

Abstract
MauG is a di-heme enzyme responsible for the posttranslational modification of two tryptophan residues to form the tryptophan tryptophylquinone cofactor (TTQ) of methylamine dehydrogenase (MADH). MauG converts preMADH, containing monohydroxylated-βTrp57, to fully functional MADH by catalyzing the insertion of a second oxygen atom into the indole ring and covalently linking βTrp57 to βTrp108. Here we report the 2.1 Å resolution X-ray crystal structure of MauG complexed with preMADH. The c-type heme irons and the nascent TTQ site are separated by long distances over which electron transfer must occur to achieve catalysis. In addition one of the hemes has an atypical His-Tyr axial ligation. The crystalline protein complex is catalytically competent, as on addition of hydrogen peroxide MauG-dependent TTQ synthesis occurs.
Diversity of the cellular proteome is created by genetically encoded polypeptides and their subsequent chemical modification. Most posttranslational modifications, such as phosphorylation, glycosylation and ubiquitination, occur at the protein surface enabling direct contact between the processing enzyme and site of modification. In contrast, protein cofactors essential for function often require in situ modification of amino acids buried within a protein. The formation of some of these is auto-catalytic, such as the fluorophore in green fluorescent protein, but others require external enzymes (1). MauG is a di-heme enzyme (2) that completes synthesis of the catalytic cofactor tryptophan tryptophylquinone (TTQ) (3) from two Trp residues in the β-polypeptide chain of methylamine dehydrogenase (MADH), a metabolic enzyme of methylotrophic and autotrophic bacteria (4). In MADH from Paracoccus denitrificans, two oxygen atoms are incorporated into the indole ring of βTrp57 and a covalent bond is formed between the indole rings of βTrp57 and βTrp108 (Scheme 1) (5).
Scheme 1.
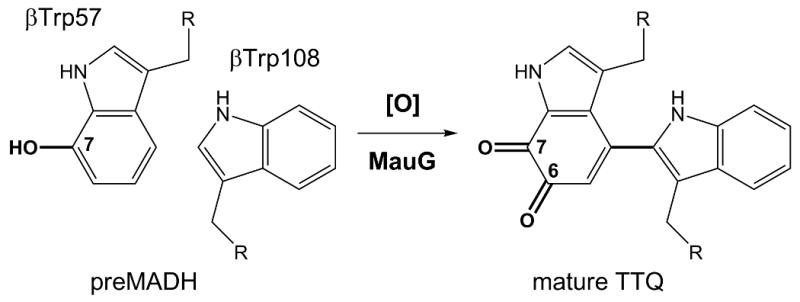
Overall reaction catalyzed by MauG
The natural substrate for MauG (preMADH) is a 119 kDa protein precursor of MADH with monohydroxylated βTrp57 and no cross-link (6, 7). PreMADH can be generated by expression of recombinant MADH in the background of a mauG deletion (6). MauG catalyzes a six-electron oxidation to complete TTQ biosynthesis (8). Oxidizing equivalents may be provided by three moles of either O2 (plus an electron donor) or H2O2 (8, 9). As such, the overall reaction can be viewed as three 2-electron oxidations (of unknown order) to catalyze (i) insertion of −OH at C6 of βTrp57, (ii) formation of the cross-link between βTrp57 and βTrp108, and (iii) oxidation of the quinol to the quinone. MauG contains two c-type hemes, with the diferric form being the resting state. The hemes are covalently bound to the protein and have a His ligand to the iron. c-Type hemes typically function as electron transfer (ET) mediators, or catalytically in certain peroxidases, and indeed there is a sequence relationship between MauG and bacterial di-heme cytochrome c peroxidases (di-heme CCPs) (10). However, unlike di-heme CCPs or other c-type hemes, MauG can activate molecular oxygen and forms an unprecedented di-heme bis-Fe(IV) intermediate that is catalytically competent (11). This intermediate is composed of an Fe(IV)=O (ferryl) heme with the second oxidizing equivalent stored as Fe(IV) at the second heme. It is an intriguing alternative to Compound I, an Fe(IV)=O heme/porphyrin cation-radical, which is the activated oxygen species required for cytochrome P450 and heme-dependent peroxidase-catalyzed chemistry (12, 13).
The X-ray crystal structure of the MauG-preMADH complex of P. denitrificans has been determined to 2.1 Å resolution (Fig. S1 and Table S1) (14). The 203.6 kDa complex consists of two MauG and one preMADH, and comprises the asymmetric unit (Fig. 1A). MADH is an α2β2 heterotetramer that contains two active sites and consequently two sites of posttranslational modification. MauG is monomeric and binds in an identical orientation to each αβ half of preMADH, with 2870 Å2 surface area buried at each interface. The preMADH structure is essentially identical to mature MADH (5) except at βTrp57 and βTrp108, the residues that are modified by MauG. As predicted by 18O isotope labeling and mass spectrometry studies, βTrp57 of preMADH is monohydroxylated at the C7 carbon of the indole ring with no crosslink between the Trp residues (Fig. 2A) (7). Significantly the two Trps are in buried positions comparable to their positions in the mature enzyme. They have no direct contact to any part of MauG, with the edge of the βTrp108 indole ring being closest to the interface (Fig. 1B). There is remarkable separation between the MauG hemes and the nascent TTQ site, with a distance of 40.1 Å between the most distant heme iron and βTrp108 (Table S2).
Figure 1.
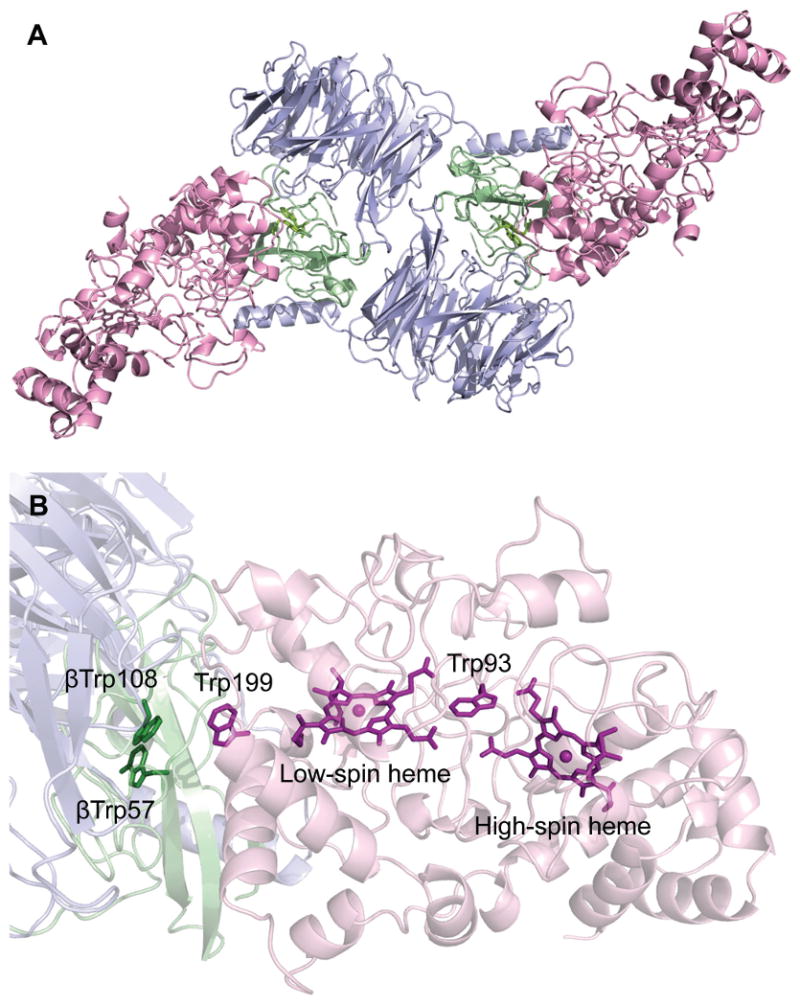
(A) Overall ribbon representation of the MauG-preMADH complex. (B) Spatial layout of potential redox groups. Color scheme is MauG (pink); preMADH α (blue) and β (green). The hemes, Trp93, Trp199 of MauG and βTrp108 and mono-hydroxylated βTrp57 of preMADH are drawn explicitly in a stick representation. Figure produced using PyMOL (http://www.pymol.org/).
Figure 2.
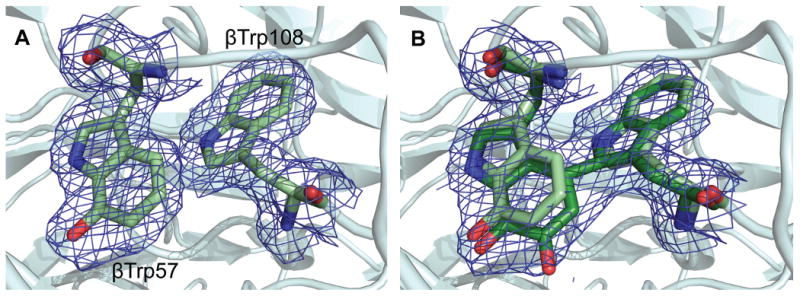
Site of TTQ formation in MADH. (A) 2Fo-Fc electron density for the MauG-preMADH complex (resolution 2.1 Å). (B) The first 2Fo-Fc electron density calculated with MauG-preMADH + H2O2 structure factors (resolution 2.1 Å) and MauG-preMADH model phases with the preMADH βTrp57 and βTrp108 side-chains omitted. Electron densities were contoured at 1 σ. Carbon coloring: preMADH, light green; preMADH + H2O2, dark green. Figure produced using PyMOL (http://www.pymol.org/).
The EPR spectrum of diferric MauG identified two distinct c-type hemes: one high-spin and one low-spin (2). Addition of H2O2 to diferric MauG resulted in formation of the unusual di-heme Fe(IV)=O/Fe(IV) reactive intermediate, as evidenced by Mössbauer and EPR spectroscopies (11). The Mössbauer spectrum of the intermediate was modeled as an Fe(IV)=O heme and a 6-coordinate Fe(IV) heme. The crystal structure reveals that the MauG heme closest to preMADH is 6-coordinate, and exhibits a rare His-Tyr axial ligation (Fig. 3A). The axial Tyr294 ligand is likely responsible for the ability of this heme to stabilize Fe(IV) without requiring an exogenous ligand. Sequence alignments of known MauGs show that the Tyr is strictly conserved (Fig. S2) (2). In contrast the di-heme CCPs, which do not stabilize the bis-Fe(IV) state, have either a Met or His in this position (Fig. S2) (2, 10).
Figure 3.
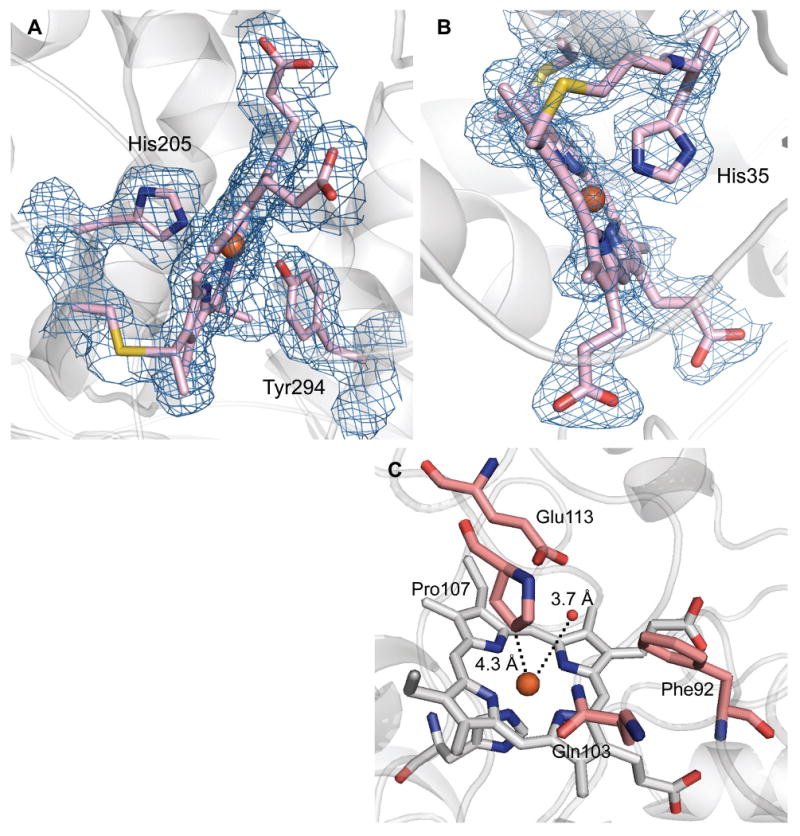
MauG hemes. (A) 6-coordinate low-spin heme (CHE600), (B) 5-coordinate high-spin heme (CHE500), and (C) residues that line the distal pocket. 2Fo-Fc electron density contoured at 1 σ. Figure produced using PyMOL (http://www.pymol.org/).
Spectroscopic data indicate that O2 binding and activation, or direct reaction with H2O2, occurs at the high-spin 5-coordinate heme observed in the crystal structure (11). This is the heme furthest from preMADH in the complex, and thus demands a catalytic mechanism involving long-range interprotein electron and radical transfer (Fig. 1B). There is no evidence of solvent ligation at the coordination site trans to the proximal His35 at this heme (Fig. 3B, C). It was previously demonstrated that the initial 2-electron oxidation of preMADH by MauG exhibits a random-binding kinetic mechanism in which the presence of preMADH neither stimulates not impedes the reaction of MauG with H2O2, O2 or CO (15). The structure of the MauG-preMADH complex is consistent with that finding because it shows that the sites of H2O2/O2 and preMADH binding are well-separated. This kinetic mechanism is in contrast to that of other enzymes that generate high-valent iron species as intermediates, such as the cytochromes P450, which have ordered mechanisms (16). In the distal pocket of the 5-coordinate high-spin heme, the ring of Pro107 is 4.3 Å from the heme iron (Fig. 3C). This is similar to the distance between the organic substrate and iron in ordered-mechanism oxygenases, such as cytochrome P450cam in complex with camphane (4.3Å) (17). The rigidity of Pro107 may afford it the structural role of the substrate in an ordered mechanism by creating a stable O2 binding site and helping to keep the reactive oxygen species localized at the iron (16). As such, the distal pocket of the 5-coordinate high-spin heme of MauG may be regarded as constitutively “on” with respect to its ability to activate O2.
The other characterized bis-Fe(IV) enzyme intermediate is the non-heme Fe(IV)2O2 (Intermediate Q) of methane monooxygenase (18). It has a diamond core structure with an Fe⋯Fe interatomic distance of 2.46 Å. In contrast, the separation of the two heme irons in MauG is 21.1 Å. Despite this distance, the Fe(IV)=O/Fe(IV) intermediate forms very rapidly (>300 s-1) and has remarkable stability, decaying at a rate of 2×10-4 s-1 in the absence of preMADH (15). Trp93 of MauG is positioned between the two hemes such that the propionates of the high-spin and low-spin hemes are 3.3 Å and 3.8 Å respectively from the Trp indole ring (Fig. 1B). This Trp is conserved in both MauGs and di-heme CCPs (2). It has been suggested to play a role in ET between the hemes in the latter. Its position in MauG also suggests a role in mediating ET between the hemes following H2O2/O2 binding and then generation of the bis-Fe(IV) species. MauG exhibits negative redox cooperativity between the c-type hemes in cycling between the diferric and diferrous forms upon successive one-electron redox events. The hemes have similar intrinsic Em values with facile ET between them. Thus, while the determined Em values are distinct (−159mV and −244mV), MauG acts as a single di-heme 2-electron cofactor, going through a valence-delocalized Fe(III)/Fe(II) state (19). In contrast, the Em values of the hemes of di-heme CCPs are separated by more than 600 mV (20), and their reactive state is a mixed-valance Fe(III)/Fe(II) species (10). In comparing the structures of MauG and di-heme CCP, the two hemes and intervening Trp overlay well (Fig. S3). A Ca2+ ion is bound in an identical position in both enzymes (Fig. S3B). Therefore, a structural change altering the spatial relationship between the hemes is not responsible for the different redox and catalytic activities of the two enzymes. The key difference in the di-heme unit of MauG is the distal Tyr294 ligand to the low-spin heme, which is a Met or His in di-heme CCPs. Therefore, Tyr294 appears to be the major determinant in the redox properties and ability of MauG to stabilize the bis-Fe(IV) state.
The catalytic competence of the MauG-preMADH complex was examined in crystallo by solving the crystal structure following treatment with excess H2O2 (Table S1) (14). The initial electron density (to 2.1 Å resolution) clearly showed that the second oxygen atom had been incorporated at C6 of βTrp57, and that a cross-link had been formed between βTrp57 and βTrp108 (Fig. 2B). It has not been possible to crystallize preMADH alone, so the catalytic requirement for MauG in H2O2-dependent TTQ synthesis was confirmed in solution under conditions that matched those of crystallization (Fig. S4) (9). The structures before and after H2O2 addition are superimposable (rmsd 0.158 Ǻ), except for the posttranslational modifications. The TTQ cofactor of mature MADH purified from source (PDB code, 2BBK) overlays well with that generated in the H2O2-treated MauG-preMADH crystals (5). Thus the complex is catalytically competent, and no major conformational rearrangements of the two proteins are required. At the center of the interface half-way between the MauG low-spin heme and preMADH βTrp108 is MauG residue Trp199 (Fig. 1B). This residue may facilitate ET across the interface through stabilization of a transient radical, though it is not conserved in other MauG sequences (Fig. S2). The catalytic competence of the crystals also indicates a processive mechanism in which MauG and preMADH do not dissociate between each of the three 2-electron oxidations. Furthermore, solvent must provide the second oxygen incorporated at βTrp57 C6, as this carbon is 43.7 Ǻ from the Fe(IV)=O. The distal side of the high-spin heme of H2O2-treated crystals has residual electron density not present in the MauG-preMADH structure. The low occupancy and complexity of the electron density prevent it from being modeled, but it is consistent with a mixture of species that could include diatomics, such as hydroperoxo, and is evident in both copies in the asymmetric unit, confirming that reaction with H2O2 occurs at this site (Fig. S5).
This structure reveals that the purpose of the high-valent intermediate in MauG is to provide an extremely high potential oxidant to extract electrons from the preMADH substrate, generating reactive radical intermediates that then acquire the oxygen atom from solvent and form the covalent crosslink between the Trp side-chains. As such the preMADH structure contributes significantly to the catalytic reaction through positioning the reacting portions of the Trps and creating the required chemical environment for TTQ synthesis. Thus the posttranslational biosynthetic reaction requires both substrate-assisted and long-range catalysis.
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM66569 to C.M.W and GM41574 to V.L.D., and a Minnesota Partnership for Biotechnology and Medical Genomics grant SPAP-05-0013-P-FY06. Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute, and we thank Can Ergenekan for his support. X-ray data were collected at the Kahlert Structural Biology Laboratory (KSBL) at The University of Minnesota and GM/CA-CAT at the Advanced Photon Source (APS), Argonne National Laboratory, Argonne, IL. GM/CA CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. DE-AC02-06CH11357. We thank Ed Hoeffner for KSBL support, and the staff at Sector 23, APS for their support, especially Michael Becker, Steve Corcoran, Venugopalan Nagarajan and Derek Yoder. We thank Sooim Shin for performing the Fig. S4 experiment. Co-ordinates and structure factors have been deposited in the Protein Data Bank with acquisition codes 3L4M for the MauG-preMADH complex, and 3L4O for the MauG-preMADH complex treated with H2O2.
References and Notes
- 1.Davidson VL. Biochemistry. 2007 May 8;46:5283. doi: 10.1021/bi700468t. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, et al. Biochemistry. 2003 Jun 24;42:7318. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 3.McIntire WS, Wemmer DE, Chistoserdov A, Lidstrom ME. Science. 1991 May 10;252:817. doi: 10.1126/science.2028257. [DOI] [PubMed] [Google Scholar]
- 4.Davidson VL. Adv Protein Chem. 2001;58:95. doi: 10.1016/s0065-3233(01)58003-1. [DOI] [PubMed] [Google Scholar]
- 5.Chen LY, et al. J Mol Biol. 1998 Feb 13;276:131. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 6.Pearson AR, et al. Biochemistry. 2004 May 11;43:5494. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 7.Pearson AR, Marimanikkuppam S, Li X, Davidson VL, Wilmot CM. J Am Chem Soc. 2006 Sep 27;128:12416. doi: 10.1021/ja064466e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, et al. J Am Chem Soc. 2005 Jun 15;127:8258. doi: 10.1021/ja051734k. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. Biochemistry. 2006 Nov 7;45:13276. doi: 10.1021/bi061497d. [DOI] [PubMed] [Google Scholar]
- 10.Pettigrew GW, Echalier A, Pauleta SR. J Inorg Biochem. 2006 Apr;100:551. doi: 10.1016/j.jinorgbio.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Li X, et al. Proc Natl Acad Sci USA. 2008 Jun 24;105:8597. [Google Scholar]
- 12.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005 Jun;105:2253. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 13.Hersleth HP, Ryde U, Rydberg P, Gorbitz CH, Andersson KK. J Inorg Biochem. 2006 Apr;100:460. doi: 10.1016/j.jinorgbio.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 14.Materials and methods are available as supporting material on Science Online.
- 15.Lee S, Shin S, Li X, Davidson V. Biochemistry. 2009 Feb 5; doi: 10.1021/bi802166c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makris TM, Davydov R, Denisov IG, Hoffman BM, Sligar SG. Drug Metab Rev. 2002 Nov;34:691. doi: 10.1081/dmr-120015691. [DOI] [PubMed] [Google Scholar]
- 17.Raag R, Poulos TL. Biochemistry. 1991 Mar 12;30:2674. doi: 10.1021/bi00224a016. [DOI] [PubMed] [Google Scholar]
- 18.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Accounts Chem Res. 2007 Jul;40:475. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Feng M, Wang Y, Tachikawa H, Davidson VL. Biochemistry. 2006 Jan 24;45:821. doi: 10.1021/bi052000n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fulop V, Watmough NJ, Ferguson SJ. Adv Inorg Chem. 2001;51:163. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.