

Abstract
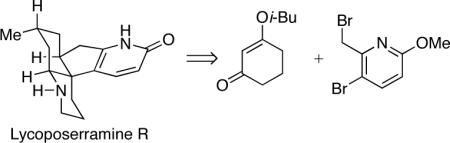
A methoxypyridine serves as a masked pyridone in a concise synthesis of the Lycopodium alkaloid lycoposerramine R, which has been prepared for the first time. The key step of the synthesis is the use of an Eschenmoser Claisen rearrangement to forge a key quaternary carbon center.
The Lycopodium alkaloid family boasts over 200 members, which possess an array of architecturally complex frameworks. The construction of these molecules has presented synthetic challenges that have inspired highly innovative strategic and tactical solutions.1 The emergence of the Lycopodium alkaloid huperzine A as a potential treatment for Alzheimer's disease has further heightened synthetic interest in this family of natural products.2 Through detailed studies that began in 1942 with the work of Manske, and later, Wiesner, MacLean, Conroy, McMaster and more recently Kobayashi and Takayama, the biosynthetic connections between many of these alkaloids continues to be uncovered.3 As a result, synthetic strategies to the Lycopodium alkaloids that exploit their structural connections are beginning to offer effective and concise avenues to a wide array of these natural products.
As a part of a program to exploit methoxypyridines in the synthesis of various alkaloids, we have reported the total syntheses of the Lycopodium alkaloid lyconadin A4 and the Galbulimima alkaloid GB 13.5 The methoxypyridine group is uniquely effective as a masked pyridone because the methoxy group significantly mitigates the basicity of the pyridine nitrogen via an inductive electron-withdrawing effect.6 This obviates the need for reversed-phase chromatographic purification, which is often associated with the synthesis of related alkaloids. Building upon our earlier studies, we have embarked on the syntheses of the Lycopodium alkaloids lycopladine A (1),7 lycoposerramine R (2)8 and lannotinidine B (3)9 with the intention of accessing all three natural products from a common intermediate.10
Key to our unified synthetic strategy is the use of a common methoxypyridine intermediate (see 4, Scheme 1). The methoxy group of the methoxypyridine moiety in 4 may be removed enroute to 1,11 demethylated to unveil the pyridone in 2 or the methoxypyridine may be unraveled at a late stage to provide 3. This communication describes our initial studies toward this overall goal, which has culminated in a concise first total synthesis of lycoposerramine R.
Scheme 1.
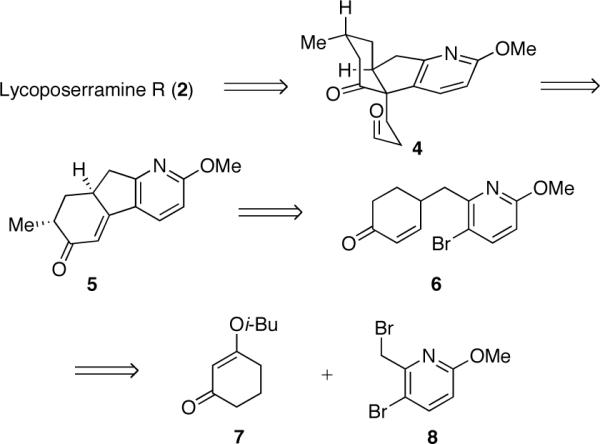
Retrosynthetic analysis of 2
Retrosynthetically, we envisioned the tetracyclic framework of 2 (Scheme 1) arising from a late-stage reductive amination of ketoaldehyde 4, which is closely related to 1. Tricycle 4 could in turn derive from enone 5 by exploiting a stereospecific pericyclic rearrangement to forge the all-carbon quaternary center. Enone 5 presented several opportunities for the diastereocontrolled intallation of the potentially challenging quaternary center (e.g., oxy-Cope, Claisen or 2,3-Wittig rearrangements). In turn, tricycle 5 could be obtained from 6 via an intramolecular Heck reaction. The potential Heck precursor 6 could arise from a union of readily available vinylogous ester 7 and dibromide 812 via a Stork-Danheiser sequence.13
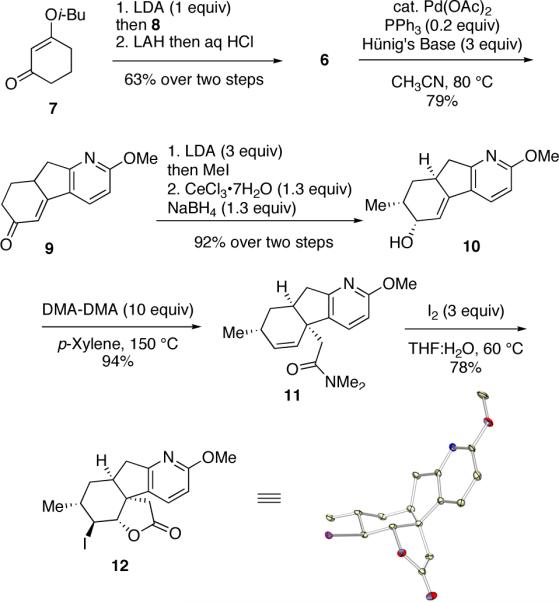
Our synthesis commenced with the coupling of the enolate of vinylogous ester 7 and picolinyl bromide 8. This was followed by reduction of the vinylogous ester of the adduct and acidic workup to give 6 in 63% yield over the two steps. Intramolecular Heck reaction of 6 proceeded without event to afford tricyclic enone 9 in 79% yield. The α-methylation of enone 9 and subsequent Luche reduction14 proceeded with excellent diastereocontrol to give 10 in 92% yield. At this stage, several variants of the Claisen rearrangement were explored. Ultimately, the Eschenmoser Claisen rearrangement15 utilizing the dimethyl acetal of N, N-dimethylacetamide (DMA-DMA) proved to be most effective, affording 11 in 94% yield. Iodolactonization of 11 yielded spiro-fused lactone 12 in 78% yield.
The structure and relative stereochemistry of iodolactone 12 was confirmed by X-ray analysis of a single crystal (see ORTEP in Scheme 2). With a robust route to tetracycle 12 secured, we next explored the installation of the piperidine ring to complete the synthesis of 2. LAH reduction of the lactone also effected cleavage of the C-I bond leading to diol 13 (Scheme 3) in 72% yield. Oxidation of 13 under Swern conditions proceeded without event to yield ketoaldehyde 14 in quantitative yield. Two routes for the selective homologation of the aldehyde group of 14 to afford 4 were explored. The first approach entailed selective Witttig reaction of the aldehyde group using the reagent derived from methoxymethylene phosphonium chloride, followed by hydrolysis of the resulting methyl enol ether to afford 4. Although this reaction worked well on small scale, the yields proved to be irreproducible on larger scale. After investigating several alternative homologation strategies, it was found that 4 could be obtained in consistently high yields from 14 using the Ohira-Bestmann reaction to install a triple bond (see 15) followed by anti-Markovnikov hydration using the procedure introduced by Grotjahn.16
Scheme 2.
Synthesis of lactone 12
Scheme 3.

Synthesis of ketoaldehyde 4
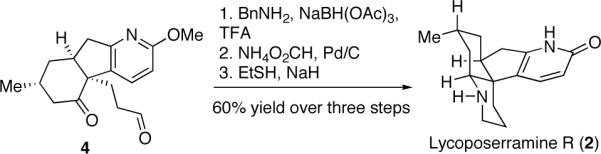
At this stage, several direct reductive amination possibilities to forge the piperidine ring in 2 were investigated. However, these were found to be low yielding. Ultimately, the piperidine moiety was best installed using benzylamine (Scheme 5) followed by hydrogenolytic cleavage of the benzyl group. A concluding methyl ether cleavage (NaSEt) gave lycoposerramine R (2). Synthetic lycoposerramine R gave spectral data (1H and 13C NMR, IR, MS) fully consistent with that reported by Takayama et al. following its isolation.17
Scheme 5.
Completion of the synthesis of 2
The synthesis of 2 proceeds in a total of 13 steps (12% overall yield) from 7 and 8. Key to the completion of the synthesis is the use of a methoxypyridine as a masked pyridone as well as an Eschenmoser Claisen reaction to install a key quaternary carbon center. Our application of these synthetic studies to the enantioselective synthesis of 2 as well as the related fawcettimine-type alkaloid 3 will be reported in due course.
Supplementary Material
Figure 1.
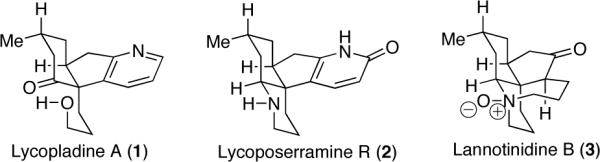
Selected Lycopodium alkaloids
Scheme 4.
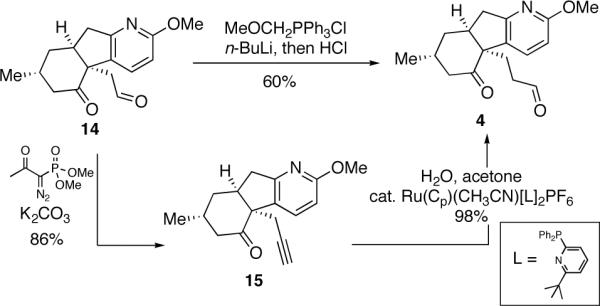
Homologation of ketoaldehyde 14
Acknowledgment
The authors are grateful to the NIGMS (RO1 GM086374-01), Amgen, Eli Lilly, Johnson and Johnson, and AstraZeneca for financial support. Dr. Antonio DiPasquale(UC Berkeley) is acknowledged for assistance with X-ray crystallography.
Footnotes
Supporting Information Available Experimental details and characterization for all new compounds are available free of charge via the internet at http://pubs.acs.org.
References
- 1.Hirasawa Y, Kobayashi J, Morita H. Heterocycles. 2009;77:679–729. [Google Scholar]
- 2.Ma X, Gang DR. Nat. Prod. Rep. 2004;21:752–772. doi: 10.1039/b409720n. [DOI] [PubMed] [Google Scholar]
- 3.Hudlicky T, Reed JW. The Way of Synthesis. 1st Ed Wiley-VCH; Weinheim: 2007. Lycopodium Alkaloids; pp. 573–602. [Google Scholar]
- 4.(a) Bisai A, West SP, Sarpong R. J. Am. Chem. Soc. 2008;130:7222–7223. doi: 10.1021/ja8028069. [DOI] [PubMed] [Google Scholar]; (b) West SP, Bisai A, Lim AD, Narayan R, Sarpong R. J. Am. Chem. Soc. 2009;131:11187–11194. doi: 10.1021/ja903868n. [DOI] [PubMed] [Google Scholar]
- 5.Larson KK, Sarpong R. J. Am. Chem. Soc. 2009;131:13244–13245. doi: 10.1021/ja9063487. [DOI] [PubMed] [Google Scholar]
- 6.This is supported by a comparison of pKas of protonated pyridine and protonated methoxypyridine, see: Joule JA, Mills K. Heterocyclic Chemistry. 4>th Ed Blackwell Science Ltd.; 2000. pp. 71–120..
- 7.Ishiuchi K, Kubota T, Morita H, Kobayashi J. Tetrahedron Lett. 2006;47:3287–3289. [Google Scholar]
- 8.Katakawa K, Kogure N, Kitajima M, Takayama H. Helv. Chim. Acta. 2009;92:445–452. [Google Scholar]
- 9.Koyama K, Morita H, Hirasawa Y, Yoshinaga M, Hoshino T, Obara Y, Nakahata N, Kobayashi J. Tetrahedron. 2005;61:3681–3690. [Google Scholar]
- 10.Lannotinidine B is especially interesting from a biological perspective because it has been shown to enhance mRNA expression for nerve growth factor (NGF) in human glial cells (Ref. 9).
- 11.For previous syntheses of 1, see: Staben ST, Kennedy-Smith J, Huang D, Corekey BK, LaLonde R, Toste FD. Angew. Chem. Int. Ed. 2006;45:5991–5994. doi: 10.1002/anie.200602035.. DeLorbe JE, Lotz MD, Martin SF. Org. Lett. 2010;12:1576–1579. doi: 10.1021/ol100273p..
- 12.Kelly SA, Foricher Y, Mann J, Bentley JM. Org. Biomol. Chem. 2003;1:2865–2876. doi: 10.1039/b305869g. [DOI] [PubMed] [Google Scholar]
- 13.Stork G, Danheiser RL. J. Org. Chem. 1973;38:1775–1776. [Google Scholar]
- 14.Luche JL. J. Am. Chem. Soc. 1978;100:2226–2227. [Google Scholar]; For a review, see:Molander GA. Chem. Rev. 1992;92:29–68..
- 15.Wick AE, Felix D, Steen K, Eschenmoser A. Helv. Chim. Acta. 1964;47:2425–2429. [Google Scholar]; For a review, see:Castro AMM. Chem. Rev. 2004;104:2939–3002. doi: 10.1021/cr020703u.
- 16.Grotjahn DB, Lev DA. J. Am. Chem. Soc. 2004;126:12232–12233. doi: 10.1021/ja046360u. [DOI] [PubMed] [Google Scholar]
- 17.We are grateful to Prof. Hiromitsu Takayama (Chiba University) for copies of 1H and 13C NMR spectra of 2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.