

Abstract
In this study, we compare the proteasome inhibition capabilities of two anticancer candidates, [Ni(LIA)2] (1) and [Zn(LIA)2] (2), where LIA- is the deprotonated form of the ligand 2,4-diiodo-6-(((2-pyridinylmethyl)amino)methyl)phenol.
Species 1 contains nickel(II), a considerably inert ion that favors covalency, whereas 2 contains zinc(II), a labile transition metal ion that favors predominantly ionic bonds.
We report on the synthesis and characterization of 1 and 2 using various spectroscopic, spectrometric, and structural methods. Furthermore, the pharmacological effects of 1 and 2, along with the salts NiCl2 and ZnCl2, were evaluated in vitro and in cultured human cancer cells in terms of their proteasome-inhibitory and apoptotic cell death-inducing capabilities. It is shown that neither NiCl2 nor 1 have the ability to inhibit the proteasome activity at any sustained levels. However, ZnCl2 and 2 showed superior inhibitory activity to the chymotrypsin-like activity of both 26S proteasome (IC50 = 5.7 and 4.4 μmol/L, respectively) and purified 20S proteasome (IC50 = 16.6 and 11.7 μmol/L, respectively) under cell-free conditions. Additionally, inhibition of proteasomal activity in cultured prostate cancer cells by 2 was associated with higher levels of ubiquitinated proteins and apoptosis. Treatment with either the metal complex or the salt was relatively non-toxic toward human normal cells. These results strengthen the current working hypothesis that fast ligand dissociation is required to generate an [MLIA]+ pharmacophore, capable of interaction with the proteasome. This interaction, possibly via N-terminal threonine aminoacids present in the active sites, renders the proteasome inactive. Our results present a compelling rationale for 2, along with its gallium(III) and copper(II) congeners to be further investigated as potential anticancer drugs that act as proteasome inhibitiors.
Introduction
Use of platinum-containing analogues has been a viable therapeutic strategy in a host of malignancies.1,2,3,4,5 However, excessive toxicity have hampered their widespread use leading to the investigation of other metal complexes and distinctive cellular apoptotic pathways.6,7
The 26S proteasome has gained substantial consideration as an anticancer target,8,9,10,11,12 and proteasomal inhibition is a mechanism for tumor cell suppression currently being investigated in our groups. The main function of the ubiquitin-proteasome pathway is to degrade damaged or misfolded proteins.13,14 Targeted proteins are tagged with a series of ubiquitin molecules which are then translocated to the 26S proteasome and subsequently degraded. 15, 16 The enzymatic activity of the 26S proteasome is mediated by the 20S proteasome core that contains three pairs of catalytic sites responsible for its chymotrypsin-, trypsin-, and caspase-like activities.17,18
The validation of the proteasome as a target for cancer therapy came with the FDA approval of bortezomib for the treatment of multiple myeloma.19,20 Studies have shown that bortezomib exerts its antitumor effects by binding and deactivating an N-terminal threonine of the chymotrypsin-like active center. Furthermore, other proteasome inhibitors, such as peptide derivatives with aldehydes, sulfones, and epoxyketones,21 have been shown to be effective in the inactivation of the 26S proteasome by similar mechanisms. Such inhibition has been a valuable approach toward cancer therapy, because it has been shown that human cancer cells are more sensitive to proteasome inhibition than normal cells.22,23
We have demonstrated that 2:1 complexes [M(LIA)2], with [NN'O]-ligands and gallium(III) or copper(II), can promote proteasome inhibition in prostate cancer24,25 and cisplatin-resistant neuroblastoma cells.26 The mechanisms of inhibition are not entirely clear and could involve the deactivation of either the 19S terminal caps, or the 20S core. Nonetheless, molecular modeling with similar complexes suggests that such species cannot dock into the proteasome satisfactorily indicating that new bonds between the inhibitor and the 20S core must be established. Experimental results with equivalent copper(II) species suggest that a 1:1 pharmacophore [M(LIA)]+ (or an aqua equivalent) is needed to allow coordination with the terminal threonine or other coordinating residues.24,25 Thus, an equilibrium [M(LIA)2] ↔ [M(LIA)]+ + LIA- for 2:1 species seems necessary. Therefore, we aim at furthering these studies by designing similar [M(LIA)2] complexes using divalent nickel(II) and zinc(II) metals. Differences due to electronic configurations of the metals lead to species with characteristic behavior for ligand dissociation as depicted in Scheme 1. Nickel(II) has a 3d8 configuration which leads to a non-zero ligand-field stabilization energies (LFSE) and, consequently, should foster slow ligand dissociation, whereas zinc(II) with a 3d10 configuration has zero LFSE, thus fostering rapid ligand dissociation.27,28,29 Slow ligand dissociation would therefore give rise to poor inhibition because no deactivation of the proteasomal active core takes place.
Scheme 1.

Suggested equilibrium of [M(LIA)2] ↔ [M(LIA)]+ + LIA- to generate the [M(LIA)]+ pharmacophore.
In this paper, we report on the synthesis and characterization of [Ni(LIA)2] (1) and [Zn(LIA)2] (2) using various spectroscopic, spectrometric, and structural methods. After careful characterization these species were evaluated as proteasome inhibitors to purified 20S proteasome and 26S proteasome in whole-cell extracts, as well as in intact prostate cancer and leukemia cells. The results follow.
Results and Discussion
Ligand Design
Our groups are interested in the development of discrete complexes of well established stoichiometry, formed between asymmetric [NN'O] ligands and transition metal ions for anticancer therapy. Such ligands are an evolution from terbutylated analogues used as biomimetic models for galactose-oxidase.30 The presence of electron-donating and -withdrawing phenol substituents (i.e. H, tBu, Cl, Br, and I) in such complexes has shown distinctive influence on the apoptosis of cisplatin-resistant neuroblastoma and prostate cancer cell lines.25,26 We are mostly engaged with the phenol-based ligand 2,4-diiodo-6-(((2-pyridinylmethyl)amino)methyl)phenol, synthesized by treatment of 2-hydroxy-3,5-diiodobenzaldehyde with 2-aminomethylpyridine followed by reduction with sodium borohydride.26 Upon deprotonation, this ligand leads to 2:1 ligand-to-metal [M(LIA)2] species with divalent ions, hence, eliminating the required charge balance by counterions. Moreover, a secondary amine in this ligand allows for the design of species with appended moieties to enhance water solubility 31,32 (currently at 4.5×10-5 mol/L for the equivalent gallium complex27) or lipophilicity. 33,34,35,36 Such changes can address concerns with future oral administration as therapeutics.
Syntheses, Spectrometric, and Spectroscopic Evaluation
Complexes [Ni(LIA)2] (1) and [Zn(LIA)2] (2) were synthesized by treatment of HLIA with the proper acetate salts in methanol and isolated in good yields (~80 %). Triethylamine was used as a base to ensure ligand deprotonation. Spectrometric evaluation of 1 and 2 in methanol using ESIMS in the positive mode led to identification with a good isotropic distribution for the main peaks m/z = 988.7 and 994.7 respectively for [1+(H+)]+ and [2+(H+)]+ (Figure 1). The 1:1 ligand-to-metal species were also detected via ESIMS. Since such species only become prominent at higher cone voltages, they were considered a direct result of fragmentation of 1 and 2. Infrared spectra confirmed the presence of the ligand and revealed the absence of peaks at ca. 1560 and 1450 cm-1 associated with potential acetate counterions. This gives further evidence that the 2:1 ligand-to-metal species are the favored products. Furthermore, elemental analyses were in excellent agreement with those expected for 1 and 2.
Figure 1.

ESI(pos) peak clusters for [1+(H+)]+ and [2+(H+)]+ in methanol. The relative abundance axis of each complex is omitted for clarity.
The 1H-NMR spectrum of the ligand shows the expected signals for protons at the pyridine and phenol rings between 7.0 and 9.0 ppm.26 Distinctive signals for methylene groups vicinal to the pyridine and phenol rings appear respectively as singlet peaks at 3.94 and 3.91 ppm. At room temperature the 1H-NMR spectrum of the zinc complex 1 is comparable to that of the ligand with further splitting of the peaks between 7-9 ppm, suggestive of two ligands with dissimilar conformation. Equally distinctive is the observation that the methylene signals become broadened and split into 6 ill-defined bands, indicating that the complex is not rigid, 37 and that at least six of the eight methylene protons are non-equivalent. By lowering the temperature to ca. -60°C, these signals coalesce into three peaks. The amine proton, originally at 3.49 ppm in the ligand, splits into two broad peaks at 1.77 and 1.18 pm and appears at 2.20 ppm at low temperature. The nickel species yielded broad and ill-defined 1H-NMR results in agreement with the paramagnetic nature of a 3d8 high spin species. A detailed investigation of the ligand dynamics38 of 1 and its gallium-containing counterpart26 using VT-NMR properties is under development.
Molecular Structural Characterization
Good crystals for X-ray diffractometric analyses were isolated for 1 and 2 in chloroform and dichloromethane, respectively, and their molecular structures were determined. The ORTEP renditions for 1 and 2 are shown in Figure 2, and selected bond lengths and angles are displayed in Table 1. Complex 1 crystallizes in a monoclinic space group P21/n composed of a nickel(II) ion coordinated to two deprotonated (LIA)- ligands, with each of them containing an [NpyNamOphen] set of donors. Both ligands are facially coordinated with the two pyridine rings (Ni - Npy ≈ 2.0Å), the two amine groups (Ni - Nam ≈ 2.09 Å), and the two phenolate rings (Ni - Ophen ≈ 2.04 Å), arranged trans to one another to yield a pseudo-octahedral geometry. Crystals of 2 appeared as colorless needles and crystallize in an orthorhombic P212121 space group, also showing a similar facial coordination of the (LIA)- ligands in a pseudo-octahedral geometry. However, whereas 1 exhibits a symmetrical all-trans environment described as [Ni<Nam1Nam2><Npy1Npy2><O(phO-)1O(phO-)2>]39 in a bent arrangement36, 2 is described as having an all-cis [Zn<Nam1O(phO-)2><Npy1Nam2><O(phO-)1 Npy2>]. As observed by the 1H-NMR spectrum with distinctive signals for methylene groups the two ligands present dissimilar conformations.
Figure 2.

ORTEP diagrams at 50 % probability level for 1(a) and 2(b).
Table 1.
Selected bond lengths (Å) and angles (deg) for 1 and 2.
| 1 | 2 |
|---|---|
| Ni(1)-O(2) = 2.036(2) | Zn(1)-O(2) = 2.025(6) |
| Ni(1)-O(1) = 2.057(2) | Zn(1)-O(1) = 2.111(5) |
| Ni(1)-N(2) = 2.088(3) | Zn(1)-N(1) = 2.149(6) |
| Ni(1)-N(3) = 2.090(3) | Zn(1)-N(4) = 2.179(6) |
| Ni(1)-N(4) = 2.096(3) | Zn(1)-N(2) = 2.181(7) |
| Ni(1)-N(1) = 2.098(3) | Zn(1)-N(3) = 2.259(7) |
| O(2)-Ni(1)-O(1) = 179.52(10) | O(2)-Zn(1)-O(1) = 92.0(2) |
| O(2)-Ni(1)-N(2) = 87.86(10) | O(2)-Zn(1)-N(1) = 90.0(2) |
| O(1)-Ni(1)-N(2) = 91.99(10) | O(1)-Zn(1)-N(1) = 99.6(2) |
| O(2)-Ni(1)-N(3) = 89.60(10) | O(2)-Zn(1)-N(4) = 92.4(2) |
| O(1)-Ni(1)-N(3) = 90.87(10) | O(1)-Zn(1)-N(4) = 88.7(2) |
| N(2)-Ni(1)-N(3) = 99.14(11) | N(1)-Zn(1)-N(4) = 171.3(3) |
| O(2)-Ni(1)-N(4) = 92.66(10) | O(2)-Zn(1)-N(2) = 168.9(2) |
| O(1)-Ni(1)-N(4) = 87.49(10) | O(1)-Zn(1)-N(2) = 89.2(2) |
| N(2)-Ni(1)-N(4) = 179.47(11) | N(1)-Zn(1)-N(2) = 79.0(2) |
| N(3)-Ni(1)-N(4) = 80.76(11) | N(4)-Zn(1)-N(2) = 98.6(2) |
| O(2)-Ni(1)-N(1) = 88.86(10) | O(2)-Zn(1)-N(3) = 87.9(2) |
| O(1)-Ni(1)-N(1) = 90.66(10) | O(1)-Zn(1)-N(3) = 166.7(2) |
| N(2)-Ni(1)-N(1) = 81.01(11) | N(1)-Zn(1)-N(3) = 93.7(2) |
| N(3)-Ni(1)-N(1) = 178.45(11) | N(4)-Zn(1)-N(3) = 78.1(3) |
| N(4)-Ni(1)-N(1) = 99.10(12) | N(2)-Zn(1)-N(3) = 93.5(2) |
We have demonstrated the role played by structural and electronic effects in a series of 3d5-10 [M(L)2] species with such asymmetric NN'O ligands. Although ligand rigidity enforces meridional coordination in similar imine ligands, electronic configuration leads to a facial coordination mode in flexible amines. The metal centers also dictate the preferential cis or trans orientation of equivalent phenolates and other donor sets in vicinal ligands, with 3d5high spin ions40 displaying a cis-arrangement and 3d6low spin and 3d7high spin ions36,37 supporting a trans-orientation. Species 1 reinforces38 the notion of an all-trans mode for 3d8 configurations, whereas 2 seems to fall within other 3d10 configurations26,41,42,43 lacking a clear preference. Another remark is that while most of the Ophenolate bond lengths for 1 and 2 are comparable at 2.02-2.05 Å, one of the Zn-Ophenolate bonds for 2 is elongated reaching ca. 2.11 Å. This longer bond length is comparable to values found in literature. 44 This is attributed to the 3d10 electronic configuration of the zinc(II) ion which favors electrostatic interactions.
Electronic Structure Calculations
A series of electronic structure calculations were carried out for the nickel-containing 1, and zinc-containing 2, as well as on isomers with alternative geometries and binding modes aiming at evaluating bond nature, delocalization and energy differences between isomers. These results allowed to gain insight on how the electronic structure of these species may foster the formation of the expected pharmacophores [MLIA]+. The optimized geometries for the electronic structures of 1 and 2 are in good agreement with the crystallographic data presented above.
Nickel species
A recent study by Thomas et. al.45 on similar nickel complexes supports a favorable trans facial coordination of the ligands over the meridional mode by approximately 5.0 kcal/mol using a comparable level of theory. Therefore. we restricted our studies to two different all-trans facial isomers, namely, the structurally characterized 1 and a hypothetical 1'. Isomer 1 displays both phenolate rings in parallel planes, whereas these same rings are perpendicular to one another in 1' (Figure 3A). The energy difference between the two structures is 3.7 kcal/mol, clearly favoring 1 as the lowest energy state. Interestingly, we recently compared similar isomers with unsubstituted phenolate rings38 and the energy difference was a mere 1.1 kcal/mol. It can be suggested that the iodine substituents play a significant role in favoring 1 instead of 1'. Furthermore, both isomers display triplet ground states with S=1, consistent with a 3d8 high spin configuration where two unpaired electrons populate the dx2-y2 (SOMO) and dz2 (SOMO-1) orbitals. (Figure 3B)
Figure 3.

A: Depiction of the two facial [Ni(LIA)2] isomers 1 and 1'. B: Selected MOs for unpaired electrons.
Because divalent zinc is a 3d10 ion, it lacks LFSE and the coordination mode is most likely the result of ligand sterics. For 2, we observed a new facial all-cis [Zn<Nam1O(phO-)2><Npy1Nam2><O(phO-)1 Npy2>] coordination mode for a doubly deprotonated species, well in contrast with the all-trans counterpart obtained by Neves et. al.44 with a non-substituted ligand. Thus two different facial isomers were explored for the zinc complex as shown in Figure 4A. The structurally characterized 2 and a hypothetical 2' that matched the geometry adopted for the nickel containing 1. Interestingly, the calculations favor 2' by a small margin of less than 2.0 kcal/mol, thus in disagreement with the observed structure. Therefore, in the absence of a LFSE the bonding is mainly ionic in nature and other factors such as solvation, crystal packing, or intermolecular force effects must control the final geometry.
Figure 4.
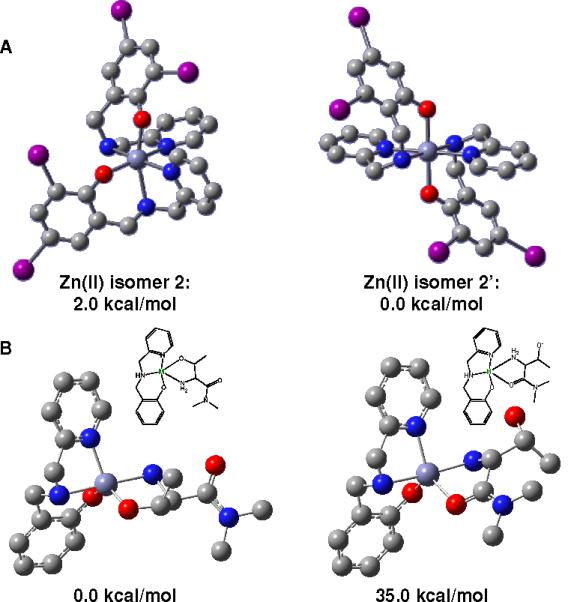
(a) Depiction of the two facial [Zn(LIA)2] isomers 2 and 2'. (b) Possible interaction between the fragment [Zn(LIA)]+ and threonine.
In order to address partial atomic charges, natural bond order (NBO) analysis was performed with both 1 and 2, the results are shown in Table 2. It is clear from these charge distributions that the interaction between the Ni(II) center in 1 and the ligand is more delocalized, i.e. covalent, than the interaction between the Zn(II) center and the ligand in 2. This can be seen by lower charges on the metal: 1.3 for 1 versus 1.6 for 2. The values for other atoms in 2 are consistently larger in magnitude reinforcing the notion of more localized, i.e. ionic bonds. These values indicate that there is less stabilization of the positive divalent metal charge unto the ligands for 2 over 1. This difference is significant, since both complexes are discrete neutral species with an overall zero charge. Thus, it is possible to conclude that an ionic nature would favor ligand dissociation in the biologic milieu for 2 and the equilibrium suggested in Scheme 1 should be facilitated.
Table 4.
Crystal data and structure refinements for [Ni(LIA)2] (1) and [Zn(LIA)2] (2)
| Formula | C29H25C19I4N4O2Ni | C29H30C16I4N4O3Zn |
|---|---|---|
| FW | 1346.89 | 1268.24 |
| Space group | P21/n | P212121 |
| a (Å) | 18.4231(5) | 13.2593(4) |
| b (Å) | 10.1797(3) | 15.4882(5) |
| c (Å) | 22.1493(6) | 19.6143(6) |
| β (deg) | 92.8120(10) | |
| V (Å3) | 4148.9(2) | 4028.0(2) |
| Z | 4 | 4 |
| Temp (K) | 100(2) | 100(2) |
| λ (Å) | 0.71073 | 0.71073 |
| Density, calcd (Mg cm-3) | 2.156 | 2.091 |
| μ (mm-1) | 4.057 | 4.109 |
| R(F)(%)a | 3.63 | 6.33 |
| Rw(F) (%)a | 5.91 | 13.18 |
R(F) = Σ||Fo|-|Fc||/Σ|Fo|; Rw(F) = [Σw(Fo2 - Fc2)2 / Σw(Fo2)2]1/2 for I>2σ(I).
Finally, initial theoretical treatment of the possible binding modes between the fragment [Zn(LIA)]+ and a deprotonated threonine residue were also performed. A simplified [Zn(L)]+ fragment with an unsubstituted ligand was used and, in order to model the terminal nature of the threonine residue, a dimethylated amide residue was incorporated, as shown in Figure 4B. Two coordination modes were probed; the first considered binding between the zinc center at the [Zn(L)]+ fragment with the terminal (and deprotonated) hydroxyl group and the secondary amine group, whereas the second focused on the binding through the secondary amine and the carbonyl group of the amide residue. The first binding mode is approximately 35 kcal/mol more stable, and although this proposition is merely speculative at this point, it suggests that terminal hydroxo/amine coordination to zinc would be favored.
Induction of Cell Death and Inhibition of Cell Proliferation
The cytotoxic effect of NiCl2, ZnCl2, 1, and 2, was tested in human leukemia Jurkat T cells treated at different concentrations for 18 h. After each treatment, trypan blue exclusion assay was performed to assess cell death (Figure 5A). Cells treated with 2 exhibited a dose-dependent activity reaching 48 %, 70 %, 95 %, and 100 % cell death at 7.5, 10, 15, and 20 μmol/L, respectively. This turned to be the single viable species, since the metal salts and 1 had marginal cell-death induction, smaller than ~10 % compared with DMSO-treated cells, at as high as 20 μmol/L.
Figure 5.

Cell death (A) and cell growth inhibition (B) on human leukemia
To further substantiate this effect, we evaluated whether 2 can suppress cell proliferation of human prostate cancer cells. C4-2B human prostate cancer cells were treated with NiCl2, ZnCl2, 1, 2, and the DMSO control at different concentrations for 18 h, followed by measurement of cell proliferation by MTT assay (Figure 5B). It was observed that cells treated with 2 suppressed cell proliferation in a dose-dependent manner (IC50 = ~6 μmol/L), reaching 100 % inhibition at 10 μmol/L. Furthermore, cells treated with 2 at 5 and 7.5 μmol/L decreased cell proliferation by ~35 % and ~85 %, respectively. Consistently, C4-2B cells treated with the metal salts or 1 showed little or no inhibitory effect even at the highest concentration tested of 20 μmol/L.
In Vitro Proteasome Inhibition
To test the proteasome inhibitory capacity of these species, a comparison of the inhibitory activity of NiCl2, ZnCl2, 1, and 2 to the 26S proteasomal activity was performed under cell-free conditions. An extract of C4-2B prostate cancer cells (Figure 6A) was used and the results indicate that both the ZnCl2 salt and 2 have the potential to inhibit the chymotrypsin-like activity of the 26S proteasome with IC50 values of 5.7 and 4.4 μmol/L, respectively. This result is consistent with our previous finding that zinc dithiocarbamate complexes can target and inhibit the protesome.46 However, extracts treated with 1 at as high as 25 μmol/L showed only ~20 % inhibition on the 26S proteasome suggesting that intrinsic distinctive mechanisms of inhibition must be present for 1 and 2. Consistent with this finding is the fact that NiCl2 at 25 μmol/L could only inhibit the proteasomal activity by ~25 %.
Figure 6.

In vitro proteasome-inhibitory activity of NiCl2, ZnCl2, 1, and 2. A: Inhibition of CT-like activity of 26S proteasome in C4-2B cell extract. B: Inhibition of the CT-like activity of purified 20S protesome.
To provide direct evidence for distinct mechanisms, we incubated a purified rabbit 20S proteasome with NiCl2, ZnCl2, 1, and 2 at various concentrations, followed by measurement of the chymotrypsin-like activity (Figure 6B). We found that this activity was significantly inhibited with the salt ZnCl2 and 2 with similar potencies (IC50 = 16.6 and 11.7 μmol/L, respectively). Although NiCl2 showed modest inhibitory activity, 1 was rather inactive. Overall, our data remain consistent with the fact that the zinc ion, both as a chloride salt and a complex with the (LIA)- ligand is able to target and inhibit the proteasome under cell-free conditions.
Proteasome Inhibition and Apoptosis Induction in Intact Cancer Cells
To confirm the ability of 2 to inhibit the proteasomal activity in intact tumor cells, C4-2B human prostate cancer cells were first treated with different concentrations (5, 10, and 25 μmol/L) of NiCl2, ZnCl2, 1, and 2 for 18 h, followed by measurement of proteasome inhibition. The values for proteasomal chymotrypsin-like activity are given as a percentage in Table 3. The C4-2B cells treated with 2 showed a dose-dependent inhibition of the proteasomal activity by 31 % inhibition at 10 μmol/L and 86 % inhibition at 25 μmol/L. Consistently, levels of ubiquitinated proteins were increased in a dose-dependent manner in C4-2B cells (Figure S1). In comparison, cells treated with either NiCl2, ZnCl2, or 1 showed negligible proteasome inhibitory effect.
Table 3.
Percentile of CT-activity after treatment with NiCl2, ZnCl2, 1, and 2
| Dose μM | C4-2B % | SDa (+-) | PC3 % | SDa (+-) | MCF 10A % | SD (+-) | |
|---|---|---|---|---|---|---|---|
| DMSO | — | 100 | 0.81 | 100 | 1.56 | 100 | 3.00 |
| NiCl2 | 25 | 92 | 1.41 | 99 | 0.10 | 98 | 1.83 |
| ZnCl2 | 25 | 99 | 0.90 | 127 | 0.45 | 116 | 1.56 |
| 5 | 84 | 0.87 | 101 | 0.01 | — | — | |
| [Ni(LIA)2] (1) | 10 | 83 | 0.96 | 81 | 1.59 | — | — |
| 25 | 80 | 2.27 | 92 | 0.85 | 89 | 1.96 | |
| 5 | 111 | 1.02 | 114 | 2.63 | 118 | 0.55 | |
| [Zn(LIA)2] (2) | 10 | 69 | 0.14 | 17 | 1.38 | 97 | 1.48 |
| 25 | 14 | 0.68 | 10 | 1.13 | 83 | 1.59 |
Standard deviation
It has been shown that proteasome inhibition can lead to decreased levels of androgen receptor (AR) expression.47,48 Therefore, a decrease in such expression should be observed assuming proteasome activity inhibition by 2. Consistently, this is the only species that down regulated significantly AR and 25 μmol/L treatment completely abrogated AR expression levels (Figure S1). These results remain consistent with the ability of 2 to inhibit the proteasome activity.
It has been shown that inhibition of the proteasomal chymotrypsin-like activity selectively in transformed cells could result in the induction of apoptosis.23 To investigate whether proteasome inhibition and androgen receptor down regulation are associated with apoptotic cell death, apoptotic-specific PARP disappearance and morphological changes were measured in the same experiment (Figure 7, Figure S1). The results show that only cells treated with 25 μmol/L of 2 were able to completely abrogate full length PARP, whereas cells treated with either NiCl2, ZnCl2, or 1 at the highest concentration tested had little visible effects. Consistently, morphological changes (detached, shrunken and apoptotic blebbing) were observed in cells treated with 25 μmol/L 2 and to a significant but lesser extent at 10 μmol/L (Figure 7). Much less aberrant morphological changes were detected in the cells treated with metal salt or 1 at the highest concentration tested (Figure 7). These results show that the induction of apoptosis in C4-2B cells by 2 is associated with inhibition of proteasomal chymotrypsin-like activity.
Figure 7.
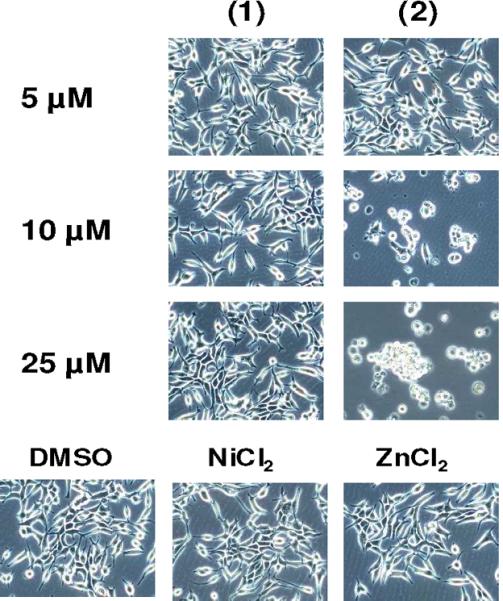
Cellular morphological effects of NiCl2, ZnCl2, 1, and 2 on C4-2B prostate cancer cells.
Upon demonstrating the ability of 2 to inhibit the proteasomal chymotrypsin-like activity in AR-dependent C4-2B prostate cancer cells, we then tested the effect of 2 on AR-independent PC-3 human prostate cancer cells. PC-3 cells were treated with 5, 10 and 25 μmol/L of 1 or 2, and their metal salt for 18 h, followed by measurement of the proteasome activity, accumulated levels of ubiquitinated proteins and apoptosis induction. Percentage of proteasomal chymotrypsin-like activity is designated in Table 3 with DMSO as control. We found that 2 could inhibit the proteasomal activity and induce apoptotic cell death in PC-3 prostate cancer cells, whereas 1, NiCl2, and ZnCl2 showed little or no effect (Table 3, Figure S2).
Kinetics of Proteasome Inhibition and Apoptosis Induction
To study the kinetic effect of proteasome inhibition, C4-2B prostate cancer cells were treated with 15 μmol/L 2 over different time points (2-18 h) and their cell lysates were used to measure the proteasomal-chymotrypsin-like activity (Figure 8A). The proteasomal chymotrypsin-like activity was inhibited by 18 %, 42 %, 57 %, and 63 %, respectively, at 2, 4, 8, and 18 h. This result was consistent with the time-dependent increase in levels of accumulated ubiquitinated proteins. Furthermore, lower levels of AR were detected in cells treated with 2 at all time points and complete abrogation of AR expression is detected in cells after 18 h treatment. Importantly, apoptosis-specific PARP down-regulation was detected at later time points with complete PARP disappearance at 18 h (Figure S3). Apoptosis induction at later time points is also typified with the appearance of aberrant morphological changes (detached, shrunken and apoptotic blebbing) (Figure 8B). These results clearly demonstrate that induction of the apoptosis occurs after proteasome inhibition. Thus, proteasome inhibition appears to be required for apoptosis induction.
Figure 8.
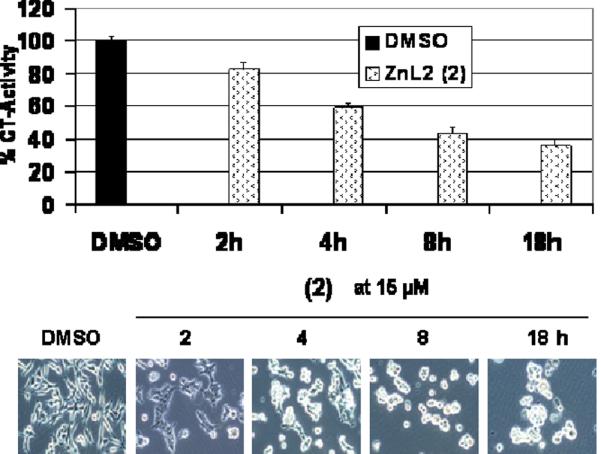
Kinetic effect of proteasome inhibition and apoptosis induction by 2 in C4-2B prostate cancer cells.
Tumor Cell Selectivity
The ability to distinguish normal from malignant cells is of paramount importance for developing clinically-relevant anticancer drugs. To determine whether inhibition of prostate cancer cellular proteasome activity achieved by 2 is selective toward malignant cells but not normal cells, we used normal-immortalized human breast cell line, MCF-10A. The MCF-10A cells were treated with different concentrations with 2 as high 25 μmol/L for 18 h, followed by measurement of proteasomal chymotrypsin-like activity and apoptosis. We found that when these nontransformed cells were treated with 2, only 17 % proteasome inhibition was detected at the highest concentrations tested (Table 3). Other treatments also had little effect on MCF-10A cells (Table 3). To determine whether the inability of 2 to inhibit the proteasome activity at sustainable levels is associated with the lack of apoptosis induction in these normal immortalized breast cells, apoptosis associated morphological changes were then assessed in the same experiment. These normal, immortalized MCF-10A cells showed only little, such cell death-related detachment after treatment with 2 up to 18 h at the highest concentration tested (Figure S4). Furthermore, the species NiCl2, ZnCl2, and 1 had little or no cytotoxic effect on normal cells. Our data suggests that 2 could inhibit the proteasome activity and induce apoptosis selectively in human cancer cells but not in normal immortalized cells, validating 2 as a promising proteasome inhibitor.
Summary and Conclusions
In this paper we report on two new coordination complexes as potential anticancer candidates, namely, [Ni(LIA)2] (1) and [Zn(LIA)2] (2). Both species were characterized by several spectroscopic, spectrometric, and structural methods and display a well established 2:1 ligand-to-metal stoichiometry. DFT calculations considering different isomers of 1 and 2 were performed and show good agreement with the nickel species, but fail to predict the appropriate geometry for the zinc-containing species. Furthermore, initial studies considering coordination of a 1:1 [Zn(L)]+ fragment with threonine suggest a favorable coordination through the terminal hydroxyl group of the amino acid.
The effects of NiCl2, ZnCl2, 1, and 2 were tested toward a purified rabbit 20S proteasome and 26S proteasome in cell extracts (in which cell membranes have been disrupted) of leukemia and human prostate cancer cell lines. The results indicate that only 2 and ZnCl2 have a direct inhibitory effect on the proteasome to any significant levels. Furthermore, when NiCl2, ZnCl2, 1, and 2 were tested on the 26S proteasome of cultured intact leukemia and human prostate cancer cells it has been evidently shown that only 2 exhibited potent anti-proliferative and cell death-inducing activity. Similarly, only 2 induced higher levels of ubiquitinated proteins, which were associated with decreased levels of proteasomal chymotrypsin-like activity. In addition, the decrease of proteasomal chymotrypsin-like activity observed for 2 is tightly associated with tumor cell apoptosis as seen by the morphological changes and the apparent disappearance of the full length PARP fragment. Species 2 also showed remarkably low cytotoxicity toward normal human breast cells.
This sharp contrast in proteasome activity inhibition between 1 and 2 is suggested to be related to the nature of the metal ion and its degree of reactivity when combined with NN'O-containing ligands. As observed in similar complexes from our group24 considerable proteasome inhibition can be attained through 1:1 ligand to metal species that is believed to be the pharmacophore in all these species. Therefore, an equilibrium [M(LIA)2] ↔ [M(LIA)]+ + LIA- seems necessary to facilitate the formation of the pharmacophore with available coordination sites capable of interaction with the 20S proteasome, likely to be via the N-terminal threonine residue. It is observed from the molecular structures and DFT calculations available that covalent interactions prevail in 1, while 2 is ionic in nature. We, therefore, propose that this intrinsic difference defines the facility of pharmacophore formation and determines the activity of these species. The lack of activity observed for ZnCl2 in intact cells reinforces the notion offered for the equivalent copper counterparts24 that the ligand (LIA)- serves as a shuttle vector to cross the cellular membrane. Taken together, we suggest that the presence of the fragment [Zn(LIA)]+ is required for proteasome inhibition. At this point it is not clear whether the ligand dissociation [Zn(LIA)2] ↔ [Zn(LIA)]+ + LIA- is intra or extracellular, and if intracellular, before or after reaching the 26S proteasome. Furthermore, it is likely that water molecules will coordinate to the zinc ion forming [Zn(LIA)(H2O)n]+ hydrated species. The investigation of these issues is a current goal of our groups and will be properly developed in the future. Nonetheless, the data observed thus far provides a compelling rationale for the clinical development of 2 as a potential anticancer drug.
Experimental Section
Materials and Methods
All reagents were obtained from commercial sources and were used as received. Methanol was distilled over CaH2. IR spectra were measured from 4000 to 400 cm-1 as KBr pellets on a Tensor 27 FTIR-spectrophotometer. ESI spectra on the positive mode were measured in methanol on a Micromass Quattro LC triple quadrupole mass spectrometer with an electrospray/APCI source and Walters Alliance 2695 LC, autosampler and photodiode array UV detector. Experimental assignments were simulated based on peak location and isotopic distributions. The 1HNMR spectra were measured in CDCl3 in a Varian Unity-300 instrument. Elemental analyses were performed by Midwest Microlab, Indianapolis, Indiana. Trypan blue exclusion dye was purchased from Sigma Aldrich (St. Louis, MO). The peptide substrate Suc-LLVY-AMC (for the proteasomal chymotrypsin-like activity) was purchased from Calbiochem, Inc (San Diego, CA). RPMI 1640, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was purchased from Aleken Biologicals (Nash, TX). Antibodies against Ubiquitin, Actin, and secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibody against human poly (ADP-ribose) polymerase (PARP) was purchased from BIOMOL International LP (Plymouth Meeting, PA).
X-ray Structural Determination for [Ni(LIA)2] (1) and [Zn(LIA)2] (2)
Diffraction data were measured on a Bruker X8APEX-II kappa geometry diffractometer with Mo radiation and a graphite monochromator. Frames were recorded for 10 s at 100 K with the detector at 40 mm and 0.3 degrees between each frame. APEX-II49 and SHELX50 software were used in the collection and refinement of the models (Table 4).
Table 2.
NBO partial atomic charges.
| Ni (1) | Zn (2) | |
|---|---|---|
| M | 1.3 | 1.6 |
| O1 | -0.87 | -0.90 |
| O2 | -0.87 | -0.92 |
| N1 | -0.56 | -0.64 |
| N2 | -0.75 | -0.77 |
| N3 | -0.56 | -0.61 |
| N4 | -0.75 | -0.80 |
Crystals of [Ni(LIA)2] (1) were colorless plates. A total of 86456 reflections were counted, which averaged to 10883 independent data. Hydrogen atoms were placed at calculated positions except for those on nitrogen which were observed. The complex crystallized with three equivalents of chloroform. All atoms occupy general positions.
Crystals of [Zn(LIA)2] (2) appeared as colorless needles. A total of 77377 reflections were measured, yielding 14112 unique data. Hydrogen atoms were placed in calculated positions. The complex crystallized with three equivalents of methylene chloride and one equivalent of water. All atoms occupy general positions.
Electronic Structure Calculations Methods
The B3LYP level of theory51 with the SDD52 basis sets was employed throughout because of the presence of iodine atoms, and all of the calculations were done using the Gaussian53 series of programs. Geometries were fully minimized, without symmetry constraints, using standard methods54. Located stationary points were characterized by computing analytic vibrational frequencies. Reported energies include zero-point correction. Cartesian coordinates of all of the optimized structures are provided in the Supporting Information.
Syntheses
The iodo-substituted ligand HLIAwas synthesized by treatment of 2-hydroxy-3,5-diiodobenzaldehyde with 2-aminomethylpyridine in methanol followed by reduction with sodium borohydride as previously published.26
[Ni(LIA)2] (1)
A 15 mL methanol solution of HLI (0.49 g, 1.1 mmol) was added dropwise to a 15 mL methanol solution of Ni(OAc)2·2H2O (0.25 g, 1.2 mmol) at 45 °C. After 45 minutes a purple precipitate was obtained, isolated by frit filtration, and washed with cold methanol and ether. The solid was recrystallized in chloroform. Yield = 0.95 g (88 %). Elemental anal. Calcd for 1 C26H22NiI4N4O2: C 31.58; H 2.24; N 5.67. Found: C, 31.47; H, 2.30; N, 5.56. IR (KBr, cm-1) 3068 ν(N-H), 1606, 1593 (C=N from pyridine), 1486 (C-O from phenyl). ESI pos. in MeOH: m/z = 988.9 for [NiL2+H+]+.
[Zn(LIA)2] (2)
A 15 mL methanol solution containing HLI (0.51 g, 1.1 mmol) was added dropwise to a 15 mL methanol solution of Zn(OAc)2· 2H2O (0.26 g, 1.2 mmol) at 45°C. A white precipitate was obtained after 1 hour, isolated by frit filtration, and washed with cold methanol and ether. The solid was recrystallized in dichloromethane. Yield = 0.85 g (77 %). Elemental anal. Calcd for 2 C26H22ZnI4N4O2: C 31.37; H 2.23; N 5.63. Found: C, 31.27; H, 2.38; N, 5.58. IR (KBr, cm-1) 3290 (N-H), 1608 (C=N from pyridine), 3079 ν(N-H). ESI pos. in MeOH: m/z = 994.9 for [ZnL2+H+]+.
Cell Cultures and Whole-cell Extract Preparation
Human prostate cancer cells, C4-2B and PC-3, and human leukemia Jurkat T cells were grown in RPMI-1640 supplemented with 10 % fetal bovine serum and maintained at 37° C and 5 % CO2. MCF-10A (normal, derived from benign human breast tissue) were obtained and cultured as previously described.55 A whole-cell extract was prepared as previously described.57
Cell Proliferation Assay
Cells were seeded in quadruplicate in a 96-well plate and grown until 70-80 % confluence, followed by treatment with indicated agents for 18 h. After that, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was done as described previously.57
Trypan Blue Assay
Jurkat T cells were treated with NiCl2, ZnCl2, 1, and 2 for 18 h at indicated concentrations followed by measurement of cell death. The trypan blue dye exclusion assay was performed by mixing 100 μl of cell suspension with 50 μl of 0.4 % trypan blue dye before injecting into a hemocytometer and counting. The number of cells that absorbed the dye and those that excluded the dye were counted, from which the percentage of nonviable cell number to total cell number was calculated.
Proteasomal Activity in Whole-cell Extract or Purified 20S proteasome
C4-2B whole-cell extract (8 μg) or a purified 20S rabbit proteasome (35 ng) were incubated with 10 μmol/L CT-substrate, Suc-LLVY-AMC, in 100 μL assay buffer [50 mmol/L Tris-HCl (pH 7.5)] in the presence of NiCl2, ZnCl2,1, and 2 at various concentrations or solvent DMSO as control. After 2 h incubation at 37°C, production of hydrolyzed AMC groups was measured using a Wallac Victor3™ multilabel counter with an excitation filter of 365 nm and an emission filter of 460 nm.
Proteasome CT-like Activity in Cells
Proteins extracted from cells after each treatment were incubated for 2 h at 37° C in 100 μl of assay buffer (50 mmol/L Tris-HCL, pH 7.5) with 10 μmol/L fluorogenic substrate Suc-LLVY-AMC as described previously.57
Western Blot Analysis
Cell extracts were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Western blot analysis was performed using specific antibodies to PARP, Ubiquitin, or AR followed by visualization using the HyGLO reagent (Denville Scientific, Metuchin, NJ).
Cellular Morphology Analysis
A Zeiss Axiovert 25 microscope was used for all microscopic imaging with phase contrast for cellular morphology. Magnification ×100.
Supplementary Material
Acknowledgements
M.F. and S.S.H. contributed equally to this publication. Q.P.D. acknowledges the Karmanos Cancer Institute of Wayne State University, the Department of Defense Breast Cancer Research Program (W81XWH-04-1-0688 and DAMD17-03-1-0175) and the National Cancer Institute (1R01CA120009). C.N.V. acknowledges the Wayne State University and partial support from the National Science Foundation (CHE-0718470). M.F. acknowledges a training grant, “Ruth L. Kirschstein National Service Research Award” (T32-CA009531).
References
- (1).Guo Z, Sadler PJ. Angew. Chem., Int. Ed. 1999;38:1512. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1512::AID-ANIE1512>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- (2).Wong E, Giandomenico CM. Chem. Rev. 1999;99:2451. doi: 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- (3).Wang D, Lippard SJ. Nat. Rev. Drug Disc. 2005;4:307. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- (4).Choi S, Vastag L, Larrabee YC, Personick ML, Schaberg KB, Fowler BJ, Sandwick RK, Rawji G. Inorg. Chem. 2008;47:1352. doi: 10.1021/ic701868b. [DOI] [PubMed] [Google Scholar]
- (5).Egger AE, Hartinger CG, Hamidane HB, Tsybin YO, Keppler BK, Dyson PJ. Inorg. Chem. 2008;47:10626. doi: 10.1021/ic801371r. [DOI] [PubMed] [Google Scholar]
- (6).Galanski M, Arion VB, Jakupec MA, Keppler BK. Curr. Pharm. Design. 2003;9:2078. doi: 10.2174/1381612033454180. [DOI] [PubMed] [Google Scholar]
- (7).Ronconi L, Giovagnini L, Marzano C, Bettìo F, Graziani R, Pilloni G, Fregona D. Inorg. Chem. 2005;44:1867. doi: 10.1021/ic048260v. [DOI] [PubMed] [Google Scholar]
- (8).Giovagnini L, Sitran S, Montopoli M, Caparrotta L, Corsini M, Rosani C, Zanello P, Dou QP, Fregona D. Inorg. Chem. 2008;47:6336. doi: 10.1021/ic800404e. [DOI] [PubMed] [Google Scholar]
- (9).Cvek B, Milacic V, Taraba J, Dou QP. J. Med. Chem. 2008;51:6256. doi: 10.1021/jm8007807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Adsule S, Barve V, Chen D, Ahmed F, Dou QP, Padhye S, Sarkar FH. J. Med. Chem. 2006;49:7242. doi: 10.1021/jm060712l. [DOI] [PubMed] [Google Scholar]
- (11).Beenen MA, An C, Ellman JA. J. Am. Chem. Soc. 2008;130:6910. doi: 10.1021/ja800829y. [DOI] [PubMed] [Google Scholar]
- (12).Paramore A, Frantz S. Nat. Rev. Drug Discovery. 2003;2:611. doi: 10.1038/nrd1159. [DOI] [PubMed] [Google Scholar]
- (13).Peters JM, Franke WW, Kleinschmidt JA. J. Biol Chem. 1994;269:7709. [PubMed] [Google Scholar]
- (14).Goldberg AL. Science. 1995;268:522. doi: 10.1126/science.7725095. [DOI] [PubMed] [Google Scholar]
- (15).Dou QP, Li B. Drug Res. Update. 1999;2:215. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- (16).Hochstrasser M. Cur. Opin. Cell Biol. 1995;7:215. doi: 10.1016/0955-0674(95)80031-x. [DOI] [PubMed] [Google Scholar]
- (17).Seemuller E, Lupas A, Stock D, Lowe J, Huber R, Baumeister W. Science. 1995;268:579. doi: 10.1126/science.7725107. [DOI] [PubMed] [Google Scholar]
- (18).Voges D, Zwickl P, Baumeister W. Ann. Rev. Biochem. 1999;68:1015. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- (19).Dou QP, Goldfarb RH. IDrugs. 2002;5:828. [PubMed] [Google Scholar]
- (20).Kane RC, Farrell AT, Sridhara R, Pazdur R. Clin. Cancer Res. 2006;12:2955. doi: 10.1158/1078-0432.CCR-06-0170. [DOI] [PubMed] [Google Scholar]
- (21).(a) Borissenko L, Groll M. Chem. Rev. 2007;107:687. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]; (b) Kisselev AF, Goldberg AL. Chem. Biol. 2001;08:739. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]; (c) McCormack TA, Cruikshank AA, Grenier L, Melandri FD, Nunes SL, Plamondon L, Stein RL, Dick LR. Biochem. 1998;37:7792. doi: 10.1021/bi980097q. [DOI] [PubMed] [Google Scholar]
- (22).Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Cancer Res. 1999;59:2615. [PubMed] [Google Scholar]
- (23).An B, Goldfarb RH, Siman R, Dou QP. Cell Death Differ. 1998;5:1062. doi: 10.1038/sj.cdd.4400436. [DOI] [PubMed] [Google Scholar]
- (24).Hindo SS, Frezza M, Tomco D, Heeg MJ, Hryhorczuk L, McGarvey BR, Dou QP, Verani CN. Metals in Anticancer Therapy: Copper(II) Complexes as Inhibitors of the 20S Proteasome. submitted to Chemistry-A European Journal. 2009 January; doi: 10.1016/j.ejmech.2009.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen D, Frezza M, Shakya R, Cui QC, Milacic V, Verani CN, Dou QP. Cancer Res. 2007;67:9258. doi: 10.1158/0008-5472.CAN-07-1813. [DOI] [PubMed] [Google Scholar]
- (26).Shakya R, Peng F, Liu J, Heeg MJ, Verani CN. Inorg. Chem. 2006;45:6263. doi: 10.1021/ic060106g. [DOI] [PubMed] [Google Scholar]
- (27).See for example Miessler GL, Tarr DA. Inorganic Chemistry. 3rd ed. Pearson-Prentice Hall; Upper Saddle River, NJ: 2004. Chapter 12; pp. 412–453.; Basolo F, Perason RG. Mechanisms of Inorganic Reactions. 2nd ed. John Wiley & Sons; New York, NY: 1967. Chapter 3; pp. 124–246.
- (28).Sekaly ALR, Murimboh J, Hassan NM, Mandal R, Ben Younes ME, Chakrabarti CL, Back MH. Environ. Sci. Technol. 2003;37:68. doi: 10.1021/es025805g. [DOI] [PubMed] [Google Scholar]
- (29).(a) Johnson DA, Nelson PG. Inorg. Chem. 1995;34:5666. [Google Scholar]; (b) Johnson DA, Nelson PG. Inorg. Chem. 1995;34:3253. [Google Scholar]
- (30).(a) Michel F, Thomas F, Hamman S, Philouze C, Saint-Aman E, Pierre J-L. Eur. J. Inorg. Chem. 2006;I-18:3684. [Google Scholar]; (b) Shimazaki Y, Huth S, Odani A, Yamauchi O. Angew. Chem., Int. Ed. 2000;39:1666. doi: 10.1002/(sici)1521-3773(20000502)39:9<1666::aid-anie1666>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]; (c) Itoh S, Taki M, Takayama S, Nagatomo S, Kitagawa T, Sakurada N, Arakawa R, Fukuzumi S. Angew. Chem., Int. Ed. 1999;38:2774. doi: 10.1002/(sici)1521-3773(19990917)38:18<2774::aid-anie2774>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (d) Neves A, Verani CN, de Brito MA, Vencato I, Mangrich A, Oliva G, Souza DDHF, Batista AA. Inorg. Chim. Acta. 1999;290:207. [Google Scholar]; (e) Vaidyanathan M, Viswanathan R, Palaniandavar M, Balasubramanian T, Prabhaharan P, Muthiah TP. Inorg. Chem. 1998;37:6418. doi: 10.1021/ic971567s. [DOI] [PubMed] [Google Scholar]; (f) Zurita D, Gautier-Luneau I, Ménage S, Pierre J-L, Saint-Aman E. J. Biol. Inorg. Chem. 1997;2:46. [Google Scholar]
- (31).(a) Storr T, Sugai Y, Barta CA, Mikata Y, Adam MJ, Yano S, Orvig C. Inorg. Chem. 2005;44:2698. doi: 10.1021/ic048528i. [DOI] [PubMed] [Google Scholar]; (b) Melchior M, Rettig SJ, Liboiron BD, Thompson KH, Yuen VG, McNeill JH, Orvig C. Inorg. Chem. 2001;40:4686. doi: 10.1021/ic000984t. [DOI] [PubMed] [Google Scholar]; (c) Clevette DJ, Nelson WO, Nordin A, Orvig C, Sjoeberg S. Inorg. Chem. 1989;28:2079. [Google Scholar]
- (32).Lord SJ, Epstein NA, Paddock RL, Vogels CM, Hennigar TL, Zaworotko MJ, Taylor NJ, Driedzic WR, Broderick TL, Westcott SA. Can. J. Chem. 1999;77:1249. [Google Scholar]
- (33).(a) Kirin SI, Dübon P, Weyhermu1ller T, Bill E, Metzler-Nolte N. Inorg. Chem. 2005;44:5405. doi: 10.1021/ic048343b. [DOI] [PubMed] [Google Scholar]; (b) Kirin SI, Happel CM, Hrubanova S, Weyhermüller T, Klein C, Metzler-Nolte N. Dalton Trans. 2004:1201. doi: 10.1039/b313634e. [DOI] [PubMed] [Google Scholar]
- (34).Shakya R, Imbert C, Hratchian HP, Lanznaster M, Heeg MJ, McGarvey BR, Allard M, Schlegel HB, Verani CN. Dalton Trans. 2006:2517. doi: 10.1039/b514190g. [DOI] [PubMed] [Google Scholar]
- (35).Shakya R, Hindo SS, Wu L, Allard MM, Heeg MJ, Hratchian HP, McGarvey BR, Da Rocha SRP, Verani CN. Inorg. Chem. 2007;46:9808. doi: 10.1021/ic7011815. [DOI] [PubMed] [Google Scholar]
- (36).Lesh FD, Hindo SS, Heeg MJ, Allard MM, Jain P, Peng B, Hryhorczuk L, Verani CN. Eur. J. Inorg. Chem. 2009:345. [Google Scholar]
- (37).Mugesh G, Singh HB, Butcher RJ. Eur. J. Inorg. Chem. 2001:669. [Google Scholar]
- (38).Coggin DK, Gonzalez JA, Kook AM, Stanbury DM, Wilson LJ. Inorg. Chem. 1991;30:1115. [Google Scholar]
- (39).The notation <A1B2> indicates that A is trans to B, with A and B corresponding to the pyridine (Npy), amine (Nam), or phenolato (OphO-) domors. Subscripts 1 and 2 designate the first or the second ligand.
- (40).Imbert C, Hratchian HP, Lanznaster M, Heeg MJ, Hryhorczuk LM, McGarvey BR, Schlegel HB, Verani CN. Inorg. Chem. 2005;44:7414. doi: 10.1021/ic050658j. [DOI] [PubMed] [Google Scholar]
- (41).Lanznaster M, Neves A, Vencato I, Bortoluzzi AJ, Gallardo H, Machado SP, Assumpcao AMC. J. Braz. Chem. Soc. 2006;17:289. [Google Scholar]
- (42).Neves A, Vencato I, Verani CN. J. Braz. Chem. Soc. 1997;8:265. [Google Scholar]
- (43).dos Anjos A, Bortoluzzi AJ, Szpoganicz B, Caro MSB, Friedermann GR, Mangrich AS, Neves A. Inorg. Chim. Acta. 2005;358:3106. [Google Scholar]
- (44).Cotton FA, Daniels LM, Murillo CA, Quesada JF. Inorg. Chem. 1993;32:4861. [Google Scholar]
- (45).Rotthaus O, Labet V, Philouze C, Jarjayes O, Thomas F. Eur. J. Inorg. Chem. 2008:4215. [Google Scholar]
- (46).Milacic V, Chen D, Giovagnini L, Diez A, Fregona D, Dou QP. Toxicol. App. Pharm. 2008;231:24. doi: 10.1016/j.taap.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lin HK, Altuwaijri S, Lin WJ, Kan PY, Collins LL, Chang C. J. Biol. Chem. 2002;277:36570. doi: 10.1074/jbc.M204751200. [DOI] [PubMed] [Google Scholar]
- (48).Yang HJ, Murthy S, Sarkar FH, Sheng S, Reddy GPV, Dou QP. J. Cell. Physiol. 2008;217:569. doi: 10.1002/jcp.21565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).APEX II collection and processing programs are distributed by the manufacturer. Bruker AXS Inc.; Madison WI, USA: 2005. [Google Scholar]
- (50).Sheldrick GM. Acta Cryst, A64, 112-122. 2008 doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- (51).(a) Becke AD. J. Chem. Phys. 1993;98:5648. [Google Scholar]; (b) Becke AD. Phys. Rev. A. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]; (c) Lee T, Yang WT, Parr RG. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (d) Gordon MS. Chem. Phys. Lett. 1980;76:163. [Google Scholar]; (e) Hariharan PC, Pople JA. Mol. Phys. 1974;27:209. [Google Scholar]; (f) Hariharan PC, Pople JA. Theo. Chim. Acta. 1973;28:213. [Google Scholar]; (g) Hehre WR, Ditchfield R, Pople JA. J. Chem. Phys. 1972;56:225. [Google Scholar]; (h) Ditchfield R, Hehre WR, Pople JA. J. Chem. Phys. 1971;54:724. [Google Scholar]
- (52).Dolg M, Wedig U, Stoll H, Preuss H. J. Chem. Phys. 1987;86:866. [Google Scholar]
- (53).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN 03. Gaussian, Inc.; Wallingford, CT: 2003. [Google Scholar]
- (54).Schlegel HB. J. Comp. Chem. 1982;3:214. [Google Scholar]
- (55).Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Breast Cancer Res. 2005;7:R897. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.