

Abstract
Glutamic acid decarboxylase GAD is a rate-limiting enzyme in the synthesis of GABA and an important autoantigen in both type 1 diabetes (T1D) and in stiff-person syndrome (SPS). Autoantibodies (GADA) to the 65kD isoform of GAD are a characteristic feature in both diseases. Approximately 30% of SPS patients develop diabetes, yet, it is unclear to which extent co-existing autoimmunity to GAD65 and other islet autoantigens determines the risk of developing T1D. In this study we monitored CD4+ T-cell responses to GAD65 and proinsulin in a patient with SPS who remained normoglycemic during the 46-month follow-up. Fluctuating but persistent T-cell reactivity to GAD65 was identified, as well as T-cell reactivity to proinsulin at one time point. The majority of the T-cell clones isolated from the SPS patient produced high levels of Th2 cytokines (IL-13, IL-5 and IL-4). We also examined levels of GADA, insulin and IA-2 autoantibodies, and epitope specificity of GADA. In both serum and cerebrospinal fluid GADA levels were high, and GADA persisted throughout the follow-up. Despite T-cell reactivity to both GAD65 and proinsulin, autoantibodies to other islet autoantigens did not develop. Further follow-up will determine whether or not the beta-cell autoimmunity observed in this patient will eventually lead to T1D.
Introduction
Stiff-person syndrome (SPS) is a rare neurological disorder characterized by muscle stiffness, hyperreflexia and elevated serum levels of creatinine kinase (CK). A key feature in the pathophysiology of SPS is the impairment of GABAergic neurotransmission.[1] Type 1 diabetes (T1D) results from destruction of insulin-producing beta cells in pancreatic islets and leads to metabolic derangement which can only be compensated by daily injections of exogenous insulin.[2–4] These two seemingly very different disorders share one pathophysiologic characteristic. A common feature in both is the presence of autoantibodies to glutamic acid decarboxylase (GAD), the rate-limiting enzyme in the synthesis of the inhibitory neurotransmitter gamma-amino butyric acid (GABA). While in T1D GAD-specific autoantibodies (GADA) are mostly considered as indicators of islet autoimmunity without any proven role in islet destruction [2–4], in SPS, a directly pathogenic role of GADA has been postulated based on the finding that they inhibit the enzymatic activity of GAD in vitro [5], and because reduced GABA levels have been detected in cerebrospinal fluid and brain of SPS patients. [5–7] Both SPS and T1D are considered to be autoimmune diseases. [3, 8–9]
GAD is expressed in two isoforms with molecular weights of 65 (GAD65) and 67 (GAD67) kDa.[10–12] While GAD65 is a common target of humoral autoimmunity in both T1D and SPS [13–16], the larger isoform is rarely recognized by autoantibodies, possibly due to differences in the enzymes’ conformations.[17] Approximately ~80% of T1D patients test positive for GADA [18–19], although the serum levels are 10–1000 fold lower than in SPS.[20] GADA in T1D patients recognize different GAD65 epitopes as compared to GADA in SPS patients [21–22], and in strong contrast with SPS-related GADA, do not inhibit GAD65 enzyme activity. [14, 22] In SPS, GADA predominantly target the linear parts of GAD65 whereas in T1D the epitopes are mainly conformation-dependent. [14, 19–20, 22] SPS patients also recognize a C-terminal epitope of GAD65, but it is not clear whether this is the same epitope as recognized by T1D patients. In fact it is unlikely that the same epitope is recognized, since the T1D-associated antibodies do not inhibit GAD65 enzyme activity.
Relatively few studies have investigated GAD65-specific cellular responses in SPS. GAD65 reactive T cells have been identified in the peripheral blood and cerebrospinal fluid of some SPS patients [23–27] although the responses have generally been weak and sometimes detectable only in T-cell lines generated by repeated stimulation with a GAD65 peptide. [26–27]. Studies comparing T-cell responses in both T1D and SPS patients have suggested differences in the T-cell epitope recognition pattern between the two diseases. [25, 28–30] However, there are no reports on monitoring of potential changes in the T-cell responses to GAD65 or other islet autoantigens in relation to clinical parameters in SPS. In this longitudinal study we evaluated the frequency and phenotype of GAD65 and proinsulin-specific T cells in a non-diabetic SPS patient using MHC class II tetramers, and correlated these findings to autoantibody status and clinical parameters.
Materials and Methods
The patient
The patient was a 32-year old Caucasian female, previously healthy other than having partial complex epilepsy. In March 2006, the patient was hospitalized because of lower back pain, walking difficulty and severe abdominal and lower lumbar muscle spasms. Serum CK levels increased 10-fold during the first 5 days in the hospital. The clinical picture raised suspicion of SPS, and very high levels of GADA were serially measured from sera (Table 1). GADA was also measured from a CSF sample drawn concomitantly with the second serum sample, and the GADA titer was more than four times higher than in serum. Clonazepam was initiated and the patient was able to walk unassisted upon release from the hospital. Brain and cervical, thoracic and lumbar spinal cord MRIs were normal. CSF leukocyte count and CSF protein concentration and IgG index were normal, but total IgG concentration in CFS was elevated to 59 mg/L (normal value < 45 mg/L). Protein electrophoresis of CFS showed three oligoclonal gamma bands, indicating immunoglobulin production in the CNS, or a compromised blood-brain barrier. No viral, Mycoplasma or Borrelia burgdorferi-antibodies were present in the CSF. IgA deficiency was diagnosed. ENMG examination was normal, and serum autoantibody titers against tissue transglutaminase and gliadin, as well as parietal cell-, nuclear, acetyl choline, insulin and IA-2 autoantibodies were all negative. Anti thyroid peroxidase antibody titre was intermediately positive 120 IU/ml (normal < 10 IU/ml). Islet-cell antibodies were of a very high titer (3033 JDFU, normal value < 2.5 JDFU) which was likely due to the high GADA titer. Blood glucose levels, monitored in every 3 to 6 months for 42 months and oral glucose tolerance test (performed twice) were normal. The clinical symptoms of SPS worsened in September 2006. Intravenous immunoglobulin treatment was contraindicated because of IgA deficiency, and she received oral prednisolone and titsanidine therapy for 3 months in addition to clonazepam. Thereafter, her SPS symptoms have been relatively stable until today (January 2010). The patient was HLA typed for class II alleles [31] and she carried the following genotype: HLA-(DR3)-DQA1*05-DQB1*02/HLA-(DR7)-DQA1*0201-DQB1*0303.
Table 1.
Autoantibody levels in the serum during the first 18 months after diagnosis of SPS
| Sample drawn | ICA (JDFU) | IAA, RU | GADA, RU | IA-2A, RU |
|---|---|---|---|---|
| 3/22/2006 | 3033 | 0.62 | 377310 | 0.13 |
| 4/26/2006 | 3033 | 0.34 | 493470 | 0.07 |
| 9/14/2006 | 3033 | 0.00 | 1100300 | 0.11 |
| 1/17/2007 | 3033 | 0.72 | 398200 | 0.09 |
| 6/12/2007 | 3033 | 0.00 | 317550 | 0.07 |
| 9/3/2007 | 1517 | 0.00 | 488210 | 0.04 |
Peripheral blood samples for this study were obtained from the patient after informed consent, and when possible they were drawn simultaneously to blood samples collected for diagnostic purposes. The use of the blood samples for immunological studies was approved by the Ethical Committee at the Hospital District of Southwestern Finland (20.5.2003, § 134).
GAD65 enzyme activity assay
GAD65 enzyme activity was measured by the 14CO2-trapping method described previously. [32] Recombinant human GAD65 (a kind donation by Amgen, Seattle, WA, USA) (100 ng) was incubated with the reaction buffer (50 mM K2HPO4, 0.03 mM PLP, 0.1 mM DTT, pH 6.8) for 1 hr at room temperature with or without the indicated amounts of isolated IgG. The enzymatic reaction was initiated by the addition of 0.56 mM L-glutamate and 0.018 μCi 14C-glutamate (Perkin Elmer, Boston MA, USA) and allowed to continue for 2 hours at 37°C with gentle agitation. During incubation, released 14CO2 was captured on filter paper (Kontes, Vineland, NJ, USA) soaked in 50 μl 1 M NaOH. After the incubation, the absorbed radioactivity was determined in a Beckman scintillation counter. The results are presented as: % residual activity = cpm in the presence of IgG/cpm in the absence of IgG × 100.
GADA assay
GADA were quantified with a radioligand assay in two different laboratories as previously described. [33–34] These two laboratories were the University of Washington laboratory (C.H.; University of Washington, WA, USA) and the University of Helsinki laboratory (M.K., University of Helsinki, Finland). GADA titers (index) presented in Figure 1 were derived using the Washington laboratory assay, while GADA titers presented in Table 1 summarize results obtained in the Helsinki laboratory.
Figure 1. GAD65Ab titer and inhibition of GAD65 enzymatic activity remains stable over time.
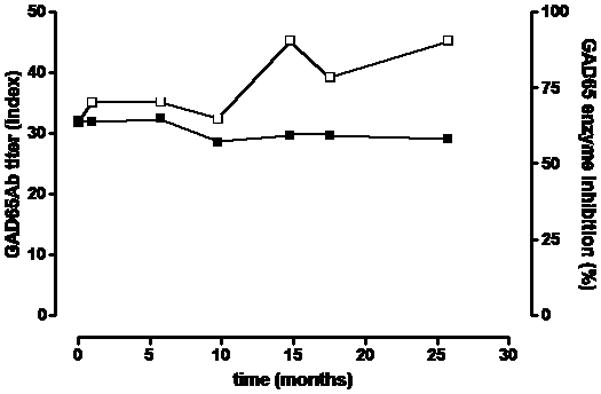
Longitudinal serum samples taken at the indicated time points were tested for their capacity to inhibit GAD65 enzymatic activity (white squares, right Y-axis) and their GAD65Ab titer (black squares, left Y-axix). GAD65 enzyme activity was measured in the absence and presence of serum samples. Inhibition of GAD65 enzyme activity is shown as percent inhibition. GAD65Ab titer was determined in a RBA and is reported as GAD65Ab index.
In the Washington assay, [33] the cut-off limit for autoantibody positivity was 0.05 (GAD65Ab index). The sensitivity and specificity for T1D in this assay were 86% and 93%, resepectively in the 2007 Diabetes Autoantibody Standardization Program (DASP) Workshop. In the Helsinki assay, [34] the cut-off limit for autoantibody positivity was 5.36 relative units (RU) representing the 99th percentile in 373 non-diabetic subjects. The sensitivity and specificity for T1D were 78% and 95%, respectively, in the 2009 DASP Workshop.
IA2-Ab and IAA assay
IA-2 antibodies were analyzed with a radioligand assay as described.[35] The cut-off limit for autoantibody positivity was 0.77 relative units (RU), representing the 99. percentile in 354 non-diabetic subjects. The sensitivity and specificity for T1D were 64% and 99%, respectively, in the 2009 DASP workshop.
IAA were measured with a radiobinding microassay as described.[36] The cut-off limit for IAA positivity was 2.80 RU representing the 99. percentile in 354 non-diabetic subjects. The sensitivity and specificity for T1D were 42% and 99%, respectively, in the 2009 DASP workshop.
Recombinant Fab fragments
Monoclonal antibodies b96.11 and b78 were derived from a patient with Autoimmune Polyendocrine Syndrome – type 2 [37], and recognize conformational epitopes formed by the 3D structure of amino acid residues 308–365 and 451–585, respectively.[38] N-GAD65mAb was raised in mice and recognized linear epitopes representing amino acid residues 4–22.[32] DPD was isolated from a T1D patient [39] and recognized epitopes that mapped between amino acid residues 483–585 and 96–173, respectively. MICA-3 was isolated from a T1D patient [40] and recognized epitopes of amino acid region 451–585.[41] All mAbs recognized GAD65 in its native conformation and do not bind GAD67.
Competition studies using rFab
The capacity of the rFab to inhibit GAD65 binding by human serum GAD65Ab was tested in a competitive radioligand-binding assay using Protein A Sepharose (PAS) (Invitrogen) as described[39]. The rFab were added at the optimal concentration (0.7–1μg/ml) as determined in competition assays using the intact mAb as a competitor. The background competition for each rFab was established in competition experiments with normal control sera. The background was subtracted prior to calculation of percent inhibition. The cutoff for specific competition was determined as >15% by using as a negative control rFab D1.3 (a kind gift from Dr. J. Foote, Arrowsmith Technologies, Seattle, WA, USA), specific to an irrelevant target, hen-egg lysozyme, at 5 μg/ml. Binding of GAD65Ab to GAD65 in the presence of rFab was expressed as follows: cpm in the presence of rFab/cpm in the absence of rFab ×100.
Peptides
Peptides used in the assay were GAD65 339–352 (TVYGAFDPLLAVAD), GAD65 247–266 (NMYAMMIARFKMFPEVKE) and ProIns B24-C36 (FFYTPMSRREVED).
Generation of tetramers
Generation of the MHC class II tetramers has been previously described. [42] Briefly, a site-specific biotinylation sequence was added to the 3′ end of the DRB1*0301 leucine zipper cassette, and the chimeric cDNA was subcloned into a Cu-inducible Drosophila expression vector. DR-A and DR-B expression vectors were co-transfected into Schneider S-2 cells, the class II monomers then purified, concentrated and biotinylated. The desired peptide was loaded for 48–72 hours, and tetramers were formed by incubating class II molecules with PE-labeled streptavidin.
Primary culture and stimulation
Peripheral mononuclear cells (PBMCs) were isolated from heparinized blood samples by Ficoll-Hypaque gradient separation (Lymphoprep, Nycomed, Oslo, Norway) and frozen. The frozen PBMC were shipped to Benaroya Research Institute in Seattle, USA on dry ice. PBMCs were thawed and cultured (2.0×106 cells/well) in RPMI media with HEPES (25mM), 15% Pooled Human Serum (PHS), 1% L-glutamine, 1% Penicillin/streptomycin and 1% Sodium Pyruvate. 10μg/mL of peptide was added to the culture and incubated for 14 days at 37°C. Human recombinant IL-2 (10U/ml, Chiron, Emeryville, CA, USA) was added on days 6 and 10.
Tetramer staining of PBMC
PBMCs were stained with a peptide loaded, phycoethrin-labeled DRB1*0301 tetramer for 1 hour at 37°C and subsequently with fluorochrome-labeled CD4 (Ebioscience, San Diego, CA, USA) antibody for 20 minutes on ice. The cells were then washed with PBS containing 1% BSA and 0.01% sodium azide and analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA, USA). The data were analyzed using FlowJo Software (Tree Star Inc, Ashland, OR, USA).
Isolation of peptide-specific T-cell clones
The peptide stimulated cells were single-cells sorted into 96-well plates using a FACSVantage cell sorter (Becton Dickinson). Sorted clones were expanded for 10 days by stimulation with irradiated unmatched PBMC (1.5 × 105/well), 5μg/ml PHA (Sigma, St. Louis, MO, USA) and 10U/ml IL-2 (Chiron, Emeryville, CA, USA) for two cycles, followed by stimulation with irradiated HLA-DR301 matched PBMC pulsed with 10 μg/ml of the peptide and 10U/ml IL-2. On day 10–12, clones were selected based on the growth for further expansion. 5 × 104 resting T cells were tested for the specificity by stimulation with irradiated HLA-DR301 matched PBMC (1 × 105/well) with and without the specific peptide in the culture. Proliferation as measured by 3H-thymidine incorporation was tested at 72 hours of culture. Supernatants were collected from culture supernatants at 48hrs and cytokines were measured by using MSD’s electrochemiluminescence MesoScale assay (Mesoscale Discovery, Gaithersburg, MD, USA) according to manufacturer’s instructions. The assay was performed using a human Th1/Th2 cytokine kit to measure interferon (IFN)-γ, IL-10, IL12p70, TNF-α, IL-5, IL-4, IL-13, IL-2 and IL-6. T cell clones were tested for tetramer binding by staining with 10μg/ml specific or negative control tetramer for 1 hour at 37 °C followed by fluorochrome conjugated CD4 Ab on ice for 20 min and analyzed by flow cytometry.
TCR sequencing of single cell clones
TCR alpha and beta chain CDR3 sequences of the CD4+ T-cell clones were determined by RT-PCR and sequencing analysis. Briefly, total RNA was isolated from CD4+ T-cell clones using an RNeasy Mini Kit (Qiagen) and cDNA was synthesized with a Taqman Reverse Transcription Kit (Applied Biosystems). A set of five multiplex PCR reactions covering a majority of the human V-beta repertoire were performed as per Akatsuka et al. [43] and two series of amplifications, first, with pooled V-alpha primers and then, with specific V-alpha primers covering the CDR3 regions, were performed as per Seitz et al.[44] PCR products were visualized on an ethidium bromide stained 2% agarose gel, sequenced using a Big Dye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) with either a TCR constant region 3′ primer or a specific variable region 5′ primer, and then run on an ABI3100 Genetic Analyzer. TCR alpha and beta CDR3 sequence data were analyzed using the IMGT/V-QUEST (http://imgt.cines.fr) web-based program from the Université Montpellier, France [34].
Results
Inhibition of GAD65 enzymatic activity by SPS sera
The manifestation of SPS symptoms was associated with high levels of GADA in both serum and cerebrospinal fluid (up to 1,1 106 RU in serum samples, Table 1). We tested the ability of the patient’s sera to inhibit GAD65 enzyme activity by incubating recombinant GAD65 with longitudinally collected serum samples and subsequently measuring GAD65 enzyme activity (Figure 1). The enzymatic activity of GAD65 was inhibited significantly at all time points by at least 57%. A slight, but insignificant decrease of inhibition (65% to 57%) was observed between months 6 and 10.
Epitope mapping of GADA
We analyzed the GADA epitope specificities present using rFab derived from GAD65-specific monoclonal antibodies (Figure 2). In our epitope-specific RBA we determine the binding of the serum sample to radiolabeled GAD65 in the presence of GAD65-specific monoclonal rFab. Competition of serum GAD65Ab with the respective rFab for a GAD65 epitope is reflected in reduced binding of the serum to radiolabeled GAD65. Significant competition (binding of serum sample to radiolabeled GAD65 is reduced by at least 15%) was observed only for rFab b96.11 and rFab b78. The longitudinal analysis showed that the competition with rFab b96.11 was stable throughout the analysis, while competition with rFab b78 increased from 3% at baseline to 20% in the final sample.
Figure 2. Epitope analysis using GAD65-specific rFab.
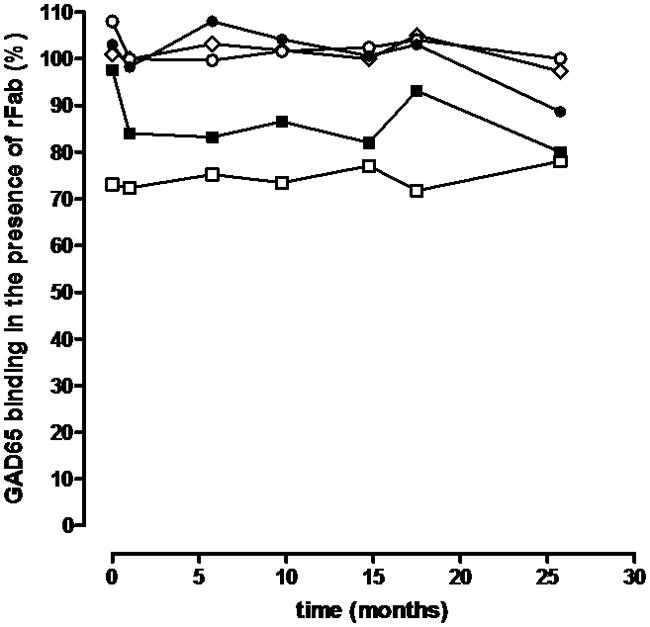
Longitudinal samples taken at the indicated time points were analyzed for their epitope binding pattern in our epitope-specific RBA using GAD65-specific rFab b78 (black squares), b96.11 (white squares), MICA-3 (white circles), DPD (diamonds), and N-GAD65 mAb (black circles). Binding to radiolabled GAD65 by serum samples was tested in the absence (100%) and presence of each of these rFab at half-maximal concentration.
T-cell reactivity with GAD and proinsulin tetramers
PBMC from four serial blood draws spanning over 40 months were stimulated with two GAD65 peptides (247–266 and 339–352) and a proinsulin peptide B24-C36, and stained with DR301 tetramer containing the same peptide as used in the stimulation. At the first time point the CD4+ T cells bound to all specific tetramers at frequencies from 2.09–6.56% (Figure 3) which was 10–32.8-fold higher than the background staining with the negative control tetramer (~0.2%). At the subsequent time points DR301- GAD339–352 tetramer-positive T cells were also identified but at much lower frequencies ranging from 1.06 to 2.97% (5.3–14.9-fold) whereas the T-cell response to proinsulin peptide B24-C36 was observed to be modestly positive (0.88%) and then disappeared. The response to the GAD339–352 peptide seemed to be the most persistent one since tetramer-binding cells were detected in all samples. All the samples were also stimulated with the DR301 binding NS1 peptide derived from flu virus as positive control, and showed a strong tetramer binding (data not shown).
Figure 3. DR301-tetramer staining of the PBMC from the SPS patient.
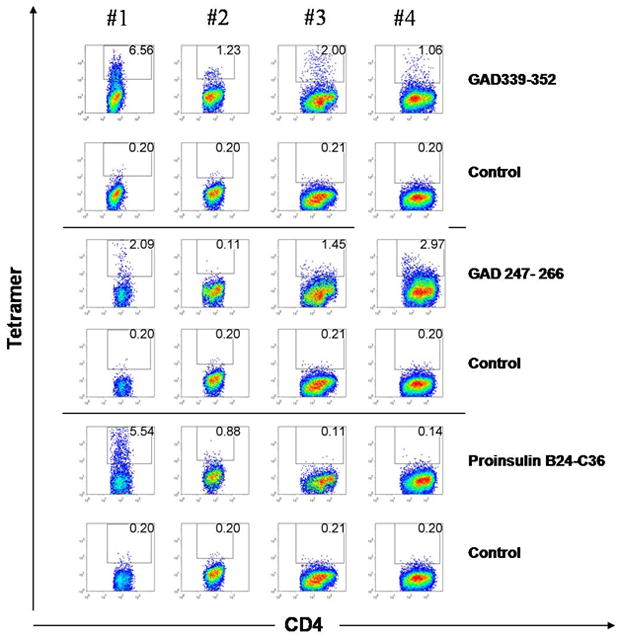
PBMC at 14-day post-stimulation with GAD65 339–352, GAD65 247–266 or proinsulin B24-C36 peptide (10 μg/ml) were stained with both specific DR301 tetramers containing the cognate peptide (upper panels in each section), and DR301-NS1 negative control tetramer (lower panel in each section). The cells were gated on live lymphocytic population and CD3+ cells. The frequency of CD4 and tetramer-positive cells is shown in the quadrant. The tetramer-positive cells were single cell sorted on 96-well plates.
Avidity and cytokine profiles of the T-cell clones
Tetramer-positive cells from the cultured CD4+ lymphocytes were single cell sorted and T-cell clones were generated. T-cell clones specific for the GAD65 339–352 peptide were isolated from the first three samples whereas the attempt to raise clones from the fourth time point was not successful. T-cell clones specific for GAD247–266 and proinsulin B24-C36 peptides were isolated only from the first blood sample. Tetramer binding when used at non-saturating concentrations has been shown to correlate with the structural avidity of TcR.[45–46] In Figure 4, tetramer staining of the GAD65 339–352-specific T-cell clones shows that high-avidity T-cell clones with a strong tetramer binding profile (77–100%) were detected at the first and third time point. However, some T-cell clones, even derived from the same sample, bound the tetramer at much lower level (34%, clone 339–1, sample #3) or, displayed lack of staining (clone 339–201, sample #2). All clones were capable of proliferating when stimulated with the specific peptide (data not shown) and produced cytokines (Figure 4) demonstrating specificity to the given T-cell epitope peptide. When the high-avidity T-cell clones derived from the first blood sample were stimulated with the GAD65 339–352 peptide robust IFN-γ and IL-13 production was observed and two of the clones (#309 and 332) also produced high levels of IL-5 and low amounts of IL-4. Three other clones isolated from the same sample displayed no tetramer binding but produced low levels of IFN-γ, IL-13 and IL-4 (50–80 pg/ml) demonstrating specificity but a low-avidity phenotype (data not shown). GAD339–352-specific T-cell clones derived from samples drawn at the later time points seemed to be biased more towards predominant IL-13 production. T cells specific for the other peptide epitopes, GAD65 247–266 and proinsulin B24-C36, also produced IL-13, and displayed low/intermediate avidity. T-cell clones specific for NS1 flu peptide were isolated from all time points (data not shown). These T cells displayed a strong tetramer binding (~100%) and significant IFN-γ production upon stimulation with the specific peptide.
Figure 4. Tetramer staining and cytokine profiles of the T-cell clones.
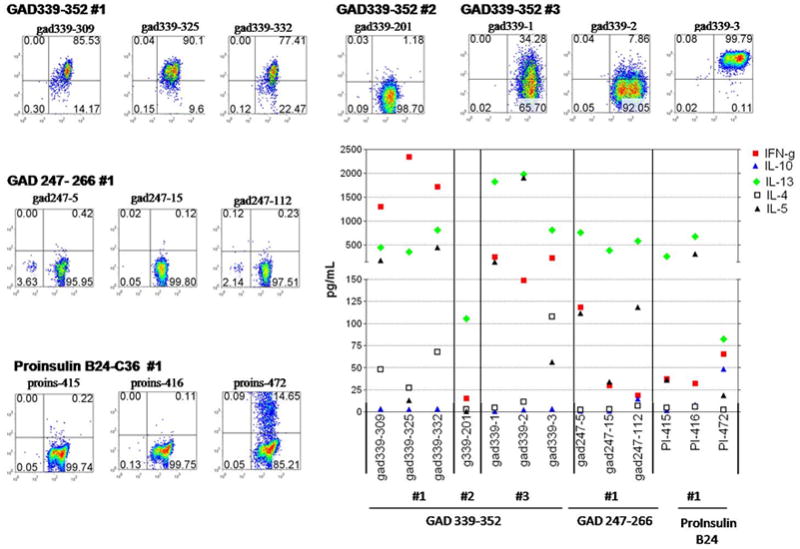
The DR301-GAD339–352 tetramer was used to stain T-cell clones single cell sorted from the PBMC samples obtained at three time points (#1–3, upper panel). Similarly, DR301-GAD 247–266 and proinsulin B24-C36 tetramers were used to stain clones derived from the first time point (middle, lower left panels). In all cases tetramer staining was evaluated on the viable lymphocytic population as gated based on the forward/side scatter profile. The frequency of CD4/tetramer-positive cells is noted in the upper right quadrant. Staining with the control tetramer containing DR301 binding irrelevant peptide (flu, NS-1) was set at 0.2%. Cytokines (pg/ml, Y axis) were determined from the supernatants collected from triplicate cultures of the T cell clones at 48hrs post stimulation with DR301+ irradiated PBMC pulsed with the cognate peptides (middle-lower right panel).
Overall, GAD65 339–352 is likely to be a predominant peptide epitope presented by DRB1*0301 in our patient since T-cell reactivity to this peptide was demonstrated in all four samples studied and, furthermore, T-cell clones with the given specificity, some with a very strong tetramer binding, were derived from three out of four samples. The T-cell clones generated at the first sampling produced high levels of IFN- γ but also IL-13 and IL-5 suggesting a Th1/Th0 profile which shifted more towards Th2 during the follow-up. T-cell responses to GAD247–266 and proinsulin B24-C36 peptides seemed to be more subdominant in nature. In addition, T-cell clones with these specificities displayed a low avidity and Th2-biased cytokine profiles and were generated only from the first time point. Sequence analysis of the complementarity determining regions demonstrated that the T-cell clones used distinct TcR-Vβ chains and that the ones derived from different time points were not identical (data not shown).
Discussion
In this study, we analyzed CD4+ T-cell responses and autoantibodies in a series of samples from a female patient who developed SPS at the age of 29. Although T-cell reactivity to GAD65 persisted throughout the follow-up, and T-cell reactivity to proinsulin emerged a year from the diagnosis of SPS, this patient did not progress to T1D during the 46-month follow-up. A meta-analysis of published case studies of SPS patients indicate that T1D may develop from one year [24] to more than a decade [26] (and B.O. Roep, personal communication to H.R.) after SPS diagnosis. In individuals genetically predisposed to T1D, the risk to develop T1D depends on both the magnitude and the type of T-cell reactivity to several autoantigens.[3] Furthermore, non-progressors are characterized by a regulatory rather than an inflammatory type of T-cell reactivity to islet autoantigens.[47]
In our patient, high GADA titers associated with the presence of GAD and proinsulin-reactive T cells. GAD-reactive T cells displayed some IFN-γ but significant IL-13 production. These findings suggest a non-inflammatory cytokine environment [47, 48] and effective B-cell antibody production with predominantly autoantibody-mediated disease pathogenesis.[5, 7–8] High titers of GADA may lead to functional GABA deficiency and typical manifestations of SPS even in a non-inflammatory cytokine environment which, in turn, may rescue islets from tissue destruction.
Despite that ~50% of SPS patients develop T1D [28, 49], there are no longitudinal studies on immune parameters in SPS patients prior to progression to clinical diabetes. T-cell responses and levels of GADA have been studied only in SPS patients who were already affected by T1D. [24, 28, 50] An early study by Hummel and co-workers demonstrated a weak but detectable T-cell response to GAD65 and IA-2 with IFN-γ production that decreased in response to immunosuppressive therapy.[24] Schloot et al. reported lack of primary T-cell response to GAD65 in a non-diabetic SPS patient. However, in vitro stimulation of the GAD65 -induced cell line generated T-cell clones responsive to GAD65 peptide 341–360 with production of the anti-inflammatory cytokine IL-10, IFN-γ and low level of IL-4.[26] Similarly, in our study T-cell clones specific for an overlapping peptide GAD65 339–352 were predominant, and increasingly Th2-biased cytokine profiles were detected during the course of the follow-up. Consistent with an anti-inflammatory environment a low efficacy of cloning from peripheral blood was observed in a recent study by Skorstad and coworkers. [27] In their study only one GAD555–565 specific T-cell clone was isolated from the PBMC of a DRB1*0408-positive individual. Interestingly, CD4+ T cells recognizing this epitope have been shown to be prevalent in DR4-positive T1D patients [46, 51] suggesting that the reported distinct T-cell epitope recognition patterns [28] in T1D and SPS patients may be associated with differences in the distribution of HLA alleles rather than the disease mechanisms.
The introduction of the tetramer technology [52] has allowed identification and immune monitoring of autoreactive T cells and their isolation at single cell level with increased efficacy and sensitivity. MHC class II tetramers have turned out to be excellent tools in studies of avidity and phenotype of GAD65-specific T cells in T1D, and these studies have revealed the existence of both low- and high-avidity T cells with various cytokine patterns. [46, 53, 54] In this study, GAD-specific T cells with a range of avidities and intermediate/low avidity (pro)insulin-specific cellular immunity in combination with high levels of GADA were demonstrated. The GADA showed SPS-characteristic inhibition of GAD65 enzymatic activity and showed reactivity towards a SPS-associated antibody epitope (b78).[17] However, reactivity to a T1D-associated GADA epitope (b96.11) [39] was also observed. This antibody recognizes a conformational epitope formed by the three-dimensional structure of amino acid residues 308–365, including the peptide sequence that dominated the T-cell response.
In summary, this longitudinal study of T-cell and antibody reactivity against GAD and two islet-antigens, insulin and IA-2, in a patient with SPS may facilitate future efforts to predict the probability of developing T1D after SPS. Although further follow-up is necessary, the transient nature of the (pro)insulin T-cell response, the lack of high avidity T-cell clones recognizing (pro)insulin and the overall Th2 biased T-cell responsiveness to GAD65 suggest slow or no progression towards induction of insulin- and other islet-specific (non-GAD65) autoantibodies and clinical manifestation of T1D. Continued follow-up will be needed to determine whether or not this patient eventually progresses to T1D, and to monitor for any apparent changes in T-cell reactivity that may accompany T1D development.
Acknowledgments
This work was supported by the Academy of Finland (A.H.), Päivikki and Sakari Sohlberg Foundation (A.H.), Medical Reseach Fund of Turku University Hospital (EVO (M. S-H.), Helsinki University Hospital (M.K.), Juvenile Diabetes Foundation (H.R.) and American Diabetes Association (H.R.). The study (C.S.H.) was performed as independent research sponsored by the National Institutes of Health (DK53004 and DK17047), and a Basic Science Award from the American Diabetes Association to C.S.H.
References
- 1.Duddy ME, Baker MR. Stiff person syndrome. Front Neurol Neurosci. 2009;26:147–65. doi: 10.1159/000212375. [DOI] [PubMed] [Google Scholar]
- 2.Jahromi MM, Eisenbarth GS. Cellular and molecular pathogenesis of type 1A diabetes. Cell Mol Life Sci. 2007;64:865–72. doi: 10.1007/s00018-007-6469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knip M, Siljander H. Autoimmune mechanisms in type 1 diabetes. Autoimmun Rev. 2008;7 :550–7. doi: 10.1016/j.autrev.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Skyler JS. Diabetes mellitus: pathogenesis and treatment strategies. J Med Chem. 2004;47 :4113–7. doi: 10.1021/jm0306273. [DOI] [PubMed] [Google Scholar]
- 5.Dinkel K, Meinck HM, Jury KM, Karges W, Richter W. Inhibition of gamma-aminobutyric acid synthesis by glutamic acid decarboxylase autoantibodies in stiff-man syndrome. Ann Neurol. 1998;44:194–201. doi: 10.1002/ana.410440209. [DOI] [PubMed] [Google Scholar]
- 6.Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology. 2001;57:780–4. doi: 10.1212/wnl.57.5.780. [DOI] [PubMed] [Google Scholar]
- 7.Levy LM, Levy-Reis I, Fujii M, Dalakas MC. Brain gamma-aminobutyric acid changes in stiff-person syndrome. Arch Neurol. 2005;62:970–4. doi: 10.1001/archneur.62.6.970. [DOI] [PubMed] [Google Scholar]
- 8.Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11:102–10. doi: 10.1007/s11940-009-0013-9. [DOI] [PubMed] [Google Scholar]
- 9.Levy LM, Dalakas MC, Floeter MK. The stiff-person syndrome: an autoimmune disorder affecting neurotransmission of gamma-aminobutyric acid. Ann Intern Med. 1999;131:522–30. doi: 10.7326/0003-4819-131-7-199910050-00008. [DOI] [PubMed] [Google Scholar]
- 10.Bu DF, Tobin AJ. The exon-intron organization of the genes (GAD1 and GAD2) encoding two human glutamate decarboxylases (GAD67 and GAD65) suggests that they derive from a common ancestral GAD. Genomics. 1994;21:222–8. doi: 10.1006/geno.1994.1246. [DOI] [PubMed] [Google Scholar]
- 11.Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7:91–100. doi: 10.1016/0896-6273(91)90077-d. [DOI] [PubMed] [Google Scholar]
- 12.Karlsen AE, Hagopian WA, Petersen JS, Boel E, Dyrberg T, Grubin CE, Michelsen BK, Madsen OD, Lernmark A. Recombinant glutamic acid decarboxylase (representing the single isoform expressed in human islets) detects IDDM-associated 64,000-M(r) autoantibodies. Diabetes. 1992;41:1355–9. doi: 10.2337/diab.41.10.1355. [DOI] [PubMed] [Google Scholar]
- 13.Bingley PJ, Bonifacio E, Williams AJ, Genovese S, Bottazzo GF, Gale EA. Prediction of IDDM in the general population: strategies based on combinations of autoantibody markers. Diabetes. 1997;46:1701–10. doi: 10.2337/diab.46.11.1701. [DOI] [PubMed] [Google Scholar]
- 14.Bjork E, Velloso LA, Kampe O, Karlsson FA. GAD autoantibodies in IDDM, stiff-man syndrome, and autoimmune polyendocrine syndrome type I recognize different epitopes. Diabetes. 1994;43:161–5. doi: 10.2337/diab.43.1.161. [DOI] [PubMed] [Google Scholar]
- 15.Solimena M, De Camilli P. Autoimmunity to glutamic acid decarboxylase (GAD) in Stiff-Man syndrome and insulin-dependent diabetes mellitus. Trends Neurosci. 1991;14:452–7. doi: 10.1016/0166-2236(91)90044-u. [DOI] [PubMed] [Google Scholar]
- 16.Vandewalle CL, Falorni A, Svanholm S, Lernmark A, Pipeleers DG, Gorus FK. High diagnostic sensitivity of glutamate decarboxylase autoantibodies in insulin-dependent diabetes mellitus with clinical onset between age 20 and 40 years. The Belgian Diabetes Registry. J Clin Endocrinol Metab. 1995;80:846–51. doi: 10.1210/jcem.80.3.7883841. [DOI] [PubMed] [Google Scholar]
- 17.Fenalti G, Rowley MJ. GAD65 as a prototypic autoantigen. J Autoimmun. 2008;31:228–32. doi: 10.1016/j.jaut.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 18.Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, De Camilli P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. [erratum appears in Nature 1990 Oct 25;347(6295):782 Note: Camilli, PD [corrected to De Camilli, P]] Nature. 1990;347:151–6. doi: 10.1038/347151a0. [DOI] [PubMed] [Google Scholar]
- 19.Ujihara N, Daw K, Gianani R, Boel E, Yu L, Powers AC. Identification of glutamic acid decarboxylase autoantibody heterogeneity and epitope regions in type I diabetes. Diabetes. 1994;43:968–75. doi: 10.2337/diab.43.8.968. [DOI] [PubMed] [Google Scholar]
- 20.Daw K, Ujihara N, Atkinson M, Powers AC. Glutamic acid decarboxylase autoantibodies in stiff-man syndrome and insulin-dependent diabetes mellitus exhibit similarities and differences in epitope recognition. J Immunol. 1996;156:818–25. [PubMed] [Google Scholar]
- 21.Daw K, Powers AC. Two distinct glutamic acid decarboxylase auto-antibody specificities in IDDM target different epitopes. Diabetes. 1995;44:216–20. doi: 10.2337/diab.44.2.216. [DOI] [PubMed] [Google Scholar]
- 22.Raju R, Foote J, Banga JP, Hall TR, Padoa CJ, Dalakas MC, Ortqvist E, Hampe CS. Analysis of GAD65 autoantibodies in Stiff-Person syndrome patients. J Immunol. 2005;175:7755–62. doi: 10.4049/jimmunol.175.11.7755. [DOI] [PubMed] [Google Scholar]
- 23.Boyton RJ, Lohmann T, Londei M, Kalbacher H, Halder T, Frater AJ, Douek DC, Leslie DG, Flavell RA, Altmann DM. Glutamic acid decarboxylase T lymphocyte responses associated with susceptibility or resistance to type I diabetes: analysis in disease discordant human twins, non-obese diabetic mice and HLA-DQ transgenic mice. Int Immunol. 1998;10 :1765–76. doi: 10.1093/intimm/10.12.1765. [DOI] [PubMed] [Google Scholar]
- 24.Hummel M, Durinovic-Bello I, Bonifacio E, Lampasona V, Endl J, Fessele S, Then Bergh F, Trenkwalder C, Standl E, Ziegler AG. Humoral and cellular immune parameters before and during immunosuppressive therapy of a patient with stiff-man syndrome and insulin dependent diabetes mellitus. Journal of Neuro Neurosurg Psychiatry. 1998;65:204–8. doi: 10.1136/jnnp.65.2.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohmann T, Londei M, Hawa M, Leslie RD. Humoral and cellular autoimmune responses in stiff person syndrome. Ann N Y Acad Sci. 2003;998:215–22. doi: 10.1196/annals.1254.024. [DOI] [PubMed] [Google Scholar]
- 26.Schloot NC, Batstra MC, Duinkerken G, De Vries RR, Dyrberg T, Chaudhuri A, Behan PO, Roep BO. GAD65-Reactive T cells in a non-diabetic stiff-man syndrome patient. J Autoimmun. 1999;12:289–96. doi: 10.1006/jaut.1999.0280. [DOI] [PubMed] [Google Scholar]
- 27.Skorstad G, Hestvik AL, Vartdal F, Holmoy T. Cerebrospinal fluid T cell responses against glutamic acid decarboxylase 65 in patients with stiff person syndrome. J Autoimmun. 2009;32 :24–32. doi: 10.1016/j.jaut.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 28.Lohmann T, Hawa M, Leslie RD, Lane R, Picard J, Londei M. Immune reactivity to glutamic acid decarboxylase 65 in stiffman syndrome and type 1 diabetes mellitus. Lancet. 2000;356:31–5. doi: 10.1016/S0140-6736(00)02431-4. [DOI] [PubMed] [Google Scholar]
- 29.Lohmann T, Leslie RD, Hawa M, Geysen M, Rodda S, Londei M. Immunodominant epitopes of glutamic acid decarboxylase 65 and 67 in insulin-dependent diabetes mellitus. Lancet. 1994;343:1607–8. doi: 10.1016/s0140-6736(94)93061-9. [DOI] [PubMed] [Google Scholar]
- 30.Lohmann T, Scherbaum WA. T cell autoimmunity to glutamic acid decarboxylase in human insulin-dependent diabetes mellitus. Horm Metab Res. 1996;28:357–60. doi: 10.1055/s-2007-979815. [DOI] [PubMed] [Google Scholar]
- 31.Hermann R, Turpeinen H, Laine AP, Veijola R, Knip M, Simell O, Sipila I, Akerblom HK, Ilonen J. HLA DR-DQ-encoded genetic determinants of childhood-onset type 1 diabetes in Finland: an analysis of 622 nuclear families. Tissue Antigens. 2003;62:162–9. doi: 10.1034/j.1399-0039.2003.00071.x. [DOI] [PubMed] [Google Scholar]
- 32.Hampe CS, Lundgren P, Daniels TL, Hammerle LP, Marcovina SM, Lernmark A. A novel monoclonal antibody specific for the N-terminal end of GAD65. J Neuroimmunol. 2001;113:63–71. doi: 10.1016/s0165-5728(00)00423-9. [DOI] [PubMed] [Google Scholar]
- 33.Hampe CS, Hammerle LP, Bekris L, Ortqvist E, Kockum I, Rolandsson O, Landin-Olsson M, Torn C, Persson B, Lernmark A. Recognition of glutamic acid decarboxylase (GAD) by autoantibodies from different GAD antibody-positive phenotypes. J Clin Endocrinol Metab. 2000;85:4671–9. doi: 10.1210/jcem.85.12.7070. [DOI] [PubMed] [Google Scholar]
- 34.Savola K, Sabbah E, Kulmala P, Vahasalo P, Ilonen J, Knip M. Autoantibodies associated with Type I diabetes mellitus persist after diagnosis in children. Diabetologia. 1998;41 :1293–7. doi: 10.1007/s001250051067. [DOI] [PubMed] [Google Scholar]
- 35.Savola K, Bonifacio E, Sabbah E, Kulmala P, Vahasalo P, Karjalainen J, Tuomilehto-Wolf E, Merilainen J, Akerblom HK, Knip M. IA-2 antibodies--a sensitive marker of IDDM with clinical onset in childhood and adolescence. Childhood Diabetes in Finland Study Group. Diabetologia. 1998;41:424–9. doi: 10.1007/s001250050925. [DOI] [PubMed] [Google Scholar]
- 36.Ronkainen MS, Hamalainen AM, Koskela P, Akerblom HK, Knip M. Pregnancy induces nonimmunoglobulin insulin-binding activity in both maternal and cord blood serum. Clin Exp Immunol. 2001;124:190–6. doi: 10.1046/j.1365-2249.2001.01506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tremble J, Morgenthaler NG, Vlug A, Powers AC, Christie MR, Scherbaum WA, Banga JP. Human B cells secreting immunoglobulin G to glutamic acid decarboxylase-65 from a nondiabetic patient with multiple autoantibodies and Graves’ disease: a comparison with those present in type 1 diabetes. J Clin Endocrinol Metab. 1997;82:2664–70. doi: 10.1210/jcem.82.8.4171. [DOI] [PubMed] [Google Scholar]
- 38.Fenalti G, Law RH, Buckle AM, Langendorf C, Tuck K, Rosado CJ, Faux NG, Mahmood K, Hampe CS, Banga JP, Wilce M, Schmidberger J, Rossjohn J, El-Kabbani O, Pike RN, Smith AI, Mackay IR, Rowley MJ, Whisstock JC. GABA production by glutamic acid decarboxylase is regulated by a dynamic catalytic loop. Nat Struct Mol Biol. 2007;14:280–6. doi: 10.1038/nsmb1228. [DOI] [PubMed] [Google Scholar]
- 39.Padoa CJ, Banga JP, Madec AM, Ziegler M, Schlosser M, Ortqvist E, Kockum I, Palmer J, Rolandsson O, Binder KA, Foote J, Luo D, Hampe CS. Recombinant Fabs of human monoclonal antibodies specific to the middle epitope of GAD65 inhibit type 1 diabetes-specific GAD65Abs. Diabetes. 2003;52:2689–95. doi: 10.2337/diabetes.52.11.2689. [DOI] [PubMed] [Google Scholar]
- 40.Richter W, Endl J, Eiermann TH, Brandt M, Kientsch-Engel R, Thivolet C, Jungfer H, Scherbaum WA. Human monoclonal islet cell antibodies from a patient with insulin-dependent diabetes mellitus reveal glutamate decarboxylase as the target antigen. Proc Natl Acad Sci U S A. 1992;89:8467–71. doi: 10.1073/pnas.89.18.8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richter W, Shi Y, Baekkeskov S. Autoreactive epitopes defined by diabetes-associated human monoclonal antibodies are localized in the middle and C-terminal domains of the smaller form of glutamate decarboxylase. Proc Natl Acad Sci U S A. 1993;90:2832–6. doi: 10.1073/pnas.90.7.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Novak EJ, Liu AW, Nepom GT, Kwok WW. MHC class II tetramers identify peptide-specific human CD4(+) T cells proliferating in response to influenza A antigen. J Clin Invest. 1999;104:R63–7. doi: 10.1172/JCI8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akatsuka Y, Martin EG, Madonik A, Barsoukov AA, Hansen JA. Rapid screening of T-cell receptor (TCR) variable gene usage by multiplex PCR: application for assessment of clonal composition. Tissue Antigens. 1999;53:122–34. doi: 10.1034/j.1399-0039.1999.530202.x. [DOI] [PubMed] [Google Scholar]
- 44.Seitz S, Schneider CK, Malotka J, Nong X, Engel AG, Wekerle H, Hohlfeld R, Dornmair K. Reconstitution of paired T cell receptor alpha- and beta-chains from microdissected single cells of human inflammatory tissues. Proc Natl Acad Sci USA. 2006;103:12057–62. doi: 10.1073/pnas.0604247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reichstetter S, Ettinger RA, Liu AW, Gebe JA, Nepom GT, Kwok WW. Distinct T cell interactions with HLA class II tetramers characterize a spectrum of TCR affinities in the human antigen-specific T cell response. J Immunol. 2000;165:6994–8. doi: 10.4049/jimmunol.165.12.6994. [DOI] [PubMed] [Google Scholar]
- 46.Reijonen H, Mallone R, Heninger AK, Laughlin EM, Kochik SA, Falk B, Kwok WW, Greenbaum C, Nepom GT. GAD65-specific CD4+ T-cells with high antigen avidity are prevalent in peripheral blood of patients with type 1 diabetes. Diabetes. 2004;53:1987–94. doi: 10.2337/diabetes.53.8.1987. [DOI] [PubMed] [Google Scholar]
- 47.Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM, Roep BO, Peakman M. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest. 2004;113:451–63. doi: 10.1172/JCI19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veldhoen M. The role of T helper subsets in autoimmunity and allergy. Curr Opin Immunol. 2009;21:606–11. doi: 10.1016/j.coi.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 49.Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N Engl J Med. 1990;322:1555–60. doi: 10.1056/NEJM199005313222202. [DOI] [PubMed] [Google Scholar]
- 50.Costa M, Saiz A, Casamitjana R, Castaner MF, Sanmarti A, Graus F, Jaraquemada D. T-cell reactivity to glutamic acid decarboxylase in stiff-man syndrome and cerebellar ataxia associated with polyendocrine autoimmunity. Clin Exp Immunol. 2002;129:471–8. doi: 10.1046/j.1365-2249.2002.01931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Endl J, Otto H, Jung G, Dreisbusch B, Donie F, Stahl P, Elbracht R, Schmitz G, Meinl E, Hummel M, Ziegler AG, Wank R, Schendel DJ. Identification of naturally processed T cell epitopes from glutamic acid decarboxylase presented in the context of HLA-DR alleles by T lymphocytes of recent onset IDDM patients. J Clin Invest. 1997;99:2405–15. doi: 10.1172/JCI119423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reijonen H, Kwok WW. Use of HLA class II tetramers in tracking antigen-specific T cells and mapping T-cell epitopes. Methods (Duluth) 2003;29:282–8. doi: 10.1016/s1046-2023(02)00350-x. [DOI] [PubMed] [Google Scholar]
- 53.Oling V, Marttila J, Ilonen J, Kwok WW, Nepom G, Knip M, Simell O, Reijonen H. GAD65- and proinsulin-specific CD4+ T-cells detected by MHC class II tetramers in peripheral blood of type 1 diabetes patients and at-risk subjects.[erratum appears in J Autoimmun. 2006 Aug;27(1):69] J Autoimmun. 2005;25:235–43. doi: 10.1016/j.jaut.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 54.Reijonen H, Kwok WW, Nepom GT. Detection of CD4+ autoreactive T cells in T1D using HLA class II tetramers. Ann N Y Acad Sci. 2003;1005:82–7. doi: 10.1196/annals.1288.009. [DOI] [PubMed] [Google Scholar]