

Abstract
Cancer stem cells (CSCs) are thought to be critical for the engraftment and long-term growth of many tumors, including glioblastoma (GBM). The cells are at least partially spared by traditional chemotherapies and radiation therapies, and finding new treatments that can target CSCs may be critical for improving patient survival. It has been shown that the NOTCH signaling pathway regulates normal stem cells in the brain, and that GBMs contain stem-like cells with higher NOTCH activity. We therefore used low-passage and established GBM-derived neurosphere cultures to examine the overall requirement for NOTCH activity, and also examined the effects on tumor cells expressing stem cell markers. NOTCH blockade by γ-secretase inhibitors (GSIs) reduced neurosphere growth and clonogenicity in vitro, whereas expression of an active form of NOTCH2 increased tumor growth. The putative CSC markers CD133, NESTIN, BMI1, and OLIG2 were reduced following NOTCH blockade. When equal numbers of viable cells pretreated with either vehicle (dimethyl sulfoxide) or GSI were injected subcutaneously into nude mice, the former always formed tumors, whereas the latter did not. In vivo delivery of GSI by implantation of drug-impregnated polymer beads also effectively blocked tumor growth, and significantly prolonged survival, albeit in a relatively small cohort of animals. We found that NOTCH pathway inhibition appears to deplete stem-like cancer cells through reduced proliferation and increased apoptosis associated with decreased AKT and STAT3 phosphorylation. In summary, we demonstrate that NOTCH pathway blockade depletes stem-like cells in GBMs, suggesting that GSIs may be useful as chemotherapeutic reagents to target CSCs in malignant gliomas.
Keywords: Cancer Stem Cell, NOTCH, Glioblastoma, γ-Secretase inhibitor
Introduction
More than 41,000 people are diagnosed with primary brain tumors each year in the U.S. [1], and glioblastomas (GBMs) are the most common malignant brain tumors in adults [2]. Even with advances in surgery, imaging, chemotherapy, and radiation therapy over the past three decades, more than 70% of GBM patients still die within 2 years of diagnosis [1-3]. New strategies to treat this deadly disease are desperately needed [1]. In this study, we focus on the potential therapeutic role of NOTCH pathway inhibition, and the requirement for ongoing NOTCH signaling in stem-like tumor cells.
There is emerging evidence that a small population of cancer stem cells (CSCs) within neoplasms is responsible for long-term tumor propagation, with initial studies focused on leukemia [4]. Several groups have subsequently demonstrated that stem-like cells exist in brain tumors, including GBMs [4-9]. CSC markers such as CD133 and side population have been used to prospectively isolate a small fraction of cells in human and rodent brain tumors with a significantly increased potential to generate tumor neurospheres and xenografts [5, 7, 8, 10, 11]. Tumor-derived neurospheres also have a genetic profile that remains closer to that of the tumor from which they are isolated than traditional adherent GBM cell lines maintained in high serum [12]. In addition, GBMs propagated as neurospheres maintain stem-like subpopulations, and more accurately replicate the infiltrating growth patterns seen in primary tumors [9, 12]. GBM neurosphere cultures thus provide an improved pathological model, and may also facilitate the discovery of new therapeutic reagents that can target the stem-like or xenograft-initiating cells required for long-term tumor growth.
The Notch locus was first described by Morgan in a strain of Drosophila with notched wing blades [13]. The gene was subsequently cloned as a cell surface receptor [14], playing a key role in the development of many different cell types and tissues, including neurons in the central nervous system [15-18]. NOTCH signaling is initiated when transmembrane ligands on one cell bind NOTCH receptors on an adjacent cell and cause the γ-secretase-mediated proteolytic release of the NOTCH intracellular domain (NICD) [19, 20]. NICD then translocates into the nucleus where it interacts with the transcriptional cofactor CBF1 and activates targets such as the HES and HEY genes, which modulate neuronal and glial differentiation [21]. In vertebrates, at least four NOTCH receptors (NOTCH 1-4), five ligands (JAG1, 2, DLL1, 3, 4), and multiple effector molecules (HES1-6, HEY1, 2 L) have been identified [21, 22]. NOTCH ligands, receptors, and targets have been found in a wide range of neoplasms, including lung, breast, cervix, head/neck, renal, and pancreas carcinoma, neuroblastoma, myeloma, melanoma, choroid plexus tumor, medulloblastoma, and GBM [20, 22-33]. In many of these tumor types, it has been shown that increased NOTCH activity promotes tumor growth, whereas NOTCH pathway blockade inhibits proliferation and/or survival. Inhibition of NOTCH signaling is therefore a promising therapeutic avenue in a wide range of cancers.
We and others have demonstrated that the NOTCH signaling pathway plays an important role in the pathogenesis of medulloblastoma and GBM [28, 34-36], and that these malignant brain tumors contain stem-like cancer cells with higher NOTCH activity [9, 12, 32]. It has recently been reported that common chemotherapeutic drugs, including temozolomide, carboplatin, paclitaxel (Taxol), and etoposide (VP16), as well as traditional radiation therapy, predominantly targeted the CD133-negative population, and spared or enriched the CD133-positive population [37, 38]. We have also observed an increase in the stem-like subpopulation in GBM-derived neurosphere lines following radiation therapy [39]. Thus, conventional chemotherapies and radiation therapies appear to effectively remove only better-differentiated cells, while leaving many GBM CSCs alive. The NOTCH pathway represents a possible target in stem-like glioma cells, as several groups have shown that GBM CSCs express NOTCH family genes, and that tumor-derived neurospheres have elevated NOTCH activity [9, 12, 32]. It is known that NOTCH regulates normal neural stem cell self-renewal and differentiation [40, 41], and that CSCs share many characteristics with their normal cognates, including the signaling pathways that regulate self-renewal. In the present study, we used both established and low-passage GBM cultures as a model to examine the effects of NOTCH pathway blockade on CSCs and tumor growth.
Materials and Methods
Cell Culture
GBM neurosphere cultures were maintained in Neurocult medium (Stem Cell Technologies, Vancouver, BC, Canada, http://www.stemcell.com) supplemented with epidermal growth factor (20 ng/ml) and fibroblast growth factor (10 ng/ml). Cell pools and stable subclones transfected with NOTCH2 intracellular domain (NICD2) were generated as previously described [34, 42]. For treatment studies, cells were plated and allowed to grow overnight in Neurocult medium; Neurocult was then replaced the next morning with medium containing γ-secretase inhibitor 18 (GSI-18) dissolved in dimethyl sulfoxide (DMSO) at the concentrations of 0.4-50 μM. RNA and protein extractions and all cell-based assays were performed 2-5 days after drug application. Cell mass was measured using CellTiter assays according to the instructions of the manufacturer (Promega, Madison, WI, http://www.promega.com). Cell number and viability were assessed using the Guava PCA and Viacount reagent according to instructions (Guava Technologies, Hayward, CA, http://www.guavatechnologies.com). All experiments were performed in triplicate.
γ-Secretase Inhibitor Synthesis
The potent γ-secretase inhibitor [11-endo]-N-(5,6,7,8,9,10-hexahydro-6,9-methanobenzo[a] [8]annulen-11-yl)-thiophene-2-sulfonamide was listed as compound 18 in the recent report by Lewis et al. [43] describing its synthesis and testing, and we refer to it as GSI-18. It was synthesized as previously described [43], and its identity and quality were confirmed by nuclear magnetic resonance and mass spectral analysis. The biological activity of this inhibitor against γ-secretase was confirmed using in vitro and cell-based assays as previously described [44]. MRK-003 was generously provided by Merck Research Laboratories (Boston, MA, http://www.merck.com) [43].
Immunocytochemistry
The GBM neurosphere lines and low-passage cultures were seeded in 12-well tissue culture plates overnight. After treating with 2 μM GSI-18 for 2-5 days, cells were washed with phosphate-buffered saline (PBS), spun down onto slides, and fixed using 4% paraformaldehyde solution in PBS at room temperature for 30 minutes. Cells were then permeabilized with 0.4% Triton X-100 in PBS for 5 minutes at room temperature, washed with PBS, and incubated in 5% bovine serum albumin (BSA)-PBS for 1 hour and then in 1:1,000 anti-NESTIN antibody (Chemicon, Temecula, CA, http://www.chemicon.com) or 1:500 anti-Ki-67 (DAKO, Glostrup, Denmark, http://www.dako.com) in 1% BSA-PBS for 2 hours. After washing with PBS, a final 60-minute incubation with a 1:300 dilution of cyanin 3 (Cy3)-conjugated goat anti-mouse and Cy2-conjugated goat anti-rabbit secondary antibodies (Jackson Immunoresearch Laboratories, West Grove, PA, http://www.jacksonimmuno.com) diluted in PBS containing 1% BSA was done. After washing with PBS, cells were then counter-stained with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, http://www.vectorlabs.com), mounted, and visualized with fluorescence microscope (Carl Zeiss, Jena, Germany, http://www.zeiss.com); 10 high-power fields were photographed and counted. For immunocytochemical evaluation of intracranial xenografts, frozen sections of cryoprotected brains were stained with anti-human antibody (1:100; Millipore, Billerica, MA, http://www.millipore.com).
Flow Cytometric Analyses
Flow cytometric analysis of CD133 was done with antibodies from Miltenyi Biotec (Auburn, CA, http://www.miltenyibiotec.com) according to the instructions of the manufacturer using a FACSCa-libur (Becton Dickinson, San Jose, CA, http://www.bd.com) with CELLQuest Version 3.3 software (Becton Dickinson, San Jose, CA, http://www.bd.com) [42]. In brief, cells were blocked in Fc receptor blocking reagent and incubated with CD133/1 (AC133)-phycoerythrin antibody (Miltenyi Biotec) for 10 minutes in the dark at 4° C. Then cells were washed and resuspended in 500 μL buffer (PBS containing 0.5% BSA and 2 mM EDTA). Cells expressing levels of CD133 higher than those seen in immunoglobulin G (IgG) controls were considered positive. Experiments measuring CD133 were repeated two to three times for each line and error bars represent the mean and SE.
Real-Time Reverse Transcription-Polymerase Chain Reaction
Quantitative reverse transcription-polymerase chain reaction (RT-PCR) for NOTCH1, NOTCH2, and HES1 was done as previously described [34, 45], with all reactions normalized to actin (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com). Commercially available Assay on Demand TaqMan (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com) primers and probes were used to measure mRNA for CD133, NESTIN, BMI1, OLIG2, HES5, and HEY1. Each quantitative RT-PCR reaction was done in triplicate and error bars represent SE.
Protein Analysis
Western blots contained 20 μg of protein per lane on a 10% Tris-glycine SDS-polyacrylamide gel electrophoresis gel (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) and were electrophoresed for several hours in 1 × Tris-glycine (TG)-SDS buffer (Amresco, Solon, OH, http://www.amresco-inc.com). Proteins were transferred to 0.45-μm Optitran nitrocellulose (Schleicher & Schuell, Keene, NH, http://www.schleicher-schuell.com) in 1 × Tris-glycine buffer (Amresco). Blots were blocked in PBS containing 5% nonfat dry milk powder and incubated overnight at 4 °C with antibodies directed against HES1 (1:2,000; kind gift of Dr. Tetsuo Sudo, Toray Industries, Tebiro, Kamakura, Japan, http://www.toray.com), cleaved caspase-3 (Asp175) (1:1,000; Cell Signaling Technology, Beverly, MA, http://www.cellsignal.com), phosphorylated Akt (Ser473) and total Akt (1:1,000; Cell Signaling Technology), phospho-STAT3 (pSer727) and total STAT3 (1:1,000; Affinity BioReagents, Golden, CO, http://www.bioreagents.com), or glyceraldehyde-3-phosphate dehydrogenase (1:20,000; Research Diagnostics, Flanders, NJ, http://www.researchd.com). Blots were then washed several times with PBS containing 0.1% Tween 20 and incubated in peroxidase-conjugated IgG diluted 1:2,000 in blocking solution. After washing several times in PBS with 0.1% Tween 20, blots were developed with enhanced chemiluminescence reagent (Pierce, Rockford, IL, http://www.piercenet.com) and exposed to film.
Flank and Intracranial Xenograft Tumors
For flank xenograft tumors, 5 × 105 viable GBM neurosphere cells were mixed 1:1 with Matrigel (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com) and a total volume of 200 μl was injected s.c. into each flank of ether-anesthetized 4-week-old female nude mice. Measurements of tumor size in the long and short axes were made weekly and estimates of volume were made using the following formula: width2 × length × 0.52 [46]. For intracranial xenografts, injection guide holes were produced in anesthetized animals by twirling an 18-gauge beveled needle to provide access to the intracranial space. Fifty thousand viable cells in 2 μl of PBS were used for intracranial injection into the right striatum (0.2 μL/minute) by stereotactic injection through a glass pipette connected to a Hamilton syringe. The following coordinates were used: antero-posterior = 0; medio-lateral = 2.5 mm; dorso-ventral = 3.5 mm [9]. The skin was sutured following injection. Fifteen days after initial injection, the NOTCH inhibitor was delivered using polymer-based beads soaked with GSI-18 into the tumor beds as described previously [47]. DMSO-soaked beads were used as controls. Tumor growth was monitored by magnetic resonance imaging (MRI) scanning at 6 weeks after GSI treatment, but survival was used as the primary endpoint in these studies. In vivo MRI studies were performed on a horizontal 9.4 T MR scanner (Bruker Biospin, Billerica, MA, http://www.bruker-biospin.com) with a triple-axis gradient and an animal imaging probe. The scanner was also equipped with a physiological monitoring system (EKG, respiration, and body temperature). Mice were anesthetized with isoflurane (1%) together with oxygen and air at 1:3 ratio via a vaporizer and a facial mask. We used a 40-mm diameter birdcage coil for the radio frequency transmitter and receiver. Temperature was maintained by a heating block built into the gradient system. Respiration was monitored throughout the entire scan and was used to synchronize the start of each acquisition. Multiple slice T2-weighted MRIs were acquired with the following parameters: fast spin echo sequence with an echo train length of 4, echo time = 35 ms, repetition time = 3,000 ms, imaging resolution = 0.1 mm × 0.1 mm per pixel, slice thickness = 0.3 mm, total imaging time of 30 minutes with respiratory triggering.
Statistical analysis
Statistical analyses were done using GraphPad Prism 4 (GraphPad Software, http://www.graphpad.com). Data graphed with error bars represent mean and SE from experiments done in triplicate unless otherwise noted. A two-sided Student’s t test was used to determine the significance of any differences.
Results
GBM Neurospheres Are Sensitive to NOTCH Pathway Blockade In Vitro
We initially studied well-characterized HSR-GBM1 neurosphere cultures (Fig. 1A), which were derived from a single tumor [9] and grown for several years as three independent sublines. The stem cell marker CD133 is expressed in these GBM-derived neurospheres, as detected using flow cytometry, in approximately 70% of the tumor cells (Fig. 1A). This line was previously shown to express markers of NOTCH activity at the time it was established [9], and we confirmed protein expression of the NOTCH pathway target HES1 in the tumor neurospheres (Fig. 1B).
Figure 1.
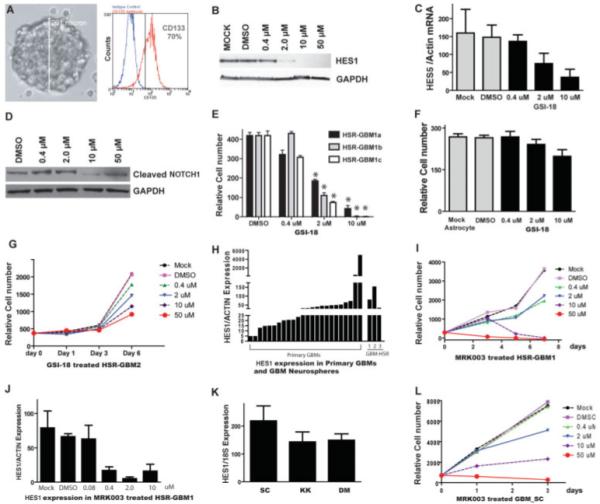
GBM neurospheres are sensitive to NOTCH pathway blockade in vitro. (A): The GBM neurosphere line HSR-GBM1 grows as large, multicellular spheres. The majority (~70%) of neurosphere cells express CD133. (B): Upon GSI-18 treatment, protein levels of HES1, a NOTCH pathway target gene, were reduced after 48 hours of GSI-18 treatment of HSR-GBM1 neurospheres. (C, D): Upon GSI-18 treatment, mRNA expression of NOTCH target gene HES5 and protein levels of cleaved active NOTCH1 intracellular domain are also reduced. (E): GSI-18 reduced total cell mass in a dose-dependent fashion in all three HSR-GBM1 sublines (*, p < .05). (F): There is no significant change in normal human astrocyte growth when treated with GSI-18. (G): Growth of a second GBM neurosphere line, HSR-GBM2, was also inhibited by GSI-18. (H): HES1 mRNA levels fell into a similar range in primary GBM and tumor-derived neurospheres grown in culture. (I): A second γ-secretase inhibitor, MRK-003, could also suppress GBM neurosphere growth. (J): MRK003 showed potency similar to GSI-18 in terms of its ability to lower HES1 mRNA levels in tumor neurospheres. (K, L): Low-passage neurospheres GBM-SC, GBM-KK, and GBM-DM express the NOTCH target gene HEY1 and had their growth inhibited by NOTCH blockade. Abbreviations: DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GBM, glioblastoma; GSI-18, γ-secretase inhibitor 18.
NOTCH pathway blockade for 48 hours using 2 μM γ-secretase inhibitor (GSI-18) decreased levels of HES1 protein in the HSR-GBM1 neurospheres by more than 80% (quantified by densitometryon the Western blot in Fig. 1B), and no HES1 was detected when 10 μM or higher concentrations of the drug were used (Fig. 1B). Because HES1 can be regulated by multiple pathways, we also examined two other markers of NOTCH signaling, HES5 expression and formation of the activated NOTCH1 intracellular domain. Both were decreased in a dose-dependent fashion by GSI-18 (Fig. 1C, 1D), providing additional confirmation of baseline NOTCH activity and pharmacologic inhibition of the pathway in tumor neurospheres. GSI-18 reduced growth of all three HSR-GBM1 sublines in vitro in a dose-dependent fashion (Fig. 1E). In contrast, the growth of normal human astrocytes was not significantly affected by the same level of GSI-18 (Fig. 1F), indicating that this agent does not nonselectively inhibit proliferation of astrocytic cells. Two slower-growing established GBM neurosphere lines (HSR-GBM2 and HSR-GBM3) were also inhibited in a dose-dependent fashion by GSI-18 (Fig. 1G and data not shown). The fact that HES1 mRNA in these GBM neurosphere lines was expressed at levels within the range seen in 24 snap-frozen GBM samples suggests they had biologically relevant levels of NOTCH activity (Fig. 1H).
To help exclude the possibility that the effects of GSI-18 were nonspecific, we also used another γ-secretase inhibitor with a significantly different structure, MRK-003. This second compound also suppressed HSR-GBM1 growth and HES1 levels in a dose-dependent fashion (Fig. 1I, 1J). Finally, we analyzed the growth of GBM neurospheres derived from four different tumors and grown for relatively few passages (6-20). High NOTCH activity in these cultures was confirmed by NOTCH target gene expression (Fig. 1K). Growth of all four lines (SC, DM, KO, and TZ) was inhibited 70% or more by 10 μM MRK-003, whereas three of the lines (SC, KO, and TZ) showed a modest growth suppression by 2 μM MRK-003 (Fig. 1L and data not shown). These studies indicate that both long-established and lower-passage GBM-derived neurospheres require ongoing NOTCH signaling for growth in culture.
Activation of NOTCH2 in GBM Neurospheres Increases Their Growth In Vitro
To examine whether activation of NOTCH signaling in GBM neurospheres is sufficient to increase tumor growth, we introduced a truncated, constitutively active form of the NOTCH2 receptor (NICD2) into GBM neurosphere lines using either stable transfection or transient expression following infection by adenovirus we generated previously [34]. We used NICD2 in these experiments as NOTCH2 receptor protein has been shown to be highly expressed in glioblastoma [48]. Following NICD2 transfection and selection, HSR-GBM1 cells had significantly increased mRNA levels of NOTCH2 and the pathway target HES1 (Fig. 2A–2C). The elevated NOTCH2 signaling following transfection of NICD2 was associated with an approximately twofold increase in growth over 5 days in culture (Fig. 2D). Transient viral delivery of NICD2 also significantly increased the growth rate of GBM-derived neurospheres, albeit to a lesser degree (Fig. 2E). This less robust induction of growth may reflect the fact that only 60%–80% of cells are infected by virus containing NICD2. These findings suggest that activation of NOTCH2 is sufficient to promote GBM neurosphere growth in vitro.
Figure 2.
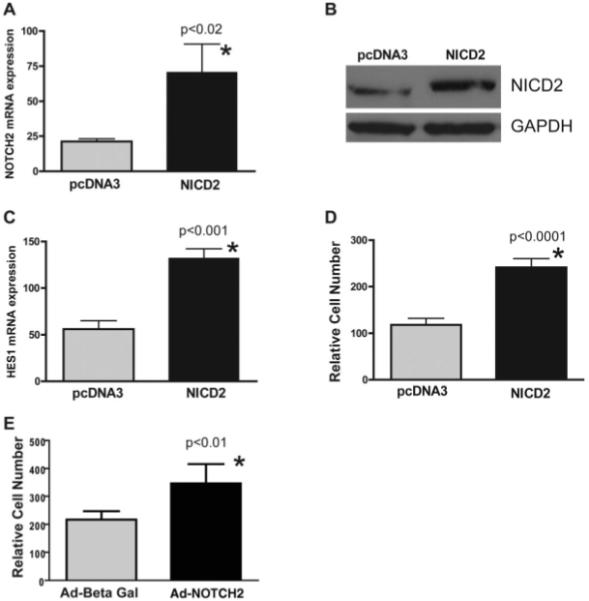
Activation of NOTCH2 in glioblastoma (GBM) neurospheres increases tumor growth in vitro. (A): When the active form of NOTCH2 receptor (NICD2) was stably transfected into HSR-GBM1a cells, receptor mRNA levels were elevated. (B): NICD2 protein expression was also increased in NICD2 stably transfected HSR-GBM1 neurospheres detected by Western blot. GAPDH was used as protein loading control. (C): The NOTCH target gene HES1 was also induced in NICD2-transfected GBM neurospheres. (D): Cell growth was significantly increased in NICD2-transfected cells. (E): Transient expression of NICD2 in HSR-GBM1 neurospheres using adenovirus vectors also increased cell growth over 2 days. Abbreviations: Ad, Adenovirus; Beta Gal, β-galactosidase; GAPDH, glyceral-dehyde-3-phosphate dehydrogenase; NICD2, NOTCH2 intracellular domain.
NOTCH Blockade Depletes CD133-Positive Cells and Inhibits Neurosphere Clonogenicity
We have previously shown that CD133 marks cells with xenograft-initiating potential [42, 49], and that NOTCH blockade can deplete stem-like cells in medulloblastoma cultures [42]. In HSR-GBM1 neurospheres, we found an approximately twofold increase in the clonogenic potential of CD133-positive cells compared with negative ones (C.G.E. and E. Bar, unpublished data). We examined CD133 mRNA levels in HSR-GBM1 cells after NOTCH pathway blockade by GSI-18, and found that this stem cell marker was almost completely suppressed by 3-μM levels of the γ-secretase inhibitor after 48 hours (Fig. 3A). We also identified a pronounced, dose-dependent decrease in the percentage of CD133-expressing cells using flow cytometric analysis, suggesting that the cancer stem cell fraction within the tumor had been depleted (Fig. 3B). Given the evidence from two recent studies showing that the CD133-negative GBM population also forms xenografts in nude mice [50, 51], suggesting that this marker may not mark all CSCs, we also examined several other stem cell markers including NESTIN, BMI1, and OLIG2. The expression of all of these was also inhibited by GSI-18 in a dose-dependent fashion, indicating a broad effect on potential CSC markers (Fig. 3C–3E). We next examined clonogenicity after treatment with 5 μM MRK-003 to document functional loss of clonogenic CSCs. Some tumor neurospheres were still present in culture after 5 days of NOTCH inhibition (Fig. 3F, upper right panel), however, when these were dissociated, and equal numbers of viable cells seeded into semisolid neurosphere media, those that had previously been exposed to NOTCH blockade could not form colonies (Fig. 3F, 3G). Similar but less pronounced effects were seen with 2-μM levels of the γ-secretase inhibitor (Fig. 3G). These data demonstrate that NOTCH pathway blockade depletes stem-like cells in GBM, and decreases tumor clonogenic growth in vitro.
Figure 3.
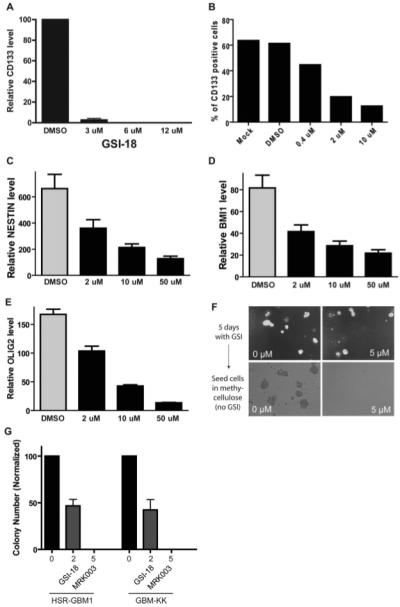
NOTCH blockade depletes CD133-positive cells and inhibits neurosphere clonogenicity. (A): CD133 mRNA levels were severely decreased following GSI-18 treatment of HSR-GBM1 cells. (B): The percentage of CD133-positive cells in these cultures was also lowered by NOTCH blockade. (C–E): mRNA levels of the stem cell markers NESTIN, BMI1, and OLIG2 were also reduced in a dose-dependent fashion following GSI-18 treatment of HSR-GBM1 neurospheres. (F, G): Growing HSR-GBM1 cultures treated for 5 days with γ-secretase inhibitor can form some neurospheres (right top panel), but when equal numbers of DMSO- and MRK003-treated cells are seeded singly in methylcellulose, the latter cannot efficiently form colonies. Abbreviations: DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GBM, glioblastoma; GSI-18, γ-secretase inhibitor 18.
NOTCH Signaling Blockade Depletes Cells Required for GBM Engraftment In Vivo
To extend our in vitro findings, we studied the effects of GSI-18 on in vivo tumor growth. First, we treated GBM neurospheres for 2 days with either GSI-18 or vehicle (DMSO) ex vivo, long enough to remove most stem-like GBM cells based on the in vitro experiments described above. We then injected equal numbers of surviving cells, which were demonstrated to be viable by trypan blue staining and vital count assays, subcutaneously into nude mice. After monitored tumor growth for more than 3 months, we found that GSI-18 pretreatment completely inhibited the growth of subcutaneous HSR-GBM1 xenografts (Fig. 4A–4D). Large tumor masses formed at all eight sites (Fig. 4A, 4B, asterisks) injected with cells in which NOTCH signaling was continuously active, including nontreated (n = 4) and vehicle (DMSO)-treated (n = 4) cells (Fig. 4A, 4B). In contrast, mice injected with an equal number of viable cells previously treated with 2 μM GSI-18 (n = 6) were unable to efficiently form tumors, with small lesions developing at only three of six injection sites (Fig. 4C). A comparison of growing tumor volumes in mock-, DMSO-, and 2 μM GSI-18-treated xenografts is shown in Figure 4D. The small xenografts that are formed from GSI-18-treated GBM cells had a microscopic appearance quite similar to those from DMSO-treated cells (Fig. 4E, 4F). They contained abundant HES1 protein (Fig. 4G), indicating that cells that survive NOTCH blockade and grow slowly into small tumors in vivo eventually re-establish expression of NOTCH pathway targets.
Figure 4.
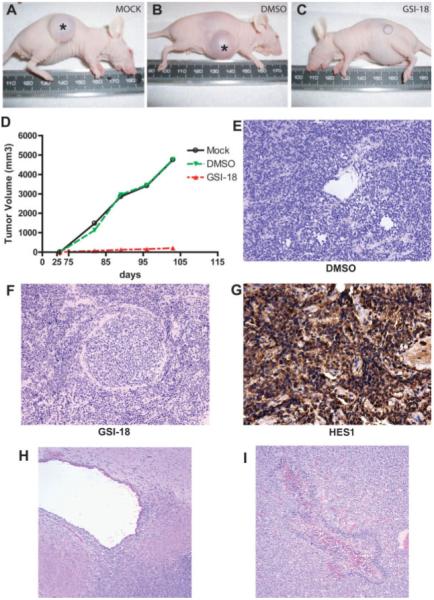
NOTCH signaling blockade depletes cells required for glioblastoma (GBM) propagation in vivo. (A–C): Large xenografts formed at all eight sites (asterisk) injected with cells in which NOTCH signaling was active, including nontreated (n = 4, A) and vehicle (DMSO)-treated (n = 4) cultures. Mice injected with equal number of viable cells previously treated with 2 μM GSI-18 (n = 6) were unable to efficiently form tumors, with small lesions developing at only three of six injection sites. (D): Tumor volume was dramatically reduced in mice engrafted with 2 μM GSI-18-pretreated cells compared with mice engrafted with mock or DMSO-pretreated cells. (E–G): The small subcutaneous xenografts that formed from cells pretreated with GSI-18 appeared similar to control tumors, and expressed abundant amounts of the NOTCH pathway target HES1 on immunohistochemical analysis. (H, I): Intracranial xenografts of HSR-GBM1 cells pretreated with DMSO or GSI-18 often invaded into the subventricular space (H), and occasionally formed necrotic foci with pseudopalisading (I). (Original magnifications both ×100.) Abbreviations: DMSO, dimethyl sulfoxide; GSI-18, γ-secretase inhibitor 18.
To determine whether downregulation of NOTCH signaling prior to injection of GBM neurospheres also affects intracranial tumor initiation, we treated HSR-GBM1 with 2 μM or 5 μM GSI-18 for 48 hours ex vivo to deplete CSCs, then stereotactically injected 50,000 viable cells into mouse brains. The same number of cells from DMSO-treated neurospheres was used as a positive control, with four mice included per group. Mice were sacrificed when they showed neurological signs suggesting tumor growth, and survival was used as the primary endpoint for statistical analysis. We found that animals injected with GSI-18-pretreated GBM neurospheres did form orthotopic xenografts and die, but had a significantly longer survival compared with those injected with DMSO-treated tumor cells (p < .05, log-rank analysis). Microscopic examination of the xenografts that formed from the GSI-18-pretreated (Fig. 4H, 4I) and DMSO-pretreated (not shown) GBM neurospheres failed to identify any significant differences in the size or histopathological appearance of the gliomas, which were all large and infiltrative. Interestingly, many of the tumor xenografts, both treated and nontreated, showed a predilection for invasion and growth in the subependymal region around the ventricles (Fig. 4H and data not shown), perhaps due to the capacity of this area to support the growth of stem and progenitor cells. In some large tumors, pseudopalisading necrosis, a common feature of glioblastoma, was also seen (Fig. 4I). The fact that tumor neurosphere cultures pretreated with γ-secretase inhibitor did form large xenografts, albeit more slowly than controls, may reflect the fact that our in vitro analysis suggests only a partial depletion of CD133-positive cells following NOTCH blockade.
Local Delivery of γ-Secretase Inhibitors Slows Growth of Intracranial GBM Xenografts
To mimic more closely the care of GBM patients, and to test the effects of longer-term delivery of γ-secretase inhibitor, we also treated small intracranial tumors using local delivery of the NOTCH inhibitor. We injected GBM neurospheres into mouse brains and waited for 15 days, a time frame during which we predict that small tumors should form based on prior xenograft time-course studies. Polymer-based beads soaked with GSI-18 or DMSO were then surgically implanted into the tumor beds as described previously [47]. To confirm that GSI-18 prevented GBM neurosphere propagation in vivo due to downregulation of NOTCH signaling pathway, we also injected cells stably transfected with NICD2, thereby constitutively activating NOTCH2 signaling and rendering the cells insensitive to γ-secretase blockade. We monitored tumor growth at 6 weeks after injection by MRI scanning in representative animals, and found that DMSO (vehicle)-treated mice form large xenografts, whereas GSI-18 treatment appeared to block formation of radiographically detectable tumors (Fig. 5A). The NICD2 transfected GBM neurospheres appeared to form larger intracranial xenografts, and were not sensitive to GSI-18 treatment (Fig. 5A). When we compared the survival of the various groups, mice treated with GSI-18 also had a significantly prolonged survival compared with DMSO-treated mice, whereas animals bearing NICD2 xenografts had shorter survival and were not sensitive to the GSI-18 treatment (Fig. 5B). Microscopic examination of brains from treated animals revealed implanted polymer beads and adjacent injection tracts but no recognizable surviving tumor cells in the multiple sections examined (Fig. 5C). Immunocytochemical analysis of intervening sections with anti-human antibodies that can recognize xenografted cells also failed to identify glioma (Fig. 5D, 5E). Thus significant tumor growth is blocked in most animals following local delivery of γ-secretase inhibitor.
Figure 5.
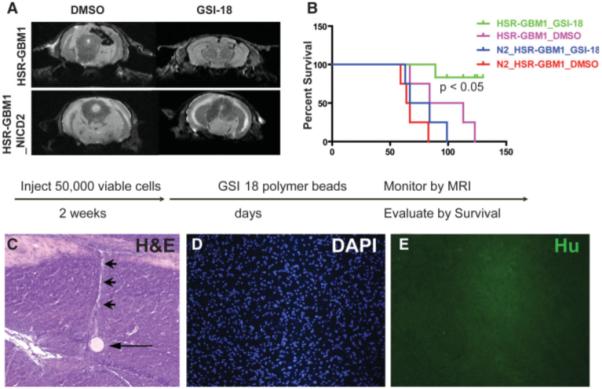
Local GSI treatment prolongs survival of mice bearing intracranial GBM xenografts. (A): Wild-type or NICD2 stably transfected GBM neurospheres were implanted into mouse brain, and GSI-18- or DMSO-soaked beads were injected into the tumor beds 2 weeks later. Tumor growth was monitored by MRI in selected animals, and large xenografts formed in DMSO- but not GSI-18-treated mice. As expected, constitutive NICD2 expression rendered xenografts insensitive to GSI-18. (B): GSI-18-treated mice have significantly prolonged survival compared with DMSO-treated mice, whereas expression of activated NOTCH2 promoted more rapid intracranial tumor growth. (C–E): Microscopic examination of a GSI-18-treated mouse brain stained with H&E reveals a round polymer bead (arrow) with a pale linear injection tract leading upward (arrowhead), but no tumor mass on this or additional step sections (original magnification ×100). Immunocytochemical stains using an antibody specific for human nuclei confirms the absence of viable tumor cells (D, E). (Original magnification ×100.) Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GBM, glioblastoma; GSI-18, γ-secretase inhibitor 18; Hu, human nuclei; MRI, magnetic resonance imaging; NICD2, NOTCH2 intracellular domain.
NOTCH Blockade Selectively Reduces Proliferation of Stem-Like GBM Cells
To examine the mechanism by which NOTCH regulates stem-like cells in GBM, we used double immunolabeling to assess proliferation in cells expressing the stem/progenitor markers. Because it has been shown that NOTCH regulates NESTIN expression in GBM [52], raising the possibility that population changes could be due to direct effects of NOTCH blockade on NESTIN levels rather than loss of stem-like cells, we used both NESTIN and CD133 as markers. Consistent with the concept that ongoing NOTCH signaling is required in poorly differentiated cells, we found that pathway blockade by GSI-18 significantly reduced the percentage of cells expressing high levels of NESTIN or CD133 in GBM neurospheres (Fig. 6A, 6B). The overall Ki-67-positive proliferating fraction of cells was also significantly reduced (Fig. 6B). Importantly, proliferation rates were most dramatically lowered by GSI-18 in the NESTIN- or CD133-bright population, whereas NESTIN- or CD133-dim cells showed a far lesser reduction in cycling cells (Fig. 6C, 6D). This suggests that NOTCH inhibition depletes the stem-like tumor subpopulation at least in part due to reduced proliferation. Although it is not entirely clear how the proliferation rate of stem-like glioma cells might affect tumor growth, it has recently been shown that the presence of a high percentage of glioblastoma cells coexpressing CD133 and Ki-67 is associated with worse clinical outcomes [53].
Figure 6.
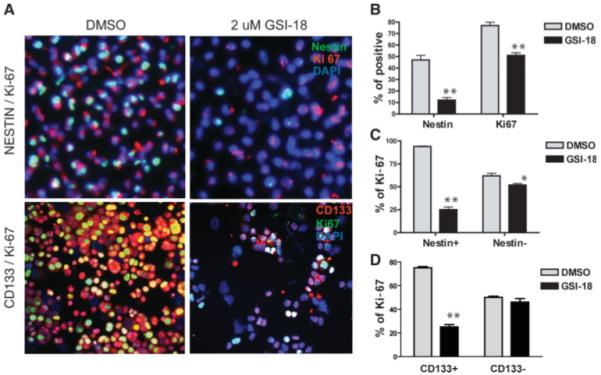
NOTCH signaling regulates proliferation of poorly differentiated GBM cells. (A): NOTCH pathway blockade by GSI-18 reduces the NESTIN- or CD133-positive population in neurosphere lines, as assessed by double immunolabeling (upper panel: NESTIN in green; Ki-67, red; DAPI, blue; lower panel: CD133 in red; Ki-67, green; DAPI, blue). (B): Both NESTIN- and Ki-67-positive populations were significantly reduced. (C, D): Ki-67 proliferation indices were significantly lowered by GSI-18 in the NESTIN- or CD133-positive cell population, whereas NESTIN- or CD133-negative cells showed a lesser reduction (p value for t tests: *, p < .05, **, p < .01). Abbreviations: DAPI, 4′,6-diamidino-2-phenylindole; DMSO, dimethyl sulfoxide; GSI-18, γ-secretase inhibitor 18.
NOTCH Signaling Regulates Phosphorylation of Akt and STAT3 in GBM Neurospheres
To address the possibility that NOTCH blockade induces cell death in GBM neurosphere cells, as well as decreasing proliferation, we examined cleavage of caspase-3 following GSI-18 treatment. An increase in the proapoptotic cleaved form of this protein was identified in cultures treated with 0.4 μM and higher levels of the NOTCH pathway inhibitor, suggesting that cell death was induced by NOTCH blockade (Fig. 7). We also wanted to identify the downstream targets that might mediate the apoptotic effects of NOTCH pathway blockade in GBM neurospheres. AKT and p53 are two key proteins that regulate apoptosis in many type of cancers [54]. We therefore examined whether protein levels or phosphorylation states of p53 and AKT were changed in GSI-18-treated GBM neurospheres. We did not observe p53 alteration in GSI-18-treated GBM cells (data not shown). However, 2 μM GSI-18 totally blocked AKT phosphorylation (pAKT-Ser473) in the GBM neurosphere line HSR-GBM1 (Fig. 7). These results suggest that phosphorylation and activation of AKT promoted by NOTCH may mediate survival of GBM CSCs. Recently, it has been shown that NOTCH signaling can regulate stem cell numbers both in vitro and in vivo through phosphorylation of STAT3 (pS727) [55]. To determine whether STAT3 might also be affected by NOTCH inhibition in brain tumors, we examined pSTAT3 protein changes in GSI-18-treated HSR-GBM1 neurospheres. We found that phosphorylation of STAT3 was significantly reduced by NOTCH pathway blockade in a dose-dependent fashion, whereas total STAT3 was essentially unchanged in GSI-18-treated HSR-GBM1 neurospheres (Fig. 7). These results suggested that NOTCH signaling may regulate GBM CSC survival through phosphorylation of AKT and STAT3.
Figure 7.
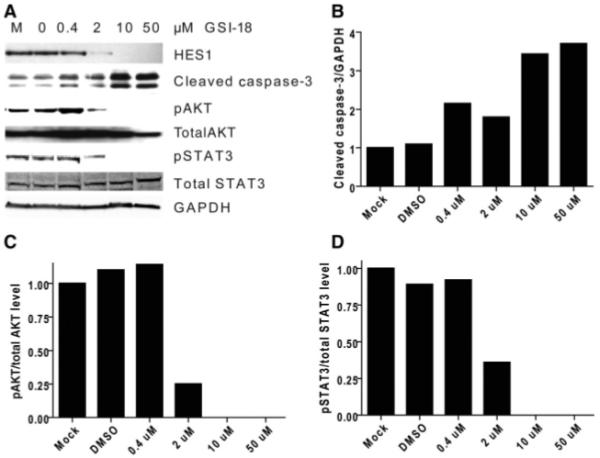
NOTCH signaling blockade promotes caspase-3 cleavage and reduces phosphorylation of AKT and STAT3. (A): Loss of AKT and STAT3 phosphorylation and activation of caspase-3 cleavage is induced by 2 μM or higher levels of GSI-18, whereas overall levels of AKT and STAT3 are unchanged in GBM neurosphere cells. GAPDH is used as a loading control. These changes were quantitated using densitometry in (B–D). Abbreviations: DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GBM, glioblastoma; GSI-18, γ-secretase inhibitor 18.
Discussion
A number of groups analyzing GBM have identified molecular changes in NOTCH pathway members, implicating activation of this developmentally important signaling cascade in the pathobiology of malignant brain tumors [9, 12, 32]. In the current study, we used neurospheres derived from human GBM specimens to examine the effects of NOTCH pathway blockade. Such neurosphere lines represent an improved model system, as when injected orthotopically into immunocompromised mice they recapitulate the pathobiology of GBM more closely than traditional adherent cell lines maintained in high serum. Both established and lower-passage neurosphere cultures were used. We found that NOTCH pathway blockade by either of two structurally distinct γ-secretase inhibitors slows tumor growth in all GBM neurosphere cultures tested, whereas overexpression of a constitutively active form of NOTCH2 increases tumor growth. In addition, we demonstrate that NOTCH pathway blockade inhibits GBM neurosphere engraftment in vivo, and significantly prolongs the survival of mice bearing intracranial xenografts. Larger preclinical studies will be needed to confirm and extend these data.
It has been suggested that NOTCH may play a particularly important role in CSCs, a subpopulation of tumor cells that have stem-like properties and are uniquely capable of enabling engraftment and long-term growth of cancers [5-10, 32, 56-58]. Although a few gain-of-function studies directly support the concept that NOTCH is important in stem-like GBM cells [52, 59], to our knowledge it has not been shown that NOTCH pathway inhibition can deplete CSCs in gliomas. We found that pharmacological NOTCH blockade using γ-secretase inhibitors reduces the percentage of cells expressing the stem/progenitor cell markers CD133 and NESTIN in GBM neurospheres. In addition, following NOTCH blockade, surviving cells are no longer able to efficiently form colonies in vitro or engraft in vivo, consistent with the concept that a key subpopulation of cells required for efficient tumor propagation is no longer present. We believe that the depletion of the GBM subpopulation expressing stem cell markers, in conjunction with the loss of clonogenic and engraftment potential, suggests that NOTCH blockade can at least partially remove CSCs from GBM neurosphere lines.
Gastrointestinal toxicity has been a major concern when using GSI, with NOTCH blockade inducing goblet cell differentiation in intestinal transit amplifying cells, leading to severe diarrhea [60-63]. To avoid such side effects, we used polymer-based beads to deliver GSI-18 locally within the brain in our in vivo experiments. This method is conceptually similar to local delivery of the chemotherapeutic agent biodegradable carmustine via polymer wafers to the tumor bed, a Food and Drug Administration (FDA)-approved strategy currently used in GBM patients [64-67]. The fact that this local delivery of GSI-18 significantly prolongs survival without any detectable gastrointestinal side effects in animals suggests a strategy for translating GSI-based therapies into neuro-oncology clinics. One additional potential concern when targeting CSCs in the brain is that NOTCH inhibition may also deplete normal neural stem cells. However, we did not find obvious neurological defects in the animal receiving GSI over several months of observation.
To maximize the efficacy of anti-CSC agents such as γ-secretase inhibitors, it will likely be necessary to give them in combination with other treatment modalities (surgery, radiation therapy, other chemotherapy). Indeed, it has recently been shown that giving a γ-secretase inhibitor in conjunction with the glucocorticoid dexamethasone can potentiate its therapeutic effect and reduce systemic side effects when treating leukemia [68]. In this regard, our discovery that the reduced proliferation and induction of apoptotic markers in GBM following NOTCH blockade is associated with phosphorylation changes to AKT and STAT3 suggests additional pathways that might be synergistically targeted.
Summary
In summary, our data support the concept that γ-secretase inhibitors can inhibit the growth of functionally heterogeneous GBM neurospheres, both in vitro and in vivo, with particularly pronounced effects on poorly differentiated tumor cells. The potential therapeutic implications of targeting of stem-like cells via NOTCH inhibition extend beyond neuro-oncology, as this signaling pathway has been shown to play a role in CSCs not only in neural tumors, but also in cancers from breast, lung, pancreas, and prostate [26, 69-72]. More studies will be needed to determine whether pharmacological NOTCH blockade, either alone or in conjunction with other therapies, will be effective in improving the survival of patients with GBM and other malignant tumors.
Acknowledgments
We acknowledge grant support to X.F. from Accelerate Brain Cancer Cure Project Award, American Brain Tumor Association Translational Grant, and Voices Against Brain Cancer Research Grant, and to C.G.E. from NIH/NS55089 and the Brain Tumor Funders Collaborative. We also acknowledge Merck and Merck scientists for providing MRK-003.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Reardon DA, Rich JN, Friedman HS, et al. Recent advances in the treatment of malignant astrocytoma. J Clin Oncol. 2006;24:1253–1265. doi: 10.1200/JCO.2005.04.5302. [DOI] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, et al. WHO Classification of Tumours of the Central Nervous System. IARC Press; Lyon, France: 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 5.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- 7.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 8.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A. 2004;101:781–786. doi: 10.1073/pnas.0307618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 10.Taylor MD, Poppleton H, Fuller C, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 13.Morgan T. The theory of the gene. Am Nat. 1917;51:513–544. [Google Scholar]
- 14.Artavanis-Tsakonas S, Muskavitch MA, Yedvobnick B. Molecular cloning of Notch, a locus affecting neurogenesis in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1983;80:1977–1981. doi: 10.1073/pnas.80.7.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Louvi A, Artavanis-Tsakonas S. Notch signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:93–102. doi: 10.1038/nrn1847. [DOI] [PubMed] [Google Scholar]
- 16.Miele L. Notch signaling. Clin Cancer Res. 2006;12:1074–1079. doi: 10.1158/1078-0432.CCR-05-2570. [DOI] [PubMed] [Google Scholar]
- 17.Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science. 2005;307:1904–1909. doi: 10.1126/science.1104815. [DOI] [PubMed] [Google Scholar]
- 18.Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci. 2005;8:709–715. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]
- 19.Mizutani T, Taniguchi Y, Aoki T, et al. Conservation of the biochemical mechanisms of signal transduction among mammalian Notch family members. Proc Natl Acad Sci U S A. 2001;98:9026–9031. doi: 10.1073/pnas.161269998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nickoloff BJ, Osborne BA, Miele L. Notch signaling as a therapeutic target in cancer: a new approach to the development of cell fate modifying agents. Oncogene. 2003;22:6598–6608. doi: 10.1038/sj.onc.1206758. [DOI] [PubMed] [Google Scholar]
- 21.Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol. 2003;194:237–255. doi: 10.1002/jcp.10208. [DOI] [PubMed] [Google Scholar]
- 22.Allenspach EJ, Maillard I, Aster JC, et al. Notch signaling in cancer. Cancer Biol Ther. 2002;1:466–476. doi: 10.4161/cbt.1.5.159. [DOI] [PubMed] [Google Scholar]
- 23.Houde C, Li Y, Song L, et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood. 2004;104:3697–3704. doi: 10.1182/blood-2003-12-4114. [DOI] [PubMed] [Google Scholar]
- 24.Pece S, Serresi M, Santolini E, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167:215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parr C, Watkins G, Jiang WG. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int J Mol Med. 2004;14:779–786. doi: 10.3892/ijmm.14.5.779. [DOI] [PubMed] [Google Scholar]
- 26.Miyamoto Y, Maitra A, Ghosh B, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–576. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 27.van Limpt V, Chan A, Caron H, et al. SAGE analysis of neuroblastoma reveals a high expression of the human homologue of the Drosophila Delta gene. Med Pediatr Oncol. 2000;35:554–558. doi: 10.1002/1096-911x(20001201)35:6<554::aid-mpo13>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 28.Purow BW, Haque RM, Noel MW, et al. Expression of Notch-1 and its ligands, Delta-like-1 And Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res. 2005;65:2353–2363. doi: 10.1158/0008-5472.CAN-04-1890. [DOI] [PubMed] [Google Scholar]
- 29.Zagouras P, Stifani S, Blaumueller CM, et al. Alterations in Notch signaling in neoplastic lesions of the human cervix. Proc Natl Acad Sci U S A. 1995;92:6414–6418. doi: 10.1073/pnas.92.14.6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leethanakul C, Patel V, Gillespie J, et al. Distinct pattern of expression of differentiation and growth-related genes in squamous cell carcinomas of the head and neck revealed by the use of laser capture microdissection and cDNA arrays. Oncogene. 2000;19:3220–3224. doi: 10.1038/sj.onc.1203703. [DOI] [PubMed] [Google Scholar]
- 31.Rae FK, Stephenson SA, Nicol DL, et al. Novel association of a diverse range of genes with renal cell carcinoma as identified by differential display. Int J Cancer. 2000;88:726–732. doi: 10.1002/1097-0215(20001201)88:5<726::aid-ijc7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 32.Ignatova TN, Kukekov VG, Laywell ED, et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 33.Dang L, Fan X, Chaudhry A, et al. Notch3 signaling initiates choroid plexus tumor formation. Oncogene. 2006;25:487–491. doi: 10.1038/sj.onc.1209074. [DOI] [PubMed] [Google Scholar]
- 34.Fan X, Mikolaenko I, Elhassan I, et al. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004;64:7787–7793. doi: 10.1158/0008-5472.CAN-04-1446. [DOI] [PubMed] [Google Scholar]
- 35.Hallahan AR, Pritchard JI, Hansen S, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 36.Fan X, Eberhart CG. Medulloblastoma stem cells. J Clin Oncol. 2008;26:2821–2827. doi: 10.1200/JCO.2007.15.2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radio-resistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 39.Bar EE, Chaudhry A, Lin A, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen Q, Goderie SK, Jin L, et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 41.Gaiano N, Fishell G. The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci. 2002;25:471–490. doi: 10.1146/annurev.neuro.25.030702.130823. [DOI] [PubMed] [Google Scholar]
- 42.Fan X, Matsui W, Khaki L, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 43.Lewis SJ, Smith AL, Neduvelil JG, et al. A novel series of potent gamma-secretase inhibitors based on a benzobicyclo[4.2.1]nonane core. Bioorg Med Chem Lett. 2005;15:373–378. doi: 10.1016/j.bmcl.2004.10.062. [DOI] [PubMed] [Google Scholar]
- 44.Li YM, Lai MT, Xu M, et al. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci U S A. 2000;97:6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan X, Wang Y, Kratz J, et al. hTERT gene amplification and increased mRNA expression in central nervous system embryonal tumors. Am J Pathol. 2003;162:1763–1769. doi: 10.1016/S0002-9440(10)64311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boehm T, Folkman J, Browder T, et al. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 47.Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 48.Sivasankaran B, Degen M, Ghaffari A, et al. Tenascin-C is a novel RBPJkappa-induced target gene for Notch signaling in gliomas. Cancer Res. 2009;69:458–465. doi: 10.1158/0008-5472.CAN-08-2610. [DOI] [PubMed] [Google Scholar]
- 49.Eberhart CG. In search of the medulloblast: neural stem cells and embryonal brain tumors. Neurosurg Clin N Am. 2007;18:59–69. doi: 10.1016/j.nec.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 50.Beier D, Hau P, Proescholdt M, et al. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 51.Ogden AT, Waziri AE, Lochhead RA, et al. Identification of A2B5+CD133− tumor-initiating cells in adult human gliomas. Neurosurgery. 2008;62:505–514. doi: 10.1227/01.neu.0000316019.28421.95. discussion 514–505. [DOI] [PubMed] [Google Scholar]
- 52.Shih AH, Holland EC. Notch signaling enhances nestin expression in gliomas. Neoplasia. 2006;8:1072–1082. doi: 10.1593/neo.06526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pallini R, Ricci-Vitiani L, Banna GL, et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin Cancer Res. 2008;14:8205–8212. doi: 10.1158/1078-0432.CCR-08-0644. [DOI] [PubMed] [Google Scholar]
- 54.Reed JC. Apoptosis-targeted therapies for cancer. Cancer Cell. 2003;3:17–22. doi: 10.1016/s1535-6108(02)00241-6. [DOI] [PubMed] [Google Scholar]
- 55.Androutsellis-Theotokis A, Leker RR, Soldner F, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- 56.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 57.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 59.Zhang XP, Zheng G, Zou L, et al. Notch activation promotes cell proliferation and the formation of neural stem cell-like colonies in human glioma cells. Mol Cell Biochem. 2008;307:101–108. doi: 10.1007/s11010-007-9589-0. [DOI] [PubMed] [Google Scholar]
- 60.Milano J, McKay J, Dagenais C, et al. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82:341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- 61.van Es JH, van Gijn ME, Riccio O, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 62.Wong GT, Manfra D, Poulet FM, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279:12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 63.Searfoss GH, Jordan WH, Calligaro DO, et al. Adipsin, a biomarker of gastrointestinal toxicity mediated by a functional gamma-secretase inhibitor. J Biol Chem. 2003;278:46107–46116. doi: 10.1074/jbc.M307757200. [DOI] [PubMed] [Google Scholar]
- 64.Brem H, Ewend MG, Piantadosi S, et al. The safety of interstitial chemotherapy with BCNU-loaded polymer followed by radiation therapy in the treatment of newly diagnosed malignant gliomas: phase I trial. J Neurooncol. 1995;26:111–123. doi: 10.1007/BF01060217. [DOI] [PubMed] [Google Scholar]
- 65.Valtonen S, Timonen U, Toivanen P, et al. Interstitial chemotherapy with carmustine-loaded polymers for high-grade gliomas: a randomized double-blind study. Neurosurgery. 1997;41:44–48. doi: 10.1097/00006123-199707000-00011. discussion 48–49. [DOI] [PubMed] [Google Scholar]
- 66.Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Westphal M, Ram Z, Riddle V, et al. Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien) 2006;148:269–275. doi: 10.1007/s00701-005-0707-z. discussion 275. [DOI] [PubMed] [Google Scholar]
- 68.Real PJ, Tosello V, Palomero T, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15:50–58. doi: 10.1038/nm.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu S, Dontu G, Wicha MS. Mammary stem cells, self-renewal pathways, and carcinogenesis. Breast Cancer Res. 2005;7:86–95. doi: 10.1186/bcr1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patrawala L, Calhoun T, Schneider-Broussard R, et al. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2− cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207–6219. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- 71.Sriuranpong V, Borges MW, Ravi RK, et al. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001;61:3200–3205. [PubMed] [Google Scholar]
- 72.Shou J, Ross S, Koeppen H, et al. Dynamics of notch expression during murine prostate development and tumorigenesis. Cancer Res. 2001;61:7291–7297. [PubMed] [Google Scholar]