. A method for using MR to evaluate the effects of cardiovascular disease on the brain: the Cardiovascular Health Study. AJNR Am J Neuroradiol. 1994;15(9):1625–1633. [PMC free article] [PubMed] [Google Scholar]
- 32.Manolio TA, Kronmal RA, Burke GL, et al. Magnetic resonance abnormalities and cardiovascular disease in older adults. The Cardiovascular Health Study. Stroke. 1994;25(2):318–327. doi: 10.1161/01.str.25.2.318. [DOI] [PubMed] [Google Scholar]
- 33.O'Leary DH, Polak JF, Kronmal RA, et al. Distribution and correlates of sonographically detected carotid artery disease in the Cardiovascular Health Study. The CHS Collaborative Research Group. Stroke. 1992;23(12):1752–1760. doi: 10.1161/01.str.23.12.1752. [DOI] [PubMed] [Google Scholar]
- 34.Radloff LS. A self-report depression scale for research in the general population. Appl Psychol Meas. 1977;1:385–401. [Google Scholar]
- 35.Eisner MD, Wang Y, Haight TJ, et al. Secondhand smoke exposure, pulmonary function, and cardiovascular mortality. Ann Epidemiol. 2007;17(5):364–373. doi: 10.1016/j.annepidem.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Hernán MA, Brumback B, Robins JM. Marginal structural models to estimate the causal effect of zidovudine on the survival of HIV-positive men. Epidemiology. 2000;11(5):561–570. doi: 10.1097/00001648-200009000-00012. [DOI] [PubMed] [Google Scholar]
- 37.Robins JM, Hernán MA, Brumback B. Marginal structural models and causal inference in epidemiology. Epidemiology. 2000;11(5):550–560. doi: 10.1097/00001648-200009000-00011. [DOI] [PubMed] [Google Scholar]
- 38.Tager IB, Haight T, Sternfeld B, et al. Effects of physical activity and body composition on functional limitation in the elderly: application of the marginal structural model. Epidemiology. 2004;15(4):479–493. doi: 10.1097/01.ede.0000128401.55545.c6. [DOI] [PubMed] [Google Scholar]
- 39.Petersen ML, Sinisi SE, van der Laan MJ. Estimation of direct causal effects. Epidemiology. 2006;17(3):276–284. doi: 10.1097/01.ede.0000208475.99429.2d. [DOI] [PubMed] [Google Scholar]
- 40.Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3(2):143–155. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- 41.Weinberg CR. Toward a clearer definition of confounding. Am J Epidemiol. 1993;137(1):1–8. doi: 10.1093/oxfordjournals.aje.a116591. [DOI] [PubMed] [Google Scholar]
- 42.Rush D, Callahan KR. Exposure to passive cigarette smoking and child development. A critical review. Ann N Y Acad Sci. 1989;562:74–100. doi: 10.1111/j.1749-6632.1989.tb21008.x. [DOI] [PubMed] [Google Scholar]
- 43.Yolton K, Dietrich K, Auinger P, et al. Exposure to environmental tobacco smoke and cognitive abilities among U.S. children and adolescents. Environ Health Perspect. 2005;113(1):98–103. doi: 10.1289/ehp.7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zlokovic BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 2005;28(4):202–208. doi: 10.1016/j.tins.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Pollard KS, van der Laan MJ. Choice of a null distribution in resampling-based multiple testing. J Stat Plan Inference. 2004;125:85–100. [Google Scholar]
- 46.van der Laan MJ, Robins J. Unified Methods for Censored Longitudinal Data and Causality. New York, NY: Springer-Verlag; 2002. [Google Scholar]
- 47.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1(1):43–46. [PubMed] [Google Scholar]
- 48.Robins JM. Association, causation, and marginal structural models. Synthese. 1999;121:151–179. [Google Scholar]
- 49.Greenland S, Robins J, Pearl J. Confounding and collapsibility in causal inference. Stat Sci. 1999;14:29–46. [Google Scholar]