

Abstract
RhoA-activated kinase (ROK) is involved in disorders of smooth muscle contraction found in hypertension model animals and patients. We examined whether the α1-adrenergic receptor agonist-induced ROK signal is perturbed in resistance small mesentery artery (SMA) of Lyon genetically hypertensive (LH) rats, using a ROK antagonist, Y27632. Smooth muscle strips of SMA and aorta were isolated from LH and Lyon normotensive (LN) rats. After Ca2+-depletion and pre-treatment with phenylephrine (PE), smooth muscle contraction was induced by serial additions of CaCl2. In LH SMA Ca2+ permeated cells to a lesser extent as compared to LN SMA, while CaCl2-induced contraction of LH SMA was greater than that of LN SMA, indicating a higher ratio of force to Ca2+ in LH SMA contraction (Ca2+ sensitization). No hyper-contraction was observed in LH aorta tissues. Treatment of LH SMA with Y27632 restored both Ca2+ permeability and Ca2+-force relationship to levels seen for LN SMA. In response to PE stimulation, phosphorylation of CPI-17, a phosphorylation-dependent myosin phosphatase inhibitor protein, and MYPT1 at Thr853, the inhibitory phosphorylation site of the myosin phosphatase regulatory subunit, was increased in LN SMA, but remained unchanged in LH SMA. These results suggest that the disorder in ROK-dependent Ca2+ permeability and Ca2+-force relationship is responsible for LH SMA hyper-contraction. Unlike other hypertensive models, the ROK-induced hyper-contractility of LH SMA is independent of MYPT1 and CPI-17 phosphorylation, which suggests that ROK-mediated inhibition of myosin phosphatase does not affect SMA hyper-contractility in LH SMA cells.
Keywords: Lyon hypertensive rats, resistance mesentery artery, α1 adrenergic contraction, Ca2+ signaling, Ca2+ sensitivity, Ca2+ influx, RhoA, ROK, CPI-17, MYPT1, Protein phosphatase
Introduction
The Lyon hypertensive rat strain (LH) is a model for genetic hypertension, which displays several phenotypes associated with elevated blood pressure, including increased mortality and body weight, and spontaneous hyperlipidemia [1]. A large number of studies in both animal models and humans indicate a link between endothelium dysfunction and hypertension [2–4]. By contrast, endothelial functions of resistance small arteries of LH are intact, compared with the control normotensive Lyon strain (LN) [5]. Enhanced constriction of small caliber resistance arteries and arterioles causes an increase in peripheral vascular resistance of LH rats, which is an important component of human essential hypertension and other animal models of hypertension [5]. Several abnormalities of Ca2+ handling have been found in LH, such as increased cellular Ca2+ levels in platelets and erythrocytes [6] and an enhanced responsiveness of renal circulation to dihydropyridine L-type Ca2+ channel openers and blockers [7]. However, little is known about the possible dysregulation of smooth muscle contraction in LH arteries.
Smooth muscle contraction is regulated via the reciprocal activities of myosin light chain kinase and phosphatase. Agonist stimulation inhibits myosin phosphatase activity, resulting in an enhanced Ca2+ sensitivity of the contraction, called Ca2+ sensitization [8, 9]. Augmented agonist-induced Ca2+ sensitization of smooth muscle contraction has been found in stroke-prone spontaneously hypertensive rats [10]. Accumulating evidence suggests that up-regulation of the RhoA/Rho-activated coiled-coil kinase (ROK) pathway is responsible for the augmented Ca2+ sensitization in several hypertensive animal models [10–19] and human patients [20, 21]. For example, the smooth muscle contraction and blood pressure of hypertensive models and patients are more sensitive to Y27632, a ROK inhibitor, compared with those of normotensive controls. ROK mediates G-protein-coupled receptor activation resulting in the inhibition of myosin phosphatase. In particular, the sustained phase of agonist-induced smooth muscle contraction is dependent on ROK activation and the phosphorylation of myosin phosphatase regulatory subunit, MYPT1, and an inhibitor protein, CPI-17 [22, 23]. Thr696 of MYPT1 is known to be an inhibitory phosphorylation site [9], but the phosphorylation level is unchanged upon agonist stimulation of arteries [24]. On the other hand, phosphorylation of MYPT1 at Thr853 and CPI-17 at Thr38 is enhanced in response to G-protein activation and is inhibited by ROK inhibitors [24, 25]. CPI-17 is also phosphorylated by PKC that is responsible for Ca2+-dependent Ca2+ sensitization at transient phase [23, 26].
In addition to its role in Ca2+ sensitization, ROK is also involved in activating a norepinephrine-induced Ca2+ entry mechanism that is distinct from Ca2+ influx via voltage- or store-operated channels in rat arteries [27]. Furthermore, Ca2+ influx induces the activation of the Rho/ROK pathway in response to agonist stimulation, suggesting a bi-directional regulation between Ca2+ and ROK [28]. In the present study we used small mesentery arteries (SMA) to test the hypothesis that ROK signaling causes dysregulation in the smooth muscle contraction of LH rat tissues. SMAs are involved in controlling blood flow by responding to α1-adrenergic receptor stimulation, which plays a critical role in blood pressure regulation [29]. Ca2+-depleted SMA strips were stimulated by re-addition of extra-cellular Ca2+ that causes Ca2+ influx through voltage-gated, receptor-operated Ca2+ channels and Na/Ca exchangers [30]. We discovered a decrease in Ca2+ influx and an increase in Ca2+-force relationship following phenylephrine (PE) stimulation in LH SMA, but not in aorta. The defect in LH SMA was improved by addition of Y27632, whereas the ROK signal is disconnected from the inhibition of myosin phosphatase.
Materials and Methods
Animals and blood pressure measurement
Male LH and LN rats (16–18 weeks old) were obtained from the laboratory of one of us (Pr. Sassard, Lyon, France), and the animal protocol was authorized by the French government (Department of Agriculture, No. 01918) for the use of laboratory animals given by. All rats were anesthetized with sodium pentobarbital (60 mg/kg, intraperitoneal) prior to any procedure. Mean arterial blood pressure was determined by direct measurement through a carotid catheter and a transducer (SensoNor® SP844, Horten, Norway).
Arterial preparation and mounting
The thoracic aorta and SMA (branch II or III, diameter 150–200 μm) were each removed from the same rat. The tissues were prepared as described previously [30]. Briefly, segments of aorta (2–3 mm in width) or SMA (1.6–2.0 mm in width) were mounted on myographs in physiological salt solution (PSS) of the following composition (in mM): NaCl 119, KCl 4.7, MgSO4 1.17, KH2PO4 0.4, NaHCO3 12.5, CaCl2 2.5 and glucose 5.5. The PSS was continuously kept at 37°C and gassed with 95% O2 and 5% CO2. The vessels were stretched under a passive wall tension of either 2g for aorta or 200mg for SMA [31]. The endothelial layer was removed immediately after dissection, either by gentle rubbing (for aorta) or by perfusion with 0.5% of 3-[(3-cholamidopropyl) dimethylammonio]-1 propane sulphonate (CHAPS, Sigma-Aldrich) in PSS, followed by repeated washing with PSS (for SMA). The absence of relaxation response to acetylcholine (ACh, 1 μM, Sigma-Aldrich) in PE (1 and 10 μM for aorta and SMA, respectively) pre-contracted vessels was taken as evidence that the vessel segments were denuded of functional endothelium.
Contraction experiments
Intracellular Ca2+ in tissues was depleted by a 15-min treatment under a nominally Ca2+-free PSS containing 0.5 mM ethylene glycol bis(β-aminoethyl ether) - N,N,N′,N′-tetraacetic acid (EGTA), followed by exposure of arteries to PE (10 and 30 μM for aortic rings and SMA segments, respectively) in Ca2+-free PSS. After approximately 1 min (or return of tension to baseline in some vessels), arteries no longer responded to PE or KCl stimulation without extra-cellular Ca2+ in the bath. Contraction was then induced by cumulative additions of CaCl2 (10 μM – 10 mM) to the bath, and the force level at plateau was recorded in the continuous presence of PE or KCl. Y27632 (10 μM) and GF109203x (3 μM) were added to the bath 15 min prior to transfer in Ca2+-free PSS and were continuously present during the whole experiment. Y27632 and GF109203x were obtained from Calbiochem and Tocris, respectively. The vehicle of GF109203X, DMSO, at concentration of 0.3 % had no effect on contractile response in the experimental conditions. The other compounds were freely dissolved in distilled water. The extent of SMA contraction was normalized in force (mN) developed per unit length (mm) of arterial segments, as previously described by Mulvany and Halpern [31].
Measurement of intracellular calcium concentration ([Ca2+]i) in SMA
[Ca2+]i and force were simultaneously recorded as described previously [30]. Briefly, SMA segments were mounted on a myograph as described above. The myograph was coupled to a dual excitation wavelength fluorometer (SPEX-AR/CM), and changes in [Ca2+]i were determined by measuring the fluorescence of trapped fura-2. The vessel segments were loaded with fura-2/AM, (5 μM) by incubation for > 2 h in PSS containing the Pluronic F127 (0.02%) and bovine serum albumin (2%). After washing, they were then left for 15 min in a nominally Ca2+-free PSS containing 0.5 mM EGTA. The vessels were then exposed to PE (30μM) for approximately 1 min. Next, Ca2+-induced contraction was induced by addition of CaCl2 to these PE-exposed vessels, using the same protocol as in contraction experiments. Due to difficulties in interpreting in situ Fura-2 calibration and subsequent conversion of [Ca2+]i absolute values in blood vessels [32], [Ca2+]i was determined as the fluorescence ratio of fura-2 excited at 340 and 380 nm (recorded at 510 nm). Contraction was simultaneously recorded. The experiments were performed either in the absence or presence of Y27632 (10 μM) added to the bath. At the end of each experiment, the maximum and minimum values of the fluorescence ratio were obtained in the presence of ionomycin (20 μM) and CaCl2 (5 mM), and EGTA (20 mM), respectively, to confirm that there were no significant differences in specimens.
Tissue preparation and Western blotting
TCA-fixed phospho-SDS samples were prepared by essentially following the procedure by Kitazawa et al [33]. Briefly, SMA segments were treated for 10 min with 0.3 mM CaCl2, in the absence and presence of PE, as described above, plus calyculin A (0.1 μM), which effectively prevented dephosphorylation during sample preparation [34]. After the treatment, segments were frozen in liquid N2, slowly thawed in TCA-acetone, and then homogenized in SDS lysis buffer (50 mM Tris-HCl, pH 8.0, supplemented with 1.2 mM sodium ortho-vanadate, 1% SDS, 1 mM EDTA, 1 μM microcystin LR, 0.4 mM Pefabloc™). The samples were homogenized by sonication, and cleared by centrifugation for 10 min at 20,000 ×g. After determination of protein concentrations by BCA method (Pierce), SDS lysates were mixed with Laemmli buffer with 2-mercaptoethanol and boiled for 5 min in a 100 °C heat block.
Total proteins (20 μg) were loaded on 10 % (for MYPT1) or 15 % (for CPI-17) polyacrylamide gels. Proteins were transferred to polyvinylidene difluoride membranes using a semidry transfer unit. The membranes were blocked in Tris-buffered saline (TBS) solution containing 0.1 % Tween-20, and 3 % Bovine Serum Albumin for 1 h at RT and then incubated overnight at 4°C, with the following primary antibodies from Millipore/Upstate: anti-CPI-17 (1:1,000 dilution), anti-P-CPI-17 (1:500), anti-MYPT1 (1:1,000), anti-P-Thr38-CPI-17 (1:1,000), and anti-P-Thr696-MYPT1 (1:1,000), anti-P-Thr853-MYPT1 (Millipore/Upstate, 1:500). After incubation with a secondary antibody (Jackson ImmunoResearch), the membranes were subjected to enhanced chemiluminescence development (Supersignal™, Pierce) and exposed to X-ray film. Band intensity was measured by Densitometer (Molecular Probes). Immunoblotting was first done with anti-phospho-specific antibodies, and the membrane was reprobed with the anti-pan antibodies, after stripping for 1 h with 0.1 M Gly-HCl, pH 1.9. The intensity of the phospho-protein signal was normalized with that of pan protein signal. Each blot was subjected to reprobing only once. Since high phosphatase activity was present in the SMA samples, 0.1 μM calyculin A was added to the bath to maintain the phosphorylation levels. PE stimulation increased the phosphorylation of CPI-17 and MYPT1 at Thr853 and the phosphorylation declined by kinase inhibitor under this condition (Figure 4).
Figure 4. Phosphorylation of CPI-17 at Thr38 (A), MYPT1 (B) at Thr696 and at Thr853 in SMA from LN and LH rats.

Phosphorylation of each protein was measured by immunoblotting method as described in Methods. The band intensity of anti-phospho-CPI-17 Thr38 (A), MYPT1-Thr696, and -Thr853 (B) was normalized with that of anti-pan CPI-17 (A), and MYPT1 (B), respectively. Data from SMA without and after a 10-min exposure to 30 μM PE and 2.5 mM CaCl2 are indicated as Ctl and PE, respectively. Y27632 (+Y) and GF109203x (+GF) were added as described in Figure 2. All samples were loaded on single gel for densitometry. The mean values ± SEM were obtained from 3 independent assays. *p < 0.01, **p < 0.05.
Expression of results and statistical analysis
Mean values ± SEM were obtained from n experiments; n represents the number of rats. Student’s t-test assuming equal distribution was used for statistical analysis, and p < 0.05 was considered to be significant.
Results
Body weight and systolic pressure
The average body weight (412±2.6 g (n=17)) and the systolic blood pressure (159±4.3 mmHg (n=17)) of LH rats under anesthesia were significantly higher (p<0.01) than those of LN rats (321±8.0 g and 125±4.4 mmHg, respectively, n=11).
Contraction of small mesentery artery
Ca2+-depleted SMA tissue was used for the contraction assay. As shown previously [30], addition of 30 μM phenylephrine (PE) to Ca2+-free medium induced a transient force along with a slight Ca2+ transient, which returned to the basal level within a few min (data not shown). Then, CaCl2 was added to the PE containing bath to induce Ca2+ influx-dependent contraction (Figure 1A). As a control, KCl (100 mM) was used to evoke depolarization (Figure 1B), instead of PE. Permeable Ca2+ influx is sufficient to induce the Ca2+ dependent contraction of SMA [27, 30]. In the presence of PE, the maximal contraction of LH SMA was reached by the addition of CaCl2 at 2.5 mM, and the contraction produced was significantly greater than that of LN SMA (Figure 1A). However, there was no significant difference in KCl-evoked contraction of LN and LH SMA (Figure 1B). Thus, the up-regulation in Ca2+ influx-dependent contraction of LH SMA was associated with the activation of α-adrenergic receptor with PE stimulation. In the presence of PE, the contraction is slightly reduced at the highest [CaCl2]. This reduction is unlikely to be due to an inhibition of Ca2+ permeability, based on the results of intracellular Ca2+ concentration measurement (Figure 3A).
Figure 1. Sustained contraction evoked by addition of extra-cellular Ca2+.

Contraction of denuded SMA strips from LH (square) and LN (circle) rats were induced by the addition of CaCl2 at the indicated concentration to the bath. Each plot indicates the maximum force value reached to plateau (mN) normalized by length of each strip (mm). Assays were done in the presence of (A) 30 μM phenylephrine (PE) and (B) 100 mM KCl. The mean values ± SEM of 8–12 experiments are shown. *p < 0.05, LN versus LH.
Figure 3. Intra-cellular Ca2+ concentration [Ca2+]i in SMA during Ca2+-induced contraction.

Fluorescence ratio of Fura2 and the smooth muscle contraction were simultaneously measured using LN (circle) and LH (square) SMA. SMA was preloaded for 2 h with a permeable Ca2+ indicator fura-2/AM and then washed extensively. Fluorescence intensities at 510 nm with excitation at 340nm and 380 nm were monitored during CaCl2 titration. (A) CaCl2-dependendent Ca2+ influx. (B) Force-[Ca2+]i relationship. Closed circle and square show the ratio of Fura2 fluorescence intensity in SMA from LN and LH, respectively. Open square indicates the fluorescence ratio of LH SMA in the presence of 10 μM Y27632. The mean values ± SEM of 4-5 experiments are shown. *p < 0.05, LN versus LH.
Figure 2 shows an involvement of PKC and ROK in Ca2+/PE-induced contraction. Both PKC and ROK are known to transduce α-adrenergic receptor signals into smooth muscle contraction. Pre-treatment with Y27632, a ROK inhibitor, reduced the contraction of LN SMA at high [Ca2+] (>2.5 mM) (Figure 2A). The inhibitory effect of Y27632 was more prominent for the augmented contraction of LH SMA (Figure 2B). The maximum contractions of LN and LH in the presence of Y27632 were of similar magnitude (LN: 1.19 ± 0.38 mN/mm, LH: 1.56 ± 0.25 mN/mm). By contrast, the PKC inhibitor (GF109203X) had little effect on the contraction of SMA from LN or LH (Figure 2C and D). Thus, the kinase sensitive to Y27632, probably ROK, is responsible for PE-induced hyper-contractility in LH SMA.
Figure 2. Effects of kinase inhibitors on Ca2+-induced sustained contraction of PE-exposed SMA.

Ca2+/PE-induced contraction was measured using SMA from LN (A, C) and LH (B, D) rats in the absence or presence of Rho-kinase inhibitor Y27632 (10 μM) (A, B) or PKC inhibitor GF109203X (3 μM) (C, D). Inhibitors were added at 15 min before CaCl2 titration was initiated. The data in the absence of inhibitors are the same as Figure 1A. The mean values ± SEM of 4–8 experiments are shown. *p < 0.05, LN versus LH.
Ca2+ permeability and Ca2+ sensitivity of LH SMA contraction
We measured the fluorescence ratio of Fura2 at 340/280 nm to estimate the intracellular Ca2+ concentration, [Ca2+]i, in LN and LH SMA. Fura-2 loading did not affect the contraction of LN and LH SMA or the potency of Y27632 (data not shown). Under Ca2+-free conditions, the fluorescence ratio in LN strips (1.07 ± 0.20) was slightly lower than that detected for LH strips (1.36 ± 0.14). In LN SMA (Figure 3A, closed circle), the addition of CaCl2 increased the fluorescence ratio, indicating a concentration-dependent elevation in [Ca2+]i, in parallel to an increase in the contraction (Figure 1). As shown in Figure 3B, the relationship between [Ca2+]i and the extent of contraction was nearly linear in LN SMA. In contrast, [Ca2+]i in LH SMA (Figure 3A, closed square) was significantly lower than that in LN SMA. Consequently, the [Ca2+]i -force curve was steeper and shifted upward, indicating a Ca2+ sensitization of LH SMA smooth muscle contraction (Figure 3B). This augmented Ca2+ sensitivity seems to be responsible for the hyper-contractility of LH SMA.
In the LH arteries, Y27632 did not significantly affect the basal fluorescence ratio (0.124 ± 0.020). However, as shown in Figure 3A, Y27632 markedly improved Ca2+ influx of LH SMA, causing an increase in [Ca2+]i to the same level as in LN SMA that restored the [Ca2+]i -force relationship of LH to that of LN (Figure 3B). These results suggest that ROK-mediated signals disrupt both Ca2+ influx and the Ca2+-sensitivity of LH SMA contraction.
Regulation of myosin phosphatase
It has been well-documented that Ca2+ sensitization occurs via the inhibition of myosin phosphatase in arteries from normotensive rats. For example, in response to α-adrenergic receptor activation, the inhibition of myosin phosphatase is mediated through phosphorylation of an inhibitor protein, CPI-17, at Thr38 and the MYPT1 regulatory subunit of myosin phosphatase, at Thr696 and Thr853 [23]. We examined the phosphorylation of CPI-17 (Figure 4, panel A) and MYPT1 (panel B) in SMA from LN and LH rats, in the absence (marked as Ctl) and the presence of CaCl2 with PE (marked as PE). The phosphatase inhibitor calyculin A was added to the bath at low concentration (0.1 μM) to prevent dephosphorylation during the sample preparation. The ratio of CPI-17 expression to myosin phosphatase (CPI-17/MYPT1) was slightly but significantly higher in LH SMA (1.39 ± 0.21), compared with LN (1.00 ± 0.05) (n=5, p < 0.05). As shown in Figure 4A, the addition of PE significantly increased the phosphorylation of CPI-17 at Thr38 in LN SMA, in agreement with previous data obtained from other artery types [23]. However, compared to LN SMA, phosphorylation levels were lower in LH SMA, and surprisingly, no increase in the phosphorylation of CPI-17 was observed in PE-treated LH SMA. Y27632 treatment did not reduce, but rather increased the phosphorylation of CPI-17 (Figure 4A). The PKC inhibitor GF109203x moderately but significantly reduced the phosphorylation of CPI-17 in LH SMA (Figure 4A). Phosphorylation of MYPT1 at Thr696 was unchanged by PE-treatment of both LN and LH SMA (Figure 4B), but the phosphorylation was significantly increased after treatment with Y27632 and GF109203x. The phosphorylation of MYPT1 at Thr853 was increased in LN SMA by PE treatment (Figure 4B). The larger error bar size is due to relatively low signal on the immunoblot (data not shown). As seen for CPI-17, PE stimulation did not enhance the phosphorylation of MYPT1 at Thr853 in LH SMA (Figure 4B). Y27632, but not GF109203x, reduced the phosphorylation of MYPT1 at Thr853 in LH SMA, although this difference was not statistically significant, due to the low phosphorylation level (Figure 4B). These results suggest that the ROK-mediated Ca2+ sensitization of LH SMA contraction is independent of myosin phosphatase inhibition mediated by CPI-17 and MYPT1 phosphorylation.
Comparison of SMA and aorta smooth muscle
Figure 5 shows the Ca2+-induced force produced by aorta strips from LN and LH, and Figure 6 summarizes the contractile data of both SMA and aorta. In contrast to SMA (Figure 1 and 2), CaCl2 induced to an equal extent the contraction of aortic rings from LN and LH (Figure 5A), suggesting that, unlike resistance artery, Ca2+ sensitivity is unchanged in LH aorta. A major difference between aorta and SMA was seen in the sensitivity of aortic tissues to Y27632, where the contraction of both LN and LH aorta was almost abolished by Y27632 pre-treatment (Figure 5B, C, and 6). The results suggest that ROK plays a dominant role in Ca2+-induced contraction in SMA. As seen in SMA, GF109203x did not alter the contractile response of aortic rings from LN rats (Figure 5D). However, the contraction of LH aorta was significantly reduced by GF100203x treatment, suggesting a difference in the signal transduction of the Ca2+-induced force, which is mediated by PKC in aorta from LH, but not from LN (Figure 6). Thus, the signal transduction pathways responsible for controlling the Ca2+ sensitivity is specific to each type of vasculature (SMA vs aorta) under a certain environment (LN vs LH).
Figure 5. Ca2+-dependent contraction of aorta smooth muscle.

Ca2+-induced contraction and the effect of kinase inhibitor were assayed with aorta ring from LN and LH rats, as described in Figure 1 and 2. The data in the absence of inhibitors are the same in LN and LH samples. Mean values ± SEM of 4–5 experiments are shown. *p < 0.05, LN versus LH.
Figure 6. Comparison of Ca2+-induced contraction of SMA and aorta from LH and LN.
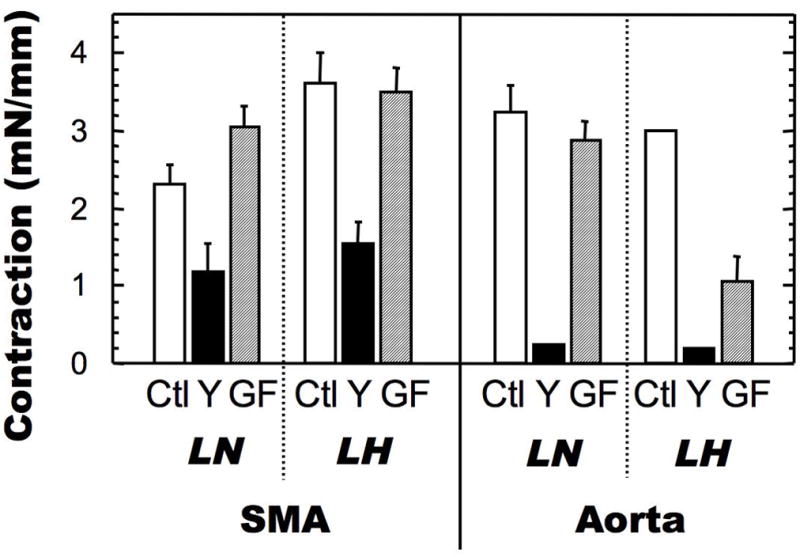
Contraction was assayed as described in Figures 2 and 5. The data for CaCl2 contraction at 2.5 mM (for SMA) and 10 mM (for aorta) in the presence of PE are shown in the bar graph. The mean values ± SEM of 4–13 experiments are shown. *p < 0.05, LN versus LH.
Discussion
Here we measured the sustained contraction of smooth muscle strips of SMA and aorta from LN and LH, elicited by re-addition of extracellular Ca2+. The results demonstrate the PE-induced hyper-contractility of LH smooth muscle, which is specific to resistance SMA. Based on previous reports, the addition of extra-cellular Ca2+ in the presence of PE triggers Ca2+ influx through voltage-gated and receptor-operated channels as well as Na/Ca exchangers that results in a sustained contraction [27, 30]. Therefore, the present study reflect the response to a tonic Ca2+ influx component rather than an initial phasic IP3-mediated Ca2+ release [35]. Previous studies show that maintenance of the sustained phase of the agonist-induced contraction depends on Ca2+-dependent activation of the RhoA/ROK pathway [28, 36]. Indeed, RhoA/ROK is activated in response to depolarization [28]. On the other hand, in this study, the hyper-contractility of SMA was seen when the arteries were exposed to PE, but not upon depolarization with KCl. Therefore, the hyper-contractility of LH SMA is caused by not only the Ca2+-induced activation of ROK, but also by other signals activated in response to α-adrenergic receptor activation. The deficiency in Ca2+ sensitivity of LH SMA was normalized by the treatment with Y27632. Generally, kinase inhibitor compounds are known to target a subset of kinases [37]. However, because accumulating evidence strongly suggests that ROK is dominantly involved in the regulation of vascular contraction, Y27632 most likely targets ROK in LH SMA and normalizes contractile dysfunction.
Ca2+ permeability appears to be greater in LH SMA, compared to LN SMA. An L-type Ca2+ channel is unlikely involved in the reduction of Ca2+ influx, because L-type channels are up-regulated in LH [7] as well as in spontaneously hypertensive rat basilar artery [16]. Because the deficiency in the Ca2+ influx is normalized by the treatment with Y27632, this suggests an involvement of ROK. The reduction of Ca2+ influx in both LN and LH SMA is more significant at higher CaCl2 concentration. We presume that the activation of ROK via Ca2+ signals causes the further inhibition in Ca2+ influx. Meanwhile, it remains unclear how ROK controls Ca2+ permeability. Recently, Iftinca et al. reported that the activation of ROK induces a reduction of T-type Ca2+ channel activity [38]. Thus, T-type Ca2+ channel and/or other unknown channels coupled with α-adrenergic receptor could be involved in altering Ca2+ influx in LH SMA.
The PE-induced Ca2+ sensitization dominates the reduction in Ca2+ influx, resulting in the hyper-contractility of LH SMA. ROK activity is known to depend on the inhibition of myosin phosphatase via phosphorylation of CPI-17 and MYPT1 [8, 39]. Sequential phosphorylation of CPI-17 at Thr38 by Ca2+-dependent PKC at early phase and then by ROK at sustained phase occurs in response to α-agonist stimulation of normotensive rabbit femoral artery [23]. In this study, the results were obtained during the sustained phase of contraction, and it is clear that CPI-17 is not the target of ROK in LH SMA. Myosin phosphatase is also inhibited through phosphorylation of MYPT1 at Thr696 and Thr853 [40]. As reported previously [23, 24], PE stimulation of the normotensive SMA increases the phosphorylation of MYPT1 at Thr853. However, the phosphorylation of MYPT1 at Thr853 in LH SMA is insensitive to PE stimulation, and as such, is unlikely to be involved in the increase of Ca2+ sensitivity of LH SMA. In contrast to the previous report of the agonist-induced phosphorylation of MYPT1 at Thr696 in hypertensive rat arteries [17], the phosphorylation of MYPT1 Thr696 is unchanged in either LN or LH, which is in agreement with the other report of the spontaneous phosphorylation at this site [24]. Thus, unlike other smooth muscle tissues from normotensive animals, it is obvious that the activation of ROK in LH SMA is linked to neither CPI-17 nor MYPT1 phosphorylation. Purified ROK can phosphorylate a subset of substrates, including myosin light chain and cytoskeletal proteins, as well as CPI-17 and MYPT1 [41]. The substrate specificity of cellular ROK is controlled by multiple factors, such as localization of active RhoA/ROK [42], and the direct binding of RhoA and MYPT1 [43]. We speculate that some factors determining the substrate specificity of cellular ROK are perturbed in LH SMA, which deny ROK to access to CPI-17 and MYPT1 and increase the contraction through the direct phosphorylation of myosin or actin binding proteins in LH SMA. Genetic mapping identified an involvement of chromosomes 2, 13, and 17 in blood pressure control of Lyon rat strain [44, 45]. Proteomic analysis is needed to elucidate alterations of a subset of gene products that disrupts ROK signaling in the regulation of Ca2+ influx and Ca2+ sensitivity in LH SMA.
There are striking differences in the Ca2+-induced contraction of SMA and aortic rings obtained from the same rats. Most importantly, PE-induced hyper-contractility did not occur in LH aorta smooth muscle, suggesting vascular type-specific alterations in ROK signaling. Another difference between SMA and aorta is in the Y27632 sensitivity of the sustained contraction. Ca2+-induced contraction of aorta smooth muscle is sensitive to ROK inhibition. Consistent with this result, Ghisdal et al. (2003) reported that Y27632 nearly abolished norepinephrine-induced contraction of aorta as a result of inhibition of both ROK-dependent Ca2+ sensitization and Ca2+ influx [27]. In addition to the effect on the contraction, it is reported that the relaxation induced by ROK inhibitors depends on the vascular tissue type, strains and experimental protocol, such that the relaxant effect of Y27632 was greater in large arteries than in SMA [11–13, 27, 28]. In contrast, PKC is partially involved in the Ca2+-induced sustained contraction of LH aorta, whereas a PKC inhibitor had no effect on the SMA contraction. Thus, the contribution of ROK and PKC to the regulation of the contraction is specific to each vascular type and probably responds to certain environmental factors. This should offer a note of caution when selecting smooth muscle tissues for studies on the regulation of contraction in hypertensive animals, and also to developing treatments for hypertension that target ROK. Furthermore, there is significant diversity in the signal transduction of hypertensive SMA between animal models, such that up-regulation of ROK activity enhances phosphorylation of MYPT1 at Thr696 causing hyper-contraction in angiotensin II-induced hypertensive rats [10, 19]. Thus, even though the involvement of ROK activity is a common theme in hypertensive rat models, the mechanisms underlying hyper-contractility of SMA are likely unique to each model, and probably between patients.
LH SMA hyper-contractility is associated with disorders in Ca2+ permeability and [Ca2+]i -force relationship. Y27632 effectively normalized the contraction of the hypertensive SMA, without affecting the signals for myosin phosphatase regulation. These observations appear counter to a large body of work demonstrating ROK-mediated inhibition of myosin phosphatase in normotensive smooth muscle. Our results encourage further studies on the mechanism of ROK-induced Ca2+ sensitization of vascular smooth muscle contraction under pathological conditions.
Acknowledgments
We thank Dr. Sharon Schendel, The Burnham Institute for Medical Research, for proofreading. This work was partially supported by grants from NHLBI HL83261, HL48807, PA Department of Health C.U.R.E. (to M.E.), C.N.R.S. and the University Louis Pasteur of Strasbourg (UMR 7034) (to J.-C.S.). M.R. Freitas was supported by a doctoral grant from Coordenacao de Aperfeicoamento de Pessoal de Nivel Superior-CAPES, Ministerio da Educacao, Brazil.
Footnotes
Conflict of interest exists in Millipore/Upstate products (ME).
References
- 1.Vincent M, Sassard J. The Lyon Strains. In: Swales T, editor. Textbook of Hypertension. Blackwell Sc. Pub; Oxford: 1994. pp. 455–7. [Google Scholar]
- 2.Luscher TF. Imbalance of endothelium-derived relaxing and contracting factors. A new concept in hypertension? Am J Hypertens. 1990;3:317–30. doi: 10.1093/ajh/3.4.317. [DOI] [PubMed] [Google Scholar]
- 3.Kang KT, Sullivan JC, Sasser JM, Imig JD, Pollock JS. Novel nitric oxide synthase--dependent mechanism of vasorelaxation in small arteries from hypertensive rats. Hypertension. 2007;49:893–901. doi: 10.1161/01.HYP.0000259669.40991.1e. [DOI] [PubMed] [Google Scholar]
- 4.Hilgers RH, Webb RC. Reduced expression of SKCa and IKCa channel proteins in rat small mesenteric arteries during angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol. 2007;292:H2275–84. doi: 10.1152/ajpheart.00949.2006. [DOI] [PubMed] [Google Scholar]
- 5.Freitas MR, Schott C, Corriu C, Sassard J, Stoclet JC, Andriantsitohaina R. Heterogeneity of endothelium-dependent vasorelaxation in conductance and resistance arteries from Lyon normotensive and hypertensive rats. J Hypertens. 2003;21:1505–12. doi: 10.1097/00004872-200308000-00014. [DOI] [PubMed] [Google Scholar]
- 6.Zicha J, Pernollet MG, Kunes J, Lacour B, Vincent M, Sassard J, et al. Alterations of cytosolic calcium in platelets and erythrocytes of Lyon hypertensive rats. Am J Hypertens. 1995;8:842–9. doi: 10.1016/0895-7061(95)00121-5. [DOI] [PubMed] [Google Scholar]
- 7.Le Roy AL, Messer-Letienne I, Bernard N, Benzoni D. Effects of L-type calcium channel activation on renal vascular resistances in the Lyon hypertensive rat. J Cardiovasc Pharmacol. 1999;33:65–9. doi: 10.1097/00005344-199901000-00010. [DOI] [PubMed] [Google Scholar]
- 8.Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. PhysiolRev. 2003;83:1325–58. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 9.Hartshorne DJ, Ito M, Erdodi F. Role of protein phosphatase type 1 in contractile functions: myosin phosphatase. JBiolChem. 2004;279:37211–4. doi: 10.1074/jbc.R400018200. [DOI] [PubMed] [Google Scholar]
- 10.Hilgers RH, Todd J, Jr, Webb RC. Increased PDZ-RhoGEF/RhoA/Rho kinase signaling in small mesenteric arteries of angiotensin II-induced hypertensive rats. J Hypertens. 2007;25:1687–97. doi: 10.1097/HJH.0b013e32816f778d. [DOI] [PubMed] [Google Scholar]
- 11.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–4. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 12.Mukai Y, Shimokawa H, Matoba T, Kandabashi T, Satoh S, Hiroki J, et al. Involvement of Rho-kinase in hypertensive vascular disease: a novel therapeutic target in hypertension. Faseb J. 2001;15:1062–4. doi: 10.1096/fj.00-0735fje. [DOI] [PubMed] [Google Scholar]
- 13.Chrissobolis S, Sobey CG. Evidence that Rho-kinase activity contributes to cerebral vascular tone in vivo and is enhanced during chronic hypertension: comparison with protein kinase C. CircRes. 2001;88:774–9. doi: 10.1161/hh0801.090441. [DOI] [PubMed] [Google Scholar]
- 14.Seasholtz TM, Zhang T, Morissette MR, Howes AL, Yang AH, Brown JH. Increased expression and activity of RhoA are associated with increased DNA synthesis and reduced p27(Kip1) expression in the vasculature of hypertensive rats. CircRes. 2001;89:488–95. doi: 10.1161/hh1801.096337. [DOI] [PubMed] [Google Scholar]
- 15.Weber DS, Webb RC. Enhanced relaxation to the rho-kinase inhibitor Y-27632 in mesenteric arteries from mineralocorticoid hypertensive rats. Pharmacology. 2001;63:129–33. doi: 10.1159/000056123. [DOI] [PubMed] [Google Scholar]
- 16.Kitazono T, Ago T, Kamouchi M, Santa N, Ooboshi H, Fujishima M, et al. Increased activity of calcium channels and Rho-associated kinase in the basilar artery during chronic hypertension in vivo. J Hypertens. 2002;20:879–84. doi: 10.1097/00004872-200205000-00022. [DOI] [PubMed] [Google Scholar]
- 17.Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, et al. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. CircRes. 2003;92:411–8. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- 18.Asano M, Nomura Y. Comparison of inhibitory effects of Y-27632, a Rho kinase inhibitor, in strips of small and large mesenteric arteries from spontaneously hypertensive and normotensive Wistar-Kyoto rats. Hypertens Res. 2003;26:97–106. doi: 10.1291/hypres.26.97. [DOI] [PubMed] [Google Scholar]
- 19.Jin L, Ying Z, Hilgers RH, Yin J, Zhao X, Imig JD, et al. Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. JPharmacolExpTher. 2006;318:288–95. doi: 10.1124/jpet.105.100735. [DOI] [PubMed] [Google Scholar]
- 20.Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension. 2001;38:1307–10. doi: 10.1161/hy1201.096541. [DOI] [PubMed] [Google Scholar]
- 21.Seasholtz TM, Wessel J, Rao F, Rana BK, Khandrika S, Kennedy BP, et al. Rho kinase polymorphism influences blood pressure and systemic vascular resistance in human twins: role of heredity. Hypertension. 2006;47:937–47. doi: 10.1161/01.HYP.0000217364.45622.f0. [DOI] [PubMed] [Google Scholar]
- 22.Sward K, Dreja K, Susnjar M, Hellstrand P, Hartshorne DJ, Walsh MP. Inhibition of Rho-associated kinase blocks agonist-induced Ca2+ sensitization of myosin phosphorylation and force in guinea-pig ileum. JPhysiology. 2000;522:33–49. doi: 10.1111/j.1469-7793.2000.0033m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dimopoulos GJ, Semba S, Kitazawa K, Eto M, Kitazawa T. Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. CircRes. 2007;100:121–9. doi: 10.1161/01.RES.0000253902.90489.df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitazawa T, Eto M, Woodsome TP, Khalequzzaman M. Phosphorylation of the myosin phosphatase targeting subunit and CPI-17 during Ca2+ sensitization in rabbit smooth muscle. J Physiol. 2003;546:879–89. doi: 10.1113/jphysiol.2002.029306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kitazawa T, Eto M, Woodsome TP, Brautigan DL. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. Journal of Biological Chemistry. 2000;275:9897–900. doi: 10.1074/jbc.275.14.9897. [DOI] [PubMed] [Google Scholar]
- 26.Eto M, Ohmori T, Suzuki M, Furuya K, Morita F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J BIOCHEM. 1995;118:1104–7. doi: 10.1093/oxfordjournals.jbchem.a124993. [DOI] [PubMed] [Google Scholar]
- 27.Ghisdal P, Vandenberg G, Morel N. Rho-dependent kinase is involved in agonist-activated calcium entry in rat arteries. JPhysiol(Lond) 2003;551:855–67. doi: 10.1113/jphysiol.2003.047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, et al. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. CircRes. 2003;93:548–56. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- 29.Christensen KL, Mulvany MJ. Location of resistance arteries. J Vasc Res. 2001;38:1–12. doi: 10.1159/000051024. [DOI] [PubMed] [Google Scholar]
- 30.Lagaud GJ, Randriamboavonjy V, Roul G, Stoclet JC, Andriantsitohaina R. Mechanism of Ca2+ release and entry during contraction elicited by norepinephrine in rat resistance arteries. AmJPhysiol. 1999;276:H300–8. doi: 10.1152/ajpheart.1999.276.1.H300. [DOI] [PubMed] [Google Scholar]
- 31.Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. CircRes. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- 32.Jensen PE, Mulvany MJ, Aalkjaer C, Nilsson H, Yamaguchi H. Free cytosolic Ca2+ measured with Ca(2+)-selective electrodes and fura 2 in rat mesenteric resistance arteries. AmJPhysiol. 1993;265:H741–6. doi: 10.1152/ajpheart.1993.265.2.H741. [DOI] [PubMed] [Google Scholar]
- 33.Kitazawa T, Gaylinn BD, Denney GH, Somlyo AP. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. JBiolChem. 1991;266:1708–115. [PubMed] [Google Scholar]
- 34.Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, et al. Calyculin A and okadaic acid: Inhibitors of protein phosphatase activity. BiochemBiophysResCommun. 1989;159:871–7. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- 35.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–25. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 36.Mita M, Yanagihara H, Hishinuma S, Saito M, Walsh MP. Membrane depolarization-induced contraction of rat caudal arterial smooth muscle involves Rho-associated kinase. BiochemJ. 2002;364:431–40. doi: 10.1042/BJ20020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. BiochemJ. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iftinca M, Hamid J, Chen L, Varela D, Tadayonnejad R, Altier C, et al. Regulation of T-type calcium channels by Rho-associated kinase. Nature Neuroscience. 2007;10:854–60. doi: 10.1038/nn1921. [DOI] [PubMed] [Google Scholar]
- 39.Hilgers RH, Webb RC. Molecular aspects of arterial smooth muscle contraction: focus on Rho. Exp Biol Med (Maywood) 2005;230:829–35. doi: 10.1177/153537020523001107. [DOI] [PubMed] [Google Scholar]
- 40.Muranyi A, Derkach D, Erdodi F, Kiss A, Ito M, Hartshorne DJ. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett. 2005;579:6611–5. doi: 10.1016/j.febslet.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 41.Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, et al. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. JBiolChem. 1997;272:12257–1260. doi: 10.1074/jbc.272.19.12257. [DOI] [PubMed] [Google Scholar]
- 42.Patil SB, Tsunoda Y, Pawar MD, Bitar KN. Translocation and association of ROCK-II with RhoA and HSP27 during contraction of rabbit colon smooth muscle cells. BiochemBiophysResCommun. 2004;319:95–102. doi: 10.1016/j.bbrc.2004.04.159. [DOI] [PubMed] [Google Scholar]
- 43.Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–28. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 44.Vincent M, Samani NJ, Gauguier D, Thompson JR, Lathrop GM, Sassard J. A pharmacogenetic approach to blood pressure in Lyon hypertensive rats. A chromosome 2 locus influences the response to a calcium antagonist. J Clin Invest. 1997;100:2000–6. doi: 10.1172/JCI119731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bilusic M, Bataillard A, Tschannen MR, Gao L, Barreto NE, Vincent M, et al. Mapping the genetic determinants of hypertension, metabolic diseases, and related phenotypes in the Lyon hypertensive rat. Hypertension. 2004;44:695–701. doi: 10.1161/01.HYP.0000144542.57306.5e. [DOI] [PubMed] [Google Scholar]