

Abstract
Pyridine-2,6-dimethanethiolate and pyridine-2,6-dithiocarboxylate form sparingly soluble NiII pincer complexes formulated as [Ni(pdmt)]2 and [Ni(pdtc)]2, respectively, with two Ni-(μ2-S)-Ni bridges. In acetonitrile reaction systems, the latter undergoes the facile bridge cleavage reactions [Ni(pdtc)]2 + 2L0,− → 2[Ni(pdtc)L]0,− with an extensive set of nucleophiles to afford planar mononuclear products with L− = halide, CN, Me3SiO−, RS− and L0 = Et3P and a N-heterocyclic carbene. [Ni(pdmt)]2 is considerably less reactive toward bridge disruption. Cleavage products support several reactions of interest leading to other mononuclear species and to di- and trinuclear complexes. [Ni(pdtc)(OSiMe3)]1− deprotonates acetonitrile and acetone to form [Ni(pdtc)(CH2R)]1− (R = CN, COMe). Reaction of [Ni(pdtc)SEt]1− with FeII yields the thiolate-bridged dimer {[Ni(pdtc)]2(SEt)}1−. Refluxing an acetonitrile solution of [Ni(pdtc)SH]1− in air results in formation of trinuclear {[Ni(pdtc)]3S]2− containing the rare unsupported Ni3(μ3-S) bridge core. Reaction of [Ni(pdtc)CN]1− with [Fe(Me6tren)(OTf)]1+ forms the complex [Ni(pdtc)CNFe(Me6tren)]1+, the only example of a single Ni-C≡N-Fe bridge within a molecule. Structures of the various types of reaction products are presented. This work demonstrates the potential utility of bridge cleavage of polynuclear NiII thiolates, an extensive family of compounds, to produce mononuclear products.
Introduction
A characteristic of transition metal thiolate complexes is the tendency to oligomerize by formation of M-(μ2-S)-M bridges.1,2 This behavior is especially pronounced with Ni(II) thiolates, one manifestation of which is the large family of cyclic homoleptic species [Ni(SR)2]n with n ≤ 12.3,4 Oligomers are frequently useful precursors to complexes of lower nuclearity by bridge cleavage reactions, as depicted in Figure 1 for selected cases. In reaction 1, dinuclear species are converted to mononuclear products which retain attacking ligand L″, often a tertiary phosphine or cyanide.5–10 Reaction 2 with cyclic trinuclear reactants proceeds similarly to yield mononuclear complexes.11–14 Linear trinuclear reactants are cleaved to mononuclear or dinuclear products in reaction 3.15,16 Reaction 4 with tertiary phosphine is a specific example of reaction 1, in which dinuclear complex 1 derived from the pincer ligand pyridine-2,6-dimethanethiolate is cleaved to mononuclear 2 (R = Et, Ph, C6H11) in unspecified yield.5 Complex 1 is sparingly soluble in most common organic solvents and has not been obtained as diffraction-quality crystals. Its 1H NMR spectrum is consistent with C2h symmetry. The indicated structure is strictly analogous to those established crystallographically for bis(1,5-dithiolato-3-thiapentane)dinickel(II)17 and bis(2,2′-dithiolatodiphenylsulfide)dinickel(II),9 both of which undergo reaction 1 with cyanide. The reactions of 1 are early (and possibly the first) examples of cleavage of Ni-(μ2-S)-Ni bridges with an external nucleophile. These were followed by demonstration of cleavage by thiolate to afford other examples of 2 (R = Ph, Et) in 40–60% yield.6
Figure 1.

Schematic representation of Ni-(μ2-S)-Ni bridge cleavage reactions leading to products of lower nuclearity. In reaction 1, L = C, N, P, or S and in reaction 2, L/L′ = N, O, S. In reactions 1–3, L″ and L are generalized ligands.
In the course of examining bridge cleavage reactions of Ni(II) thiolates as a means of obtaining complexes of interest in the biomimetic chemistry of nickel in this laboratory,15,18,19 we have further explored the reactivity of 1. The complex does not react cleanly or to any significant extent with excess alkoxides, aryloxides, silyloxide, halides, azide, N-heterocyclic carbenes, and pyridine and other N-bases at ambient temperature. Consequently, we have examined the influence of ligand structure, based on the observations of Espinet et al.20 that the Pd(II) complex of pyridine-2,6-dithiocarboxylate exhibits reactivity with a wider variety of nucleophiles. As will be evident, [Ni(pdtc)]2 is amenable to cleavage with a broad range of ligands.
Experimental Section
Preparation of Compounds
Unless otherwise stated, all reactions and manipulations were performed under a dinitrogen atmosphere using either Schlenk techniques or an inert atmosphere box. Volume reduction and drying steps were performed in vacuo. Commercial grade chemicals were used without further purification. Acetonitrile, tetrahydrofuran, dichloromethane, ethanol, and diethylether were purified by an Innovative Technology or MBraun solvent purification system and degassed before use. Chloroform was freshly distilled from CaH2 under a dinitrogen atmosphere. Acetone was dried over 3 Å molecular sieves for six hours and degassed before use. NMR data do not include cation resonances. Representative compounds were analyzed (Kolbe Microanalytical Laboratory, Mülheim, Germany). Others were identified by spectroscopic and/or crystallographic characterization. Complexes are numerically designated as 1–21 and abbreviations are defined in the Chart.
Chart.
Abbreviations and Designation of Complexes
| [Ni(pdmt)]2 | 15 |
| [Ni(pdmt)L]0,1− | 25,6 |
| [M(pdmt)(SH)]1− | M = Ni 3, Pd 4 |
| [Ni(pdtc)]2 | 5 |
| {[Ni(pdtc)]3(DMF)3} | 6 |
| [Ni(pdtc)X]1− | X = Cl 7, Br 8, I 9 |
| OAc 10, OSiMe3 11 | |
| SH 12 SEt 13 | |
| {[Ni(pdtc)]2SEt}1− | 14 |
| {[Ni(pdtc)]3S}2− | 15 |
| [Ni(pdtc)X]1− | X = CH2CN 16, CH2COMe 17, CN 18, Pri2NHCMe2 19, PEt3 20 |
| [Ni(pdtc)CNFe(Me6tren)]1+ | 21 |
acac, acetylacetonate(1-); Me6tren, tris(N,N-dimethyl-2-aminoethyl)amine; pdtc, pyridine-2,6-dithiocarboxylate; pdmt, pyridine-2,6-dimethanethiolate; Pri2NHCMe2, 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene; OTf, triflate
(Bu4N)[Ni(pmdt)(SH)]
To a suspension of 100 mg (0.22 mmol) of 15 in 40 mL of acetonitrile was added 121 mg (0.44 mmol) of (Bu4N)(SH). The reaction mixture was stirred for 90 min and solvent was removed. The residue was washed thoroughly with ether to afford the product 185 mg (84%) of orange solid. 1H NMR (CD3CN): δ −3.56 (s, 1), 3.91 (s, 4), 6.95 (d, 2), 7.35 (t, 1). The Et4N+ salt was prepared in comparable yield by an analogous procedure.
(Bu4N)[Pd(pmdt)(SH)]
To a suspension of 100 mg (0.20 mmol) of [Pd(pmdt)]25 in 50 mL of acetonitrile was added 110 mg (0.40 mmol) of (Bu4N)(SH). The reaction mixture was stirred for 2 h and solvent was removed. The residue was washed thoroughly with ether to afford the product 181 mg (86%) of light orange solid. 1H NMR (CD3CN): δ −2.39 (s, 1), 4.27 (s, 4), 7.12 (d, 2), 7.46 (t, 1). The Et4N+ salt was prepared in comparable yield by an analogous procedure.
[Ni(pdtc)]2
To a green solution of [Ni(acac)2] (0.593 g, 2.31 mmol) in THF (70 mL) was added slowly a solution of H2pdtc21 (0.460 g, 2.31 mmol) in THF (10 mL). The mixture was stirred at room temperature for 20 h to deposit a solid, which was washed with THF (10 mL) and dried in vacuo to afford the product as a brown solid (0.473 g, 80%). The compound is insoluble in common organic solvents.
{[Ni(pdtc)]3(DMF)3}·DMF
Compound 5 (20 mg, 0.039 mmol) was treated with DMF (1 mL) to give a deep red solution. Diffusion of ether into this solution gave the product as dark red block-like crystals (17 mg, 61%). Anal. Calcd for C33H37N7Ni3O10S6: C, 37.39; H, 3.52; N, 9.25. Found: C, 37.20; H, 3.44; N, 9.15.
(Et4N)[Ni(pdtc)Cl]
To a suspension of compound 5 (138 mg, 0.27 mmol) in acetonitrile (1 mL) was added a solution of Et4NCl (89 mg, 0.54 mmol) in acetonitrile (3 mL). The mixture was stirred for 1 h and filtered to remove a small amount of brown precipitate. Diffusion of ether into the filtrate caused separation of the product as red block-like crystals (184 mg, 81%). Absorption spectrum (CH2Cl2): λmax(εM) 310 nm (5930), 345 (5900), 432 (4640), 517 (1180) nm. 1H NMR (CDCl3): δ 7.58 (d, 2), 7.92 (t, 1).
(Bu4N)[Ni(pdtc)Br]
The preceding method was followed with use of Bu4NBr (161 mg, 0.50 mmol) and gave the product as a red crystalline solid (228 mg, 79%). Absorption spectrum (CH2Cl2): λmax(εM) 313 (6340), 359 (4520), 438 (5120), 525 (1180) nm. 1H NMR (CDCl3): δ 7.61 (d, 2), 7.91 (t, 1).
(Bu4N)[Ni(pdtc)I]
Compound 5 (150 mg, 0.293 mmol) and Bu4NI (216 mg, 0.586 mmol) were mixed and stirred in acetone (18 mL) for 40 min. The red solution was filtered and solvent was removed to leave a red paste, which was dissolved in a small amount of THF and the solution was filtered. Ether was added to the filtrate to deposit a red oil. The oil was dissolved in acetone (3 mL) and ether was added to make a saturated solution, which was allowed to stand at room temperature for 30 min. The product was isolated as dark red crystals. The filtrate was treated with ether and the procedure repeated until no more crystals were obtained (combined yield 220 mg, 60%). 1H NMR (CDCl3): δ 7.68 (d, 2), 7.96 (t, 1).
(Bu4N)[Ni(pdtc)(OAc)]
Compound 5 (19 mg, 0.037 mmol) and Bu4N(OAc) (22 mg, 0.074 mmol) were mixed and stirred in acetonitrile (2 mL) for 1 h. The solution was filtered and the filtrate was diffused with ether to deposit the product as plate-like orange-red crystals (22 mg, 53%). Absorption spectrum (CH2Cl2): λmax(εM) 325 (3850), 426 (2320), 507 (710) nm. 1H NMR (CDCl3): δ 2.02 (s, 3), 7.50 (d, 2), 7.82 (t, 1).
(Et4N)[Ni(pdtc)(OSiMe3)]
To a solution of compound 7 (160 mg, 0.38 mmol) in dichloromethane was added a solution of NaOSiMe3 (43 mg, 0.38 mmol) in dichloromethane (2 mL). The mixture was stirred for 90 min and filtered to give a dark-red solution. Hexane (18 mL) was added to deposit a red paste which was taken up in dichloromethane (5 mL). The solution was saturated with ether and stored at −30°C overnight to deposit red needle crystals, which were collected by decantation. The process was repeated until no further product was obtained (combined yield 81 mg, 45%). Absorption spectrum (CH2Cl2): λmax(εM) 327 (5860), 444 (5140), 534 (2200) nm. 1H NMR (CDCl3): δ −0.03 (s, 9), 7.43 (d, 2), 7.79 (t, 1).
(Et4N)[Ni(pdtc)(SH)]
Compound 5 (143 mg, 0.28 mmol) and Et4N(SH) (91 mg, 0.56 mmol) were mixed and stirred in acetonitrile (7 mL) for 1 h. The solution was filtered and diffused with ether to deposit deep-red crystals (169 mg, 72%). Absorption spectrum (CH2Cl2): λmax(εM) 461 (9200), 548 (3940) nm. 1H NMR (CDCl3): δ −1.99 (s, 1), 7.69 (d, 2), 7.91 (t, 1); (CD3CN): δ −2.45 (s, 1), 7.63 (d, 2), 8.03 (t, 1). Anal. Calcd for C15H24N2NiO2S3: C, 42.97; H, 5.77; N, 6.68; S, 22.94. Found: C, 43.66; H, 5.54; N, 6.90; S, 22.59.
(Et4N)[Ni(pdtc)SEt]
Compound 5 (60 mg, 0.12 mmol) and Et4N(SEt) (45 mg, 0.23 mmol) were mixed and stirred in acetonitrile (2 mL) for 90 min. The solution was filtered and the filtrate was saturated with ether. The red solution was stored at −30°C overnight to deposit dark red crystals. This material was collected; ether was diffused into the filtrate to produce a second crop of crystals (combined yield 68 mg, 65%). Absorption spectrum (CH2Cl2): λmax(εM) 315 nm (10700), 441 (8950) nm. 1H NMR (CD3CN): δ 1.44 (t, 3), 1.99 (m, 2), 7.64 (d, 2), 8.05 (t, 1).
(Et4N){[Ni(pdtc)]2SEt}·CH2Cl2
(Et4N)[13] (38 mg, 0.085 mmol) and [Fe(Me6tren)OTf](OTf)22 (50 mg, 0.085 mmol) were mixed and stirred in acetonitrile (3 mL) for 2.5 h. The solution was filtered and ether (18 mL) added to deposit a red paste, which was washed with THF (2 × 5 mL) and dissolved in dichloromethane (6 mL) to give a red solution. Diffusion of ether into this solution caused separation of the product as a red crystalline solid (18 mg, 54%). Absorption spectrum (CH2Cl2): λmax(εM) 314 (15200), 441 (12800) nm. 1H NMR (CDCl3): δ 1.48 (t, 3), 2.12 (m, 2), 7.68 (d, 4), 7.94 (t, 2).
(Et4N)2{[Ni(pdtc)]3S})
Method A
To a suspension of compound 5 (115 mg, 0.23 mmol) in acetone (25 mL) was added dropwise a solution of 25% Et4NOH in methanol (0.40 ml, 0.58 mmol). The mixture was stirred for 2 h to give a red solution. The solution was filtered and the filtrate was reduced to a dark red oil, which was washed with chloroform (3 × 5 mL) and taken up in acetonitrile (5 mL). The solution was diffused with ether to deposit dark red block-like crystals (33 mg, 21%). Absorption spectrum (CH2Cl2): λmax(εM) 478 (14000) nm. 1H NMR (CD3CN): δ 7.57 (d, 6), 7.97 (t, 3). Anal. Calcd for C37H49N5Ni3O6S7: C, 41.91; H, 4.66; N, 6.60. Found: C, 40.49; H, 4.41; N, 6.37.
Method B
A solution of (Et4N)[12] (40 mg, 0.021 mmol) in acetonitrile (20 mL) was refluxed in air for 8 h. The solution volume was reduced to 5 mL. Diffusion of ether into this solution caused separation of dark red block-like crystals (26 mg, 78%) whose spectroscopic properties and cell parameters are identical to those of Method A.
(Et4N)[Ni(pdtc)(CH2CN)]
To a solution of (Et4N)[7] (41 mg, 0.097 mmol) in acetonitrile (2 mL) was added a solution of NaOSiMe3 (11 mg, 0.097 mmol) in acetonitrile (1 mL). The mixture was stirred for 2 h and filtered. Ether (18 mL) was added to the dark red filtrate to deposit a brown precipitate, which was dissolved in acetonitrile (2 mL) and diffused with ether to give product as dark red crystals (26 mg, 63%). Absorption spectrum (CH2Cl2): λmax(εM) 454 (7790) nm. 1H NMR (CDCl3): δ 0.67 (s, 2), 7.67 (d, 2), 7.89 (t, 1). Anal. Calcd for C17H25N3NiO2S2: C, 47.90; H, 5.91; N, 9.86. Found: C, 47.32; H, 5.38; N, 9.66.
(Et4N)[Ni(pdtc)(CH2COMe)]
The previous procedure was followed with acetone instead of acetonitrile. The reaction mixture was stirred for 50 min. The product was isolated as a dark red microcrystalline solid (36 mg, 84%). Absorption spectrum (CH2Cl2): λmax(εM) 467 (8900) nm. 1H NMR (CDCl3): δ 1.70 (s, 2), 2.11 (s, 3), 7.64 (d, 2), 7.86 (t, 1).
(Et4N)[Ni(pdtc)(CN)]
Compound 5 (100 mg, 0.20 mmol) and Et4N(CN) (61 mg, 0.39 mmol) were mixed and stirred in acetonitrile (3 mL) for 2 h. The red solution was filtered; diffusion of ether into the filtrate yielded the product as red block-like crystals (121 mg, 75%). IR (KBr): νCN 2115 cm−1. Absorption spectrum (CH2Cl2): λmax(εM) 402 (3700), 421 (3600), 544 (470) nm. 1H NMR (CD3CN): δ 7.65 (d, 2), 8.06 (t, 1). Anal. Calcd for C16H23N3NiO2S2: C, 46.62; H, 5.62; N, 10.19. Found: C, 46.63; H, 5.38; N, 10.22. E½ 1−,2− = −1.36 V (acetonitrile).
[Ni(pdtc)(Pri2NHCMe2)]·0.25THF
Compound 5 (36 mg, 0.070 mmol) and Pri 2NHCMe2 23 (25 mg, 0.140 mmol) were mixed and stirred in THF (6 mL) for 65 h. The solution was filtered and the filtrate was saturated with hexane to give a red solution with a small amount of red oil, which was removed by filtration. The filtrate was stored at −30°C for 3 d to deposit the product as red crystals (18 mg, 28%). Absorption spectrum (CH2Cl2): λmax(εM) 310 nm (3740), 404 (3550), 423 (3550), 548 (500) nm. 1H NMR (CDCl3): δ 1.65 (d, 12), 2.17 (s, 6), 6.16 (m, 2), 7.79 (d, 2), 8.00 (t, 1). E½ 0,1− = −1.37 V (acetonitrile).
[Ni(pdtc)(PEt3)]
To a suspension of compound 5 (42 mg, 0.082 mmol) in dichloromethane (2 mL) was added a solution of PEt3 (19 mg, 0.164 mmol) in dichloromethane (1 mL). The mixture was stirred for 30 min to give a red solution. The solution was filtered and the filtrate was layered with hexane to deposit red block-like crystals (44 mg, 72%). Absorption spectrum (CH2Cl2): λmax(εM) 398 (5100), 421 (4700), 553 (700) nm. 1H NMR (CDCl3): δ 1.28 (m, 9), 1.76 (m, 6), 7.83 (d, 2), 8.04 (t, 1). 31P NMR (CDCl3): δ 26.03. E½ 0,1− = −1.21 V (acetonitrile).
[Ni(pdtc)CNFe(Me6tren)](OTf)·CH2Cl2
To a solution of (Et4N)[18] (40 mg, 0.097 mmol) in dichloromethane (3 mL) was added dropwise a solution of [Fe(Me6-tren)(OTf)](OTf) (57 mg, 0.097 mmol) in dichloromethane (2 mL). The mixture was stirred at room temperature for 3 h and filtered to give a deep-red solution. Addition of ether (15 mL) led to a red precipitate, which was washed with chloroform (5 mL) and dissolved in dichloromethane (10 mL). The solution was filtered and the filtrate reduced in volume to give the product as a red crystalline solid (24 mg, 31%). IR (KBr): νCN 2140 cm−1. Absorption spectrum (CH2Cl2): λmax(εM) 306 nm (17400), 382 (11500), 402 (10300), 522 (1270) nm.
X-ray Structure Determinations
The structures of the eleven compounds in Tables 1 and 2 were determined; for brevity compounds are referred to by their anion or cation number. Compounds were crystallized as follows: 6, DMF/ether; 8, acetone/ether; 11, 14, dichloromethane/ether; 3, 4, 12, 16, acetonitrile/ether; 15, dichloromethane/chloroform; 19, THF/hexane; 21, dichloromethane. Diffraction data were collected on a Bruker CCD area detector diffractometer equipped with an Oxford 700 low temperature apparatus. Single crystals were coated with Paratone-N oil and mounted on a nylon loop. Structures were solved by direct methods using the SHELX program package.24 Solvate molecules in 14, 15, 19, and 21 were disordered and were modeled over two positions with occupancies varying from 0.15 to 0.85. Two of the six independent coordinated DMF molecules in 6 were modeled as disordered over two positions. In 11, the methyl carbon atoms of the -OSiMe3 group were disordered over three sites with occupancies of 0.5, 0.3, and 0.2. In 14, three of the four Ni(pdtc) units were disordered over two positions. In 12, the hydrogen atom of the -SH group was located from a difference Fourier map and refined isotopically. Crystal data and refinement details are given in Tables 1 and 2.25
Table 1.
| (Et4N)[3] | (Et4N)[4] | 6·DMF | (Bu4N)[8] | (Et4N)[11] | (Et4N)[12] | |
|---|---|---|---|---|---|---|
| formula | C15H28N2NiS3 | C15H28N2PdS3 | C33H37N7O10S6Ni3 | C23H39N2O2S2BrNi | C18H32N2O3SiS2Ni | C15H24N2O2S3Ni |
| M | 391.28 | 438.97 | 1060.19 | 578.30 | 475.38 | 419.25 |
| crystal system | monoclinic | monoclinic | monoclinic | monoclinic | triclinic | monoclinic |
| space group | Cc | Cc | P21/c | C2/c | P1̄ | P21/n |
| a, Å | 11.799(1) | 11.925(2) | 46.973(4) | 13.552(1) | 8.907(1) | 6.966(1) |
| b, Å | 12.874(1) | 12.822(2) | 12.405(1) | 22.893(2) | 8.977(1) | 21.928(2) |
| c, Å | 12.475(1) | 12.559(2) | 15.338(1) | 9.296(1) | 15.290(1) | 12.270(1) |
| α, deg | 90 | 90 | 90 | 90 | 86.703(4) | 90 |
| β, deg | 92.029(2) | 92.653(2) | 96.912(1) | 111.464(1) | 76.782(4) | 100.084(1) |
| γ, deg | 90 | 90 | 90 | 90 | 85.129(4) | 90 |
| V, Å3 | 1893.6(3) | 1918.3(5) | 8872.1(1) | 2684.0(3) | 1185.0(1) | 1845.4(3) |
| Z | 4 | 4 | 8 | 4 | 2 | 4 |
| μ, mm−1 | 1.351 | 1.290 | 1.602 | 2.389 | 1.065 | 1.400 |
| unique data | 2452 | 2758 | 20365 | 3209 | 4181 | 4400 |
| observed data [F≥4σ (F)] | 2260 | 2738 | 14330 | 2925 | 2893 | 3735 |
| refined parameters | 198 | 198 | 1171 | 145 | 311 | 216 |
| R1c, wR2d [F≥4σ(F)] | 0.0279, 0.0481 | 0.0166, 0.0413 | 0.0790, 0.2053 | 0.0306, 0.0790 | 0.0449, 0.1102 | 0.0303, 0.0723 |
| R1, wR2 (all data) | 0.0313, 0.0498 | 0.0167, 0.0414 | 0.1067, 0.2231 | 0.0335, 0.0814 | 0.0724, 0.1264 | 0.0377, 0.0772 |
MoKα radiation (λ = 0.71013 Å), 193(2) K.
Salts designated by anion number.
R1 = Σ||Fo| − | Fc||/Σ|Fo|.
wR2 = {Σ[w(Fo2 − Fc2)2/(Fo2)2]}½.
Table 2.
Crystallographic Dataa for compounds (Et4N)[14]·CH2Cl2, 15·CH2Cl2, 16, 19·0.25THF, and [21](OTf) ·CH2Cl2.b
| (Et4N)[14] ·CH2Cl2 | 15·CH2Cl2 | 16 | 19·0.25THF | [21](OTf) ·CH2Cl2 | |
|---|---|---|---|---|---|
| formula | C25H33N3O4S5Cl2Ni2 | C38H51N5O6S7Cl2Ni3 | C17H25N3O2S2Ni | C19H25N3O2.25S2Ni | C22H35N6O5F3S3Cl2FeNi |
| M | 788.16 | 1145.29 | 426.23 | 454.25 | 802.20 |
| crystal system | triclinic | triclinic | monoclinic | monoclinic | triclinic |
| space group | P1̄ | P1̄ | P21/c | P21/c | P1̄ |
| a, Å | 9.784(1) | 11.409(1) | 11.812(2) | 23.468(1) | 9.358(1) |
| b, Å | 13.494(2) | 12.371(1) | 13.794(2) | 26.356(1) | 12.801(1) |
| c, Å | 13.750(2) | 17.786(2) | 13.019(2) | 14.037(1) | 14.588(1) |
| α, deg | 77.248(2) | 93.198(2) | 90 | 90 | 94.966(2) |
| β, deg | 70.317(2) | 99.185(2) | 111.730(2) | 99.714(1) | 94.132(2) |
| γ, deg | 79.757(2) | 104.338(2) | 90 | 90 | 97.477(2) |
| V, Å3 | 1656.5(4) | 2389.0(4) | 1970.5(5) | 8557.9(2) | 1720.0(3) |
| Z | 2 | 2 | 4 | 16 | 2 |
| μ, mm−1 | 1.648 | 1.637 | 1.212 | 1.122 | 1.363 |
| unique data | 7737 | 11686 | 5274 | 19652 | 6218 |
| observed data [F≥4σ(F)] | 6286 | 8547 | 3815 | 15668 | 4702 |
| refined parameters | 402 | 576 | 230 | 1330 | 413 |
| R1c, wR2d [F≥4σ(F)] | 0.0427, 0.1076 | 0.0517, 0.1446 | 0.0438, 0.0938 | 0.0547, 0.1494 | 0.0564, 0.1405 |
| R1, wR2 (all data) | 0.0541, 0.1185 | 0.0722, 0.1583 | 0.0687, 0.1062 | 0.0694, 0.1600 | 0.0771, 0.1598 |
MoKα radiation (λ = 0.71013 Å), 193(2) K.
Salts designated by anion number.
R1 = Σ||Fo| − | Fc||/|Fo|.
wR2 = {Σ [w(Fo2 − Fo2)2/(Fo2)2]}½.
Other Physical Measurements
1H NMR spectra were recorded on a Varian AM-400 instrument. Infrared spectra were recorded with a Nicolet 5PC FT-IR spectrometer. Absorption spectrum spectra were recorded on a Varian Cary 50 Bio spectrophotometer. Cyclic voltammetric measurements were carried out on a BASi Epsilon-EC Bioanalytical System with glassy-carbon, Pt wire and saturated calomel functioning as the working, counter and reference electrodes, respectively. Cyclic voltammetry was carried out on 10−3 M solutions of the complexes at 298 K under a dinitrogen atmosphere with 0.1 M (Bu4N)(PF6) supporting electrolyte. All potentials are quoted vs. the SCE.
Results and Discussion
In an attempt to provide an X-ray structure proof of complex 5, the starting material for the bridge cleavage reactions set out in Figure 2, the complex was treated with DMF to give a deep red solution. Addition of ether resulted in formation of dark red crystals. The isolated product is not a simple dimer but rather the trinuclear species 6 whose structure is shown in Figure 3. The structure consists of two planar Ni(pdtc) units of normal dimensions (vide infra) bridged through Ni-(μ2-S)-C units to a central octahedral Ni(pdtc)(DMF)3 portion. Utilization of thioester sulfur atoms in the bridges leads to a relatively weak ligand field at the central nickel atom and the attendant tendency to adopt six-coordination in the presence a coordinating solvent. Throughout we assume for the pdtc complex 5 the same structure as has been proposed for pdmt complex 1.5,6
Figure 2.

Scheme summarizing the bridge cleavage reactions of [Ni(pdtc)]2 (5) to yield mononuclear (7–13, 16–20) and trinuclear (6 and 15) products. Dinuclear 14 and 21 are derived from mononuclear products.
Figure 3.

Structures of dinuclear and trinuclear complexes and selected metric data (Å; deg). {[Ni(pdtc)]3(DMF)3} (6, upper): Ni(1,3)-N 1.902[4], Ni(1,3)-S 2.17[1], Ni(1,3)-S(3,4) 2.195[6], Ni(2)-N 1.977(4), Ni(2)-O(3,4) 2.02[1], Ni(2)-O(7–9) 2.03[1] Å; 8 angles at Ni(1,3) 88.3(1)-92.6(1)°, N-Ni(2)-O(3,4) 77.5[1]°, 10 angles at Ni(2) 85.0(2)-96.3(2)°, Ni(1)-S(3)-C 101.5(2)°, Ni(3)-S(4)-C 108.9(2)°. {[Ni(pdtc)]2SEt}1− (14, center): Ni-N 1.904[2], Ni-S(1–4) 2.18[1], Ni-S(5) 2.190[5], Ni(1)-Ni(2) 3.35 Å; 8 angles at Ni(1,2) 88.05(7)-94.17(3)°, Ni(1)-S(5)-Ni(2) 99.97(3)°. {[Ni(pdtc)]3S}2− (15, lower): Ni-N 1.906[1], Ni-S(1–6) 2.182[8], Ni-S(7) 2.198[6], Ni···Ni 3.51–3.61 Å; 12 angles at Ni(1–3) 88.0(1)-95.33(4)°, Ni-S(7)-Ni 105.73(4)-110.2(4)°.
Comparative Structures of pdmt and pdtc Complexes
To provide comparative structures of the two types of complexes as bridge cleavage products, both were reacted with an arbitrary common ligand, hydrosulfide. Products 3 (84%) and 12 (72%) were isolated as Et4N+ salts; PdII complex 4 (86%) was obtained in a similar manner. These complexes are readily recognized by the upfield chemical shifts of the -SH ligand, −3.56 (3), −2.39 (4), and −2.45 ppm (12) in acetonitrile. Similar upfield shifts occur for diamagnetic NiII and PtII hydrosulfide complexes.26,27 The +2 to −4 ppm range is considered characteristic of coordinated hydrosulfide.28 As seen in Figure 4, complexes 3 and 12 are isostructural and nearly isometric. There are no significant differences in the coordination spheres. In particular, the mean Ni-S(C) distances are indistinguishable (2.186[1] vs. 2.173[1] Å); by implication Ni-(μ2-S) bonds lengths in the putative dimers are likely to nearly identical. There is no molecular structural basis for a large difference in reactivity of 1 and 5.
Figure 4.

Structures and metric comparisons of [Ni(pmdt)(SH)]1− (3) and [Ni(pdtc)(SH)]1− (12). Metric data (Å; deg) for 3/12: Ni-S(1,2) 2.165(1), 2.202(1)/2.172(1), 2.181(1); Ni-N 1.904(3)/1.893(1); Ni-S(3) 2.192(1)/2.166(1) Å; N(1)-Ni-S(1,2) 88.73(9), 88.28(9)/88.71(4), 88.54(5); S(3)-Ni-S(1,2) 92.08(4), 91.16(4)/92.95(1), 89.93(2); S(1)-Ni-S(2) 173.11(4)/176.25(2); N(1)-Ni-S(3) 177.64(9)/176.21(4)°.
Bridge Cleavage and Related Reactions
These reactions proceed under the stoiochiometry of reaction 5 in 2–3 hrs at room temperature. In addition to 12 (Figure 4), the
| (5) |
structures of 8 and 19 demonstrating mononuclear cleavage are given in Figure 5.29 Isolated product yields are 45–81%; the carbene Pri2NHCMe2 gave 19 in 28% yield. These results serve to demonstrate the generality of bridge cleavage of 5. Reaction with excess Et4NOH (ca. 3–6 equiv) in acetone also resulted in cleavage. In the 6 equiv system, the only identifiable product is [Ni(pdtc)2]2−, a redox-active octahedral complex previously reported.30
Figure 5.

Structures of mononuclear complexes obtained by bridge cleavage, ligand substitution, or deprotonation and selected metric data (Å; deg). [Ni(pdtc)Br]1− (8, imposed mirror symmetry): Ni-S 2.176(1), Ni-N 1.889(3), Ni-Br 2.311(6); N-Ni-S 88.94(2), S-Ni-Br 91.06(2). [Ni(pdtc)(Pri2NHCMe2)] (19, one of four independent molecules of nearly identical dimensions, Ni-S 2.170[8], Ni-N 1.897(2), Ni-C 1.879(3); 4 angles at Ni 89.00(8)-91.3(1). [Ni(pdtc)(OSiMe3)]1− (11): Ni-S 2.177[5], Ni-N 1.874(3), Ni-O 1.831(3); 4 angles at Ni 88.6(1)-92.0(1), Ni-O-Si 134.6(2). [Ni(pdtc)(CH2CN)]1− (16): Ni-S 2.170[5], Ni-N 1.901(2), Ni-C 1.952(2); 4 angles at Ni 88.51(6)-91.94(9), Ni-C-C 108.8(2).
Several additional reactions are of interest. The failure to isolate a terminal hydroxide complex, one of main goals of this work, led to substitution reaction 6 affording silyloxide complex 11 (45%). This complex supports proton transfer reaction 7 to form Ni-C bonds in 16 (R = CN, 63%) and 17 (R = COMe, 84%). Both of these reactions appear capable of extension to other ligands and acidic carbon substrates.
| (6) |
| (7) |
Polynuclear Complexes
In certain of the foregoing bridge cleavage reactions, a dark red minority product was observed. It was subsequently found that this product could be obtained by the reaction of 5 as a suspension in acetone with ca. 3 equiv of Et4NOH or in much better yield (78%) by refluxing an acetonitrile solution of 12 in air. The red product was identified by a structure determination as trinuclear 15 (Figure 3), in which three Ni(pdtc) units are bridged by a μ3-S atom that derived from the pdtc ligand by base hydrolysis or most likely from hydrosulfide in the case of 12. The three Ni(pdtc) units are arrayed around the bridging atom such that the dihedral angles between coordination planes are 84.2–97.9° and Ni···Ni separations are 3.51–3.61 Å. The Ni3(μ3-S) unit is pyramidal; the sulfur atom is positioned 0.79 Å out of the Ni3 plane. This bridged core structure, in which three nickel atoms are connected through only a single sulfur atom (no other sulfur bridge interactions) is extremely rare. It has been encountered previously only in [Cp2Ni3S(PEt3)2] in which, however, there are direct Ni-Ni interactions supporting the structure.31 Among trinuclear nickel-sulfide complexes, the core motifs Ni3(μ3-S)2 and Ni3(μ3-S)(μ2-SR)3 are relatively common.32
In an attempt to form bridged Ni-Fe complexes, thiolate complex 13 was reacted with one equiv of [Fe(Me6tren)(OTf)]1+ in acetonitrile. Workup resulted in isolation of the red dinuclear complex 14 (54%) containing the Ni-S(Et)-Ni bridge unit (Figure 3) with a bridge angle of 99.97° and a Ni···Ni distance of 3.35 Å. Evidently, the FeII reactant removed the terminal thiolate ligand from 13, leading to product formation. Complexes 13 and 14 are distinguishable in acetonitrile solution by small differences in chemical shifts. Reaction of the same iron complex with cyanide complex 18 in dichloromethane proceeded with formation of the bridged complex 21, whose structure is set out in Figure 6. Positions of the bridge carbon and nitrogen atoms were not determined from the X-ray structure. The reasonable assumptions were made that the Ni-C bond was retained at the low-spin NiII site and that a Fe-N bond was formed at labile high-spin FeII.22 The Ni-C≡N bridge portion is essentially linear and the Fe-N≡C part somewhat bent (166°). The bridge links the planar Ni(pdtc) and trigonal bipyramidal [Fe(Me6tren)]2+ components in an entity that is the only molecular example of a single Ni-C≡N-Fe bridge. Of the 15 species assigned to this atom sequence in the Cambridge Structural Database,31 nearly all are derived from [Ni(CN)4]2− and all are polymeric. Similarly, ca. 100 compounds are reported with the Ni-N≡C-Fe bridge, many of which are based on the [Fe(CN)6]4− and all of which contain more than one bridge, often at the same FeII atom.
Figure 6.
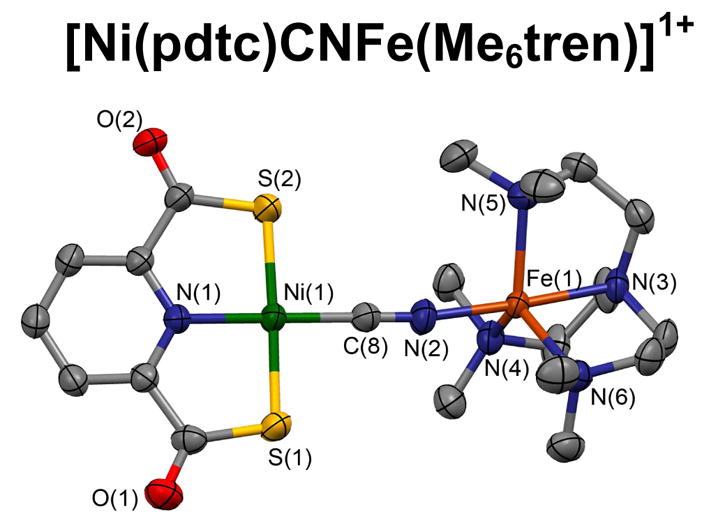
Structure and metric data (Å, deg) for [Ni(pdtc)CNFe(Me6tren)]1+ (21): Ni-S 2.168[6], Ni-N 1.890(4), Ni-C 1.834(5), C(8)-N(2) 1.148(6), Fe-N(2) 2.042(4), Fe-N(3–6) 2.16(2); 4 angles at Ni 89.0(1)-91.4(2), Ni-C-N 178.3(5), Fe-N(2)-C(8) 166.2(4), N(2)-Fe-N(3) 177.2(2), N(3)-Fe-N(4–6) 81.7(2)-82.3(2), N(4–6)-Fe-N(4–6) 115.5(2)-121.1(2).
This work provides an extensive demonstration of Ni-(μ2-S)-Ni bridge cleavage in a constant structure with a broad range of nucleophiles, and emphasizes the utility of this reaction type in synthesis. Bridge cleavage has also been applied to [Pd(pdtc)]2 and its derivatives,20,33 but not with as broad a range of ligand types. The method does not require preisolation of a dinuclear complex. It has been found that reaction of equimolar Ni(acac)2, and substituted pyridine-2,6-dithiocarboxylic acids, followed by addition of substituted pyridine and isonitrile ligands proceeds in situ to give the desired mononuclear products.34 Given the heterogeneous reaction systems in acetonitrile, the cause of the increased reactivity of pdtc complex 5 over pmdt complex 1 may be due to solid state structural differences. The choice of acetonitrile facilitated product isolation. Possibly the more nucleophilic sulfur atoms in 1 promote Ni-S intermolecular interactions and diminish reactivity. As noted at the outset, the crystal structures of these compounds have not yet been determined.
Supplementary Material
Acknowledgments
This research was supported by NIH Grant GM 28856.
Footnotes
Supporting Information Available. Crystallographic data for 11 compounds in Tables 1 and 2. This material is available free of charge via the Internet at http://pubs/acs.org.
References
- 1.Dance IG. Polyhedron. 1986;5:1037–1104. [Google Scholar]
- 2.Blower PJ, Dilworth JR. Coord Chem Rev. 1987;76:121–185. [Google Scholar]
- 3.Ivanov SA, Kozee MA, Merrill WA, Agarwal S, Dahl LF. J Chem Soc, Dalton Trans. 2002:4105–4114. [Google Scholar]
- 4.Zhang C, Takada S, Kölzer M, Matsumoto T, Tatsumi K. Angew Chem Int Ed. 2006;45:3768–3772. doi: 10.1002/anie.200600319. [DOI] [PubMed] [Google Scholar]
- 5.op den Brouw PM, van der Linden JGM. Inorg Nucl Chem Lett. 1977;13:149–151. [Google Scholar]
- 6.Krüger HJ, Holm RH. Inorg Chem. 1989;28:1148–1155. [Google Scholar]
- 7.Sellman D, Prechtel W, Knoch F, Moll M. Inorg Chem. 1993;32:538–546. [Google Scholar]
- 8.Mizra SA, Pressler MA, Kumar M, Day RO, Maroney MJ. Inorg Chem. 1993;32:977–987. [Google Scholar]
- 9.Sellman D, Häussinger D, Heinemann FW. Eur J Inorg Chem. 1999:1715–1725. [Google Scholar]
- 10.Cerruda E, Falvello LR, Hursthouse MB, Laguna M, Luquín A, Pozo-Gonzalo C. Eur J Inorg Chem. 2002:826–833. [Google Scholar]
- 11.Baidya N, Olmstead MM, Mascharak PK. Inorg Chem. 1989;28:3426–3432. [Google Scholar]
- 12.Sellman D, Haeussinger D, Knoch F, Moll MJ. Am Chem Soc. 1996;118:5368–5374. [Google Scholar]
- 13.Sellman D, Geipel F, Heinemann FW. Eur J Inorg Chem. 2000:271–279. [Google Scholar]
- 14.Sellman D, Prakash R, Geipel F, Heinemann FW. Eur J Inorg Chem. 2002:2138–2146. [Google Scholar]
- 15.Rao PV, Bhaduri S, Jiang J, Holm RH. Inorg Chem. 2004;43:5833–5849. doi: 10.1021/ic040055s. [DOI] [PubMed] [Google Scholar]
- 16.Ito M, Kotera M, Song Y, Matsumoto T, Tatsumi K. Inorg Chem. 2009;48:1250–1256. doi: 10.1021/ic801967e. [DOI] [PubMed] [Google Scholar]
- 17.Barclay GA, McPartlin EM, Stephenson NC. Acta Crystallogr. 1969;B25:1262–1273. [Google Scholar]
- 18.Rao PV, Bhaduri S, Jiang J, Hong D, Holm RH. J Am Chem Soc. 2005;127:1933–1945. doi: 10.1021/ja040222n. [DOI] [PubMed] [Google Scholar]
- 19.Sun J, Tessier C, Holm RH. Inorg Chem. 2007;46:2691–2699. doi: 10.1021/ic062362z. [DOI] [PubMed] [Google Scholar]
- 20.Espinet P, Lorenzo C, Miguel JA, Bois C, Jeannin Y. Inorg Chem. 1994;33:2052–2055. [Google Scholar]
- 21.Hildebrand U, Ockels W, Lex J, Budzikiewicz H. Phosphorus and Sulfur. 1983;16:361–364. [Google Scholar]
- 22.Britovsek GJP, England J, White AJP. Inorg Chem. 2005;44:8125–8134. doi: 10.1021/ic0509229. [DOI] [PubMed] [Google Scholar]
- 23.Kuhn N, Kratz T. Synthesis. 1993:561–562. [Google Scholar]
- 24.Sheldrick GM. Acta Crystallogr. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 25.See paragraph at the end of this article for Supporting Information available.
- 26.Vicic DA, Jones WD. J Am Chem Soc. 1999;121:4070–4071. [Google Scholar]
- 27.Morton MS, Lachicotte RJ, Vicic DA, Jones WD. Organometallics. 1999;18:227–234. [Google Scholar]
- 28.Kuwata S, Hidai M. Coord Chem Rev. 2001;213:211–305. [Google Scholar]
- 29.Because the chelate ring dimensions of other complexes reported here are practically identical with those of 12, metric data are reported as appropriate in terms of mean distances and ranges of values of angles (Figures 3, 5, and 6).
- 30.Krüger HJ, Holm RH. J Am Chem Soc. 1990;112:2955–2963. [Google Scholar]
- 31.Denninger U, Schneider JJ, Wilke G, Goddard R, Krüger C. Inorg Chim Acta. 1993;213:129–140. [Google Scholar]
- 32.Allen FH. Acta Crystallogr. 2002;B58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 33.Espinet P, Garcia-Orodea E, Miguel JA. Inorg Chem. 2009;39:3645–3651. doi: 10.1021/ic0001457. [DOI] [PubMed] [Google Scholar]
- 34.Espinet P, Garcia-Orodea E, Miguel JA. Chem Mater. 2004;16:551–558. doi: 10.1021/ic0001457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.