

Abstract
Status epilepticus is a neurological emergency that results in mortality and neurological morbidity. It has been postulated that the reduction of inhibitory transmission during status epilepticus results from a rapid modification of GABAA receptors. However, the mechanism(s) that contributes to this modification has not been elucidated. We report, using an in vitro model of status epilepticus combined with electrophysiological and cellular imaging techniques, that prolonged epileptiform bursting results in a reduction of GABA-mediated synaptic inhibition. Furthermore, we found that constitutive internalization of GABAA receptors is rapid and accelerated by the increased neuronal activity associated with seizures. Inhibition of neuronal activity reduced the rate of internalization. These findings suggest that the rate of GABAA receptor internalization is regulated by neuronal activity and its acceleration contributes to the reduction of inhibitory transmission observed during prolonged seizures.
Keywords: GABAA receptor, endocytosis, status epilepticus, neuronal activity, plasticity, synapse
Introduction
Seizures are paroxysmal events characterized by the excessive, synchronous bursting of a group of neurons. The majority of seizures terminate spontaneously within a short period of time because of endogenous inhibitory mechanisms. Some seizures do not stop, which results in a life-threatening neurological emergency called status epilepticus (SE). There are estimated to be between 126,000 and 195,000 cases of SE each year in the United States (DeLorenzo et al., 1995). In the 30 d following SE, a mortality as high as 25% has been reported, a rate that approaches the deaths from gunshot wounds, major head trauma, and myocardial infarction (Towne et al., 1994). Those who survive SE are at risk for neurological morbidity, such as injury to the hippocampus and the development of chronic epilepsy (Jackson et al., 1998; VanLandingham et al., 1998; Schulz and Ebner, 2001).
Single and recurrent seizures result in a reduction in GABA-mediated inhibition. Animal models of SE resulting from continuous hippocampal stimulation, cholinergic stimulation, and excitatory amino acid stimulation also demonstrate a loss of inhibition during SE (Kapur and Macdonald, 1996). Thus, it has been suggested that SE is a self-reinforcing condition in which there is a progressive, use-dependent reduction of GABA-mediated inhibition of principal neurons. Reduced inhibition allows the seizure to continue, causing yet an additional decrease in inhibition and continuation of the seizure. The pathophysiological mechanism responsible for this progressive reduction of inhibition during SE has not been completely elucidated. However, recordings of whole-cell GABAA receptor (GABAR) currents from acutely dissociated principal neurons of the hippocampus obtained from rats in SE (Kapur and Coulter, 1995; Kapur and Macdonald, 1997) suggest that this reduced inhibition is attributable, in part, to an alteration of the population of postsynaptic GABARs.
GABARs are known to undergo a continuous cycle of internalization and insertion into the cell membrane (Borden et al., 1984). The internalization occurs predominantly via a clathrin-dependent endocytic process (Tehrani and Barnes, 1993, 1997; Connolly et al., 1999; Kittler et al., 2000) and can be triggered by calcium-phospholipid dependent protein kinase C (Chapell et al., 1998; Filippova et al., 2000) and brain-derived neurotrophic factor (Cheng and Yeh, 2003; Jovanovic et al., 2004). Internalization of the surface GABARs correlates with a reduced response to GABA, whereas inhibition of internalization results in an increased amplitude of synaptic GABAR currents (Kittler et al., 2000).
An activity-dependent rapid internalization of GABARs from the cell surface is an attractive mechanism that could partially explain the reduction in GABA-mediated inhibition observed during the prolonged seizures of SE. Because the number of receptors at a synapse is an important determinant of synaptic strength (Edwards et al., 1990; Maconochie et al., 1994; Nusser et al., 1997, 1998), a reduction of the total number of postsynaptic GABARs could result in a decline of GABAergic inhibition. Although the endocytosis of AMPA receptors is known to be activity dependent (Ehlers, 2000; Lin et al., 2000), it is not known whether the rate of GABAR internalization is modulated by neuronal activity.
Materials and Methods
Treatment of animals followed guidelines set by the University of Virginia Health Sciences Center Animal Research Committee. All efforts were made to minimize animal stress and discomfort.
Hippocampal neuronal cultures. The methods for obtaining and maintaining the hippocampal cultures used in our laboratory have been described previously (Mangan and Kapur, 2004) and are modified from those of Goslin et al. (1998). In these experiments, both high-density and low-density cultures were used. For high-density cultures, 100,000 cells are platted on a poly-l-lysine-coated coverslip. For low-density cultures, 10,000 cells are platted.
Electrophysiology. Whole-cell patch-clamp recording of GABAA receptor currents from cultured neurons were performed using standard techniques described previously (Hamill et al., 1981; Mtchedlishvili and Kapur, 2003; Mangan and Kapur, 2004). Thick-walled 1.5 mm (outer diameter) × 0.86 mm (inner diameter) borosilicate patch electrodes were pulled on a P-97 Flaming-Brown horizontal puller (Sutter Instruments, Novato, CA) using a three-stage pull to a final resistance of 3-8 MΩ. Patch electrodes were filled with an internal solution containing the following (in mm): 153.3 CsCl, 1.0 MgCl2, 10.0 HEPES, 5.0 EGTA, with a pH of 7.30 (with sterile filtered CsOH and osmolarity of 280-295 mOsm). ATP disodium salt, in a final concentration of 4 mm, was added to the solution used to fill the electrode shank just before recording.
The standard, control external medium used for electrophysiological recordings and immunocytochemical experiments contained the following (in mm): 146 NaCl, 2.5 KCl, 2.0 MgCl2, 2.0 CaCl2, 10.0 glucose, and 10.0 HEPES, with a pH of 7.4 (osmolarity of 305-320 mOsm). For experiments performed in a nominally magnesium-free external environment (0 [Mg2+]o), MgCl2 was replaced by an osmotically equivalent concentration of NaCl. For experiments performed in a high-potassium external environment (10 [KCl]o), the 2.5 mm KCl was replaced with 10 mm KCl. For miniature IPSC (mIPSC) recordings, glutamate receptor-mediated synaptic currents were blocked by adding 50 μm d(-)-2-amino-5-phosphonovaleric acid (d-APV) and 20 μm 6,7-dinitroquinoaxaline-2,3-dione (DNQX) to the external solution. Action potentials were blocked by adding 1 μm tetrodotoxin (TTX) to the external solution.
Cultured neurons 14-21 d in vitro were viewed on the stage of an inverted Nikon (Tokyo, Japan) microscope. Current-clamp recordings were performed by injecting current to alter the membrane potential between -50 and -100 mV. Mangan and Kapur (2004) have previously characterized the membrane potential for action potential firing for these cultured neurons in both the standard, control external medium and 0 [Mg2+]o medium and demonstrated that the bursting that occurs in 0 [Mg2+]o is a property of the network. Voltage-clamp recordings were performed at a holding potential of -60 mV. Membrane voltage and currents were recorded with an Axopatch 200A amplifier (Axon Instruments, Union City, CA) and low-pass filtered at 2-3 kHz with an eight-pole Bessel filter before digitization, storage, and display. Currents were recorded for off-line analysis on a Pentium II personal computer using the Axoscope software program (Axon Instruments) digitized at 400 Hz. Resistance and capacitance were compensated for each neuron. Each neuron was recorded for up to 60 min.
Off-line analysis of mIPSC amplitude, 10-90% rise time, decay, and frequency was performed using MiniAnalysis software (Synaptosoft, Decatur, GA). The threshold for mIPSC detection was set at three times the root mean square of baseline noise. The accuracy of detection was visually confirmed. The amplitude of all mIPSCs greater than the detection criteria were included in the amplitude analysis. Because the distribution of mIPSC amplitudes is skewed, a median mIPSC amplitude was calculated for each neuron, and the mean of the medians is reported. The 10-90% rise time, the time interval between the last data point with a value of 10% and the first data point with a value of 90% of the peak amplitude, was determined for all selected events during the recording period. The decay phase was calculated by fitting individual mIPSCs with a 10-90% rise time <3 ms to a two-exponential function characterized by two time constants (τ1 and τ2) and accepted if r2 > 0.70. For each neuron, the decay phase was based on the first 20 current traces meeting these criterion. Decay phase, 10-90% rise time, and frequency values are reported as mean ± SEM. Error bars represent the SEM. Statistical comparisons were performed using a two-tailed Student's t test.
Fluorescence internalization assay. Cultures of living hippocampal neurons 14-18 d in vitro were incubated at 4°C for 1 h in the presence of a high concentration (20 μg/ml) of an anti-GABAR antibody (Upstate Biotechnology, Lake Placid, NY) directed against the extracellular epitopes of the β2/3 subunits (1-3 N-terminal, clone 62-3G1) (de Blas et al., 1988). Because low temperature is known to inhibit endocytosis, antibody-bound receptors stay on the surface during this incubation. After this incubation, the neurons were washed with ice-cold 1× PBS to remove the unbound antibody, before incubation in an antibody-free external medium allowing antibody-bound receptors to undergo internalization, followed by fixation for 15 min with 4% paraformaldehyde/4% sucrose. After fixation, the neurons were exposed to a super-saturating concentration (20 μg/ml) of the first of two secondary antibodies (Alexa Fluor 488 goat anti-mouse; Molecular Probes, Eugene, OR) for 2 h under a nonpermeabilized condition before permeabilization (0.25% Triton X-100 for 10 min) and incubation with a second secondary antibody (Alexa Fluor 594 goat anti-mouse, 4 μg/ml) for 1 h. This process permits antibody-tagged surface receptors to be independently identified from those antibody-tagged receptors that have been internalized before fixation. The super-saturating concentration of the first secondary antibody is required to limit the possibility of the second secondary antibody recognizing unlabeled antibody-tagged surface receptors. The coverslips were mounted on slides with Gel/Mount (Biomeda, Foster City, CA). The edge of each coverslip was sealed with clear nail polish, and slides were stored at -20°C.
Fluorescent images of cells were captured on a Roper Scientific (Tucson, AZ) Photometrics CoolSNAPcf CCD camera mounted on a Nikon Eclipse TE200 fluorescent microscope equipped with a mercury lamp, with excitation and emission filters optimized for separation of 488 and 594 emission spectra, driven by the MetaMorph imaging software system (Universal Imaging Corporation, Downington, PA). Before imaging, the quality of the experiment was assessed using a low-power objective to assess neuronal morphology and fluorescent variability. Only those coverslips in which neuronal morphology was intact were used. Although variability in staining was observed, most neurons on a single coverslip had similar fluorescence intensity. Morphologically intact neurons, independent of fluorescent intensity, were randomly chosen for imaging. High-resolution digital images (16-bit) were acquired of each fluorochrome using a 60× 1.4 numerical aperture lens. Image exposure times ranged from 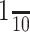 to 2 s depending on the brightness of the fluorochrome. The investigator was not blind to the treatment condition. Off-line analysis of immunoreactivity was performed using the MetaMorph imaging system. For each captured neuron, the immunoreactive area of surface and internalized antibody-tagged receptors was determined by thresholding the image so that no weak diffuse background labeling was visible. Each experiment was replicated three times for a total of 15 neurons per time point per condition. All values are reported as mean ± SEM. Error bars represent the SEM. Statistical comparisons were performed using a two-way ANOVA.
to 2 s depending on the brightness of the fluorochrome. The investigator was not blind to the treatment condition. Off-line analysis of immunoreactivity was performed using the MetaMorph imaging system. For each captured neuron, the immunoreactive area of surface and internalized antibody-tagged receptors was determined by thresholding the image so that no weak diffuse background labeling was visible. Each experiment was replicated three times for a total of 15 neurons per time point per condition. All values are reported as mean ± SEM. Error bars represent the SEM. Statistical comparisons were performed using a two-way ANOVA.
Results
Cultures of hippocampal pyramidal neurons on glass coverslips were prepared per the methods of Goslin et al. (1998). Neuronal activity in cultures 14-18 d in vitro was assessed by current-clamp recordings in the whole-cell recording mode with a ruptured patch. Neurons were incubated in an extracellular medium containing either 2 mm MgCl2 (n = 8) or 0 mm MgCl2 (n = 20). In the presence of MgCl2 (Fig. 1A), occasional spontaneous action potentials were observed (n = 7). In two of these seven neurons, there were depolarizations with two or three superimposed action potentials. These bursts were always brief, never longer than 1 s, and were rare during the 5 min recording period. In one neuron, no action potentials were observed during the 5 min recording period, despite injecting current to vary the membrane potential between -50 and -100 mV. In the absence of extracellular MgCl2, there was recurrent epileptiform bursting in these neurons (Fig. 1B), characterized by prolonged depolarizations with multiple superimposed action potentials. This recurrent sustained bursting is similar to that described previously (Mangan and Kapur, 2004) and was observed in 80% of the recordings.
Figure 1.

Recurrent paroxysmal epileptiform bursting in a nominally magnesium-free extracellular medium. A, B, Current-clamp recordings in the whole-cell mode with ruptured membrane were obtained from cultured hippocampal neurons 14-21 d in vitro. The recording was performed with the neurons bathed in either a standard, control external medium containing (in mm) 146 NaCl, 2.5 KCl, 2.0 MgCl2, 2.0 CaCl2, 10.0 glucose, and 10.0 HEPES (A) or in an extracellular medium devoid of magnesium (B). Removal of magnesium from the extracellular medium resulted in the induction of recurrent bursts consisting of a prolonged depolarization with multiple superimposed action potentials.
Mangan and Kapur (2004) demonstrated previously that the recurrent bursting observed in these neuronal cultures did not alter the resting membrane potential or input resistance of the neuron. To confirm this finding, input resistance was measured as described previously (Mangan and Bertram, 1998; Mangan and Kapur, 2004). In the presence of MgCl2, input resistance was 156.2 ± 15.99 MΩ (n = 11). Input resistance was not significantly altered by exposure to the nominally magnesium-free medium (168.1 ± 23.15 MΩ; n = 13; t test, p > 0.05). Although these values are slightly higher than those reported previously, they are still in accord with that study and other studies using patch electrodes (Spruston and Johnston, 1992).
Recurrent bursting in a nominally magnesium-free extracellular medium is commonly used to study the mechanisms underlying seizures and seizure-induced plasticity (Sombati and Delorenzo, 1995; Heinemann et al., 1996; Gibbs et al., 1997; Engel et al., 2000; Kohling et al., 2000). For the purpose of this study, these cultures provide the unique advantage that cell-surface and intracellular pools of native GABARs can be readily examined and distinguished using fluorescent immunocytochemical techniques.
Inhibitory synaptic transmission after prolonged epileptiform bursting
The impact of prolonged bursting on inhibitory synaptic transmission was determined by comparing the characteristics of mIPSCs recorded from bursting-treated neurons to those recorded from control neurons 14-21 d in vitro. Bursting-treated neurons were exposed for 2 h or longer to a nominally magnesium-free external medium (0 [Mg2+]o), whereas control neurons were exposed to an extracellular medium that contained 2 mm MgCl2. Recordings from the bursting-treated neurons commenced during the period of exposure to 0 [Mg2+]o to ensure that bursting was occurring. Before recording mIPSCs, the bursting was terminated by the addition of 1 μm TTX, 50 μm d-APV, and 20 μm DNQX. Whole-cell ruptured patch-clamp recordings of mIPSCs in control and bursting-treated neurons were performed in an isotonic chloride environment in the presence of TTX, d-APV, and DNQX with the neuron voltage clamped to -60 mV. The addition of 10 μm bicuculline abolished the recorded events, demonstrating that they were mediated by GABARs (data not shown).
mIPSCs recorded from bursting-treated neurons were distinct from those recorded from controls. mIPSC amplitude was lower in bursting-treated neurons compared with control neurons. Figure 2A displays “averaged” mIPSCs obtained by averaging the mIPSCs (≥100, during a 5-15 min recording) recorded from one control neuron and one bursting-treated neuron. The mean peak amplitude recorded from the control neuron was -59.0 pA and that recorded from the bursting-treated neuron was -36.6 pA. Because the distribution of individual mIPSC amplitudes recorded from a neuron tends to be skewed by larger-amplitude mIPSCs that bias the mean, the median value is used because it is more representative of the distribution (Edwards et al., 1990). The amplitude distribution histograms shown in Figure 2B are for the control and bursting-treated neurons displayed in Figure 2A. The median amplitude of mIPSCs recorded from the control neuron was 50.0 pA and that recorded from the bursting-treated neuron was 34.0 pA. Recordings were obtained from 13 control neurons and nine bursting-treated neurons. A population mean of the median values for the control neurons was 53.9 ± 1.6 pA and that for the bursting-treated neurons was 36.2 ± 1.5 pA (p < 0.01, t test).
Figure 2.

Diminished GABAergic synaptic transmission in bursting-treated neurons. A, Averaged mIPSC traces from a control cultured hippocampal neuron and from a bursting-treated neuron after a 3 h exposure to 0 [Mg2+]o. The control and bursting-treated traces were obtained from different cells voltage clamped to a holding potential of -60 mV. B, Amplitude distribution histogram for mIPSCs recorded from the control (crosshatched bars) and bursting-treated (black bars) neurons displayed in A.
Compared with controls, the decay phase of mIPSCs was prolonged in bursting-treated neurons. The mean τ1 and mean τ2 of the mIPSCs recorded from the control neuron displayed in Figure 2A were 24 and 91 ms, respectively; those recorded from the bursting-treated neuron were 54 and 118 ms, respectively. For the control and bursting-treated neurons, a population mean was obtained by averaging the mean decay constants obtained for each neuron. The population means of mIPSCs decay constants recorded from the control neurons were τ1 = 30.3 ± 2.4 and τ2 = 84.3 ± 4.4, and those for the bursting-treated neurons were τ1 = 42.0 ± 4.3 and τ2 = 120.6 ± 6.5 (p < 0.01, t test).
Given the finding that amplitude was smaller but decay was longer in the bursting-treated neurons than in control neurons, total charge transfer was calculated and used as the measure of mIPSC inhibition. Total charge transfer was smaller in bursting-treated neurons compared with controls. For each neuron, total charge transfer was calculated as the integral over 300 ms for the average mIPSC. For the control and bursting-treated neurons, a population mean was obtained by averaging the total charge transfer obtained for each neuron. The mean charge transfer for control neurons (n = 13) during this time interval was 3789 ± 274 pC and for bursting-treated neurons (n = 9) was 2901 ± 125.8 pC (p < 0.05, t test).
Together, the finding of a reduced mIPSC amplitude and a corresponding reduction in total charge transfer for bursting-treated neurons demonstrates a reduction in synaptic GABA-mediated inhibition in bursting-treated neurons. Because prolonged seizures are known to activate excitotoxic mechanisms resulting in neuronal injury and death, one possible mechanism that could explain these results is a selective loss of GABAergic synapses located proximal to the soma. When the recording pipette is attached to the soma, synapses located proximal to the soma generate signals that appear larger and decay faster than signals from distal synapses (for review, see Spruston et al., 1994). Thus, if prolonged bursting resulted in the selective loss of synapses located close to the soma, the remaining mIPSCs would be smaller and decay more slowly. This possibility was supported by the observation that mIPSC frequency was reduced in bursting-treated neurons (0.20 ± 0.05 Hz; n = 9) compared with control neurons (0.38 ± 0.15 Hz; n = 13; t test, p < 0.01). However, a reduction in the contribution of proximal synapses should also result in a slower rise time because longer rise times tend to be associated with distal synapses. We compared the 10-90% rise time in the 13 control and nine bursting-treated neurons. No change in rise time occurred. The 10-90% rise time for the control neurons was 2.9 ± 0.1 ms and for the bursting-treated neurons 3.1 ± 0.1 ms (p = 0.1194, t test). Therefore, the reduction in mIPSC amplitude does not appear to be related to a selective loss of proximal synapses.
Previous studies (Kapur and Coulter, 1995; Kapur and Macdonald, 1997) have suggested that the postsynaptic GABAR population is rapidly modified during SE and that one of the consequences of this rapid modification is a reduction in GABA-mediated inhibition. The finding that the amplitude and charge transfer of mIPSCs recorded from bursting-treated neurons were reduced compared with those recorded from controls is in agreement with this hypothesis. We hypothesized that an increase in the rate of GABAR intracellular accumulation, which resulted in a reduction in the number of functional GABARs on the cellular surfaces, was partly responsible for the reduction of GABA-mediated inhibition observed in bursting-treated neurons.
Measurement of GABAR intracellular accumulation using an immunocytochemical technique
To assess the rate at which the cell-surface GABAR pool is redistributed to the intracellular compartment, an antibody-feeding technique (Connolly et al., 1999; Lin et al., 2000) that permits the cell-surface and the intracellular pools of native GABARs to be distinguished was used. Cell-surface GABARs on living cultured hippocampal neurons 14-18 d in vitro were tagged with antibodies directed against the extracellular N terminus of the β2/3 subunits (clone 62-3G1) (de Blas et al., 1988). This incubation was performed at 4°C to minimize endocytosis of tagged receptors during the incubation process. After tagging, the neurons (control neurons) were incubated in antibody-free external control medium at 37°C, allowing antibody-tagged receptors to undergo endocytosis at the basal rate. The cultures were incubated at 37°C for durations of 0, 10, 20, 30, or 60 min before fixation. After fixation, the neurons were exposed to a supersaturating concentration of the first of two fluorescent secondary antibodies before permeabilization and incubation with a second fluorescent secondary antibody. This process permits antibody-tagged surface GABARs to be independently identified from those antibody-tagged GABARs that have been internalized.
Immediate fixation after incubation in anti-β2/3 subunit antibody resulted in robust staining of GABARs present on the surface of the cell with visualization of punctate regions (Fig. 3, green), which represent clusters of GABARs containing the β2/3 subunit. At this time point (0 min), there was only faint staining of intracellular, antibody-tagged GABARs (Fig. 3, red). Incubating the cells in the antibody-free external medium at 37°C for longer periods of time resulted in a diminution of surface reactivity and an increased intracellular GABAR immunoreactivity (Fig. 3).
Figure 3.
Rapid Intracellular accumulation of GABARs. A-O, GABARs containing the β2/3 subunits were tagged with a primary antibody directed against the N terminus of these subunits. After tagging, the cells were incubated in an antibody-free standard, control external medium (see Materials and Methods) at 37°C for 0 min (A-C), 10 min (D-F), 20 min (G-I), 30 min (J-L), and 60 min (M-O) before fixation. The neurons were then sequentially exposed to secondary antibodies before and after permeabilization, allowing antibody-tagged surface (A, D, G, J, M) and internalized (B, E, H, K, N) receptors to be independently identified. Color-merged images of the antibody-tagged surface and internalized receptors are displayed in C, F, I, L, and O.
The intracellular accumulation of GABARs was quantified by measuring the cell-surface and internalized GABAR immunoreactivity. The GABAR intracellular accumulation ratio was defined as the ratio of immunoreactivity resulting from intracellular receptors (Fig. 3, red) to total immunoreactivity, in which total immunoreactivity is the sum of antibody-tagged surface (Fig. 3, green) and intracellular receptor immunoreactivity. The GABAR intracellular accumulation ratio obtained for living hippocampal cultures at 37°C in the control extracellular medium (basal activity) for 0, 10, 20, 30, and 60 min is displayed in Figure 4J (filled circles). Each data point was obtained by averaging the GABAR intracellular accumulation ratio for 15 neurons selected randomly from three replicates. The intracellular accumulation appears to plateau at ∼50% after 30 min, suggesting that a new equilibrium between the surface and intracellular compartments had been reached.
Figure 4.
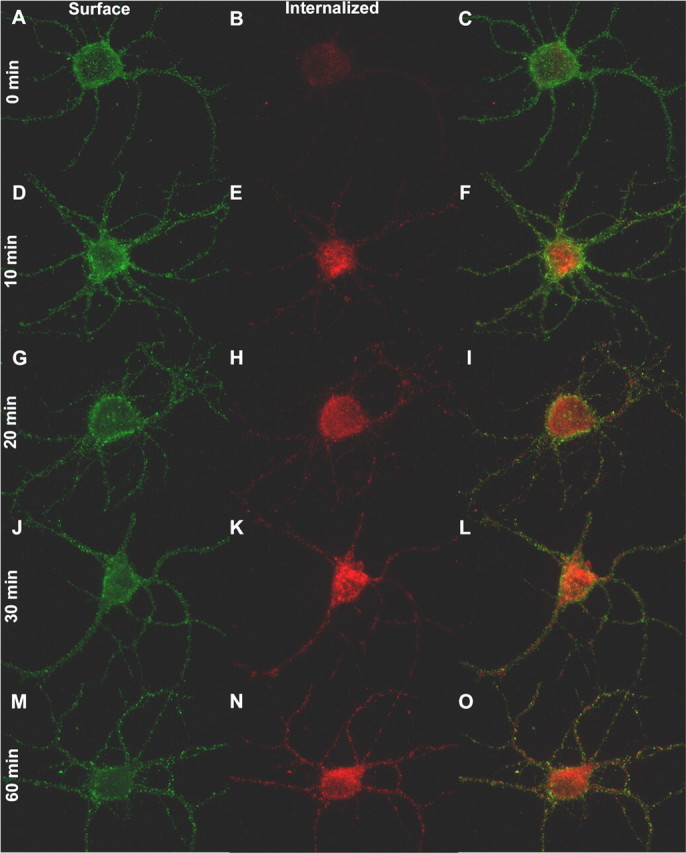
GABAR intracellular accumulation is inhibited at a reduced temperature and in a hyperosmolar extracellular medium. A-I, Neurons incubated in standard, control external medium at 37°C (A-C), at 24°C (D-F), or in standard, control external medium supplemented with 350 mm sucrose at 37°C (G-I) for 30 min after antibody tagging of the GABARs. Images of antibody-tagged surface (green; A, D, G) and internalized (red; B, E, H) receptors are merged in the images in C, F, and I. J, Quantification of the GABAR intracellular accumulation for neurons incubated in standard, control external medium at either 37°C or 24°C for 0, 10, 20, 30, and 60 min after antibody tagging of the GABARs The intracellular accumulation ratio was defined as the ratio of intracellular (red) to total immunoreactivity (red plus green). The ratio at each time point was obtained by pooling data from 15 neurons (3 replicates). The time course was fitted by the least-squares method to a single-exponential growth equation with rate constant K. Solid line, Time course of GABAR intracellular accumulation at 37°C (r2 = 0.85). Dotted line, Time course of GABAR intracellular accumulation at 24°C (r2 = 0.79). *p < 0.01. K, Quantification of the GABAR intracellular accumulation for neurons incubated at 37°C in either standard, control external medium or in standard, control external medium supplemented with 350 mm sucrose for 30 min after antibody tagging of the GABARs. *p < 0.01. Incubating at 24°C or in the presence of 350 mm sucrose reduced intracellular accumulation.
To determine the rate at which GABARs were redistributed to the intracellular compartment, the GABAR intracellular accumulation ratio was fitted to the following single-exponential growth equation: GABAR intracellular accumulation ratio (time) = Ymin + [Ymax - Ymin][1 - e(K × time)], where Ymin is the minimum derived GABAR intracellular accumulation ratio at time 0, Ymax is the maximum derived GABAR intracellular accumulation ratio, and K represents the rate constant of GABAR intracellular accumulation in minutes-1. The rate constant describes the rate at which GABARs are removed from the surface and accumulate in the intracellular compartment. Dividing 0.693 (ln 2) by the rate constant (0.693/K) provides the time for the intracellular receptor population to increase to 50% of the Ymax [half-life (τ1/2)]. At 37°C, the rate constant for GABAR intracellular accumulation was 0.072 ± 0.002 min-1. This rate constant corresponds to a τ1/2 of 9.6 min.
To validate this technique, the GABAR intracellular accumulation ratio was calculated under two conditions expected to inhibit the basal rate of GABAR endocytosis: (1) reduced temperature and (2) hyperosmolar medium (Hansen et al., 1993; Kittler et al., 2000). At room temperature (24°C), GABARs did redistribute to the intracellular compartment (Fig. 4D-F). However, at 24°C, there was an ∼25-30% decrease (n = 15 from 3 replicates; ANOVA, p < 0.01) in the GABAA receptor intracellular accumulation ratio at each time point (10, 20, 30, and 60 min) and a corresponding decrease in the rate constant (0.051 ± 0.004 min-1) compared with controls (Fig. 4A-C, J). The effect of hyperosmolar sucrose on GABAR redistribution was studied by measuring the GABAR intracellular accumulation ratio after 30 min of incubation in a 350 mm sucrose external medium at 37°C. Whereas hyperosmolar sucrose at this concentration did not result in complete inhibition (Fig. 4G-I), there was a 43% reduction (n = 15 from 3 replicates; ANOVA, p < 0.01) in the GABAR intracellular accumulation ratio at 30 min compared with controls (Fig. 4A-C, K).
The rate of GABAR intracellular accumulation correlated with neuronal activity
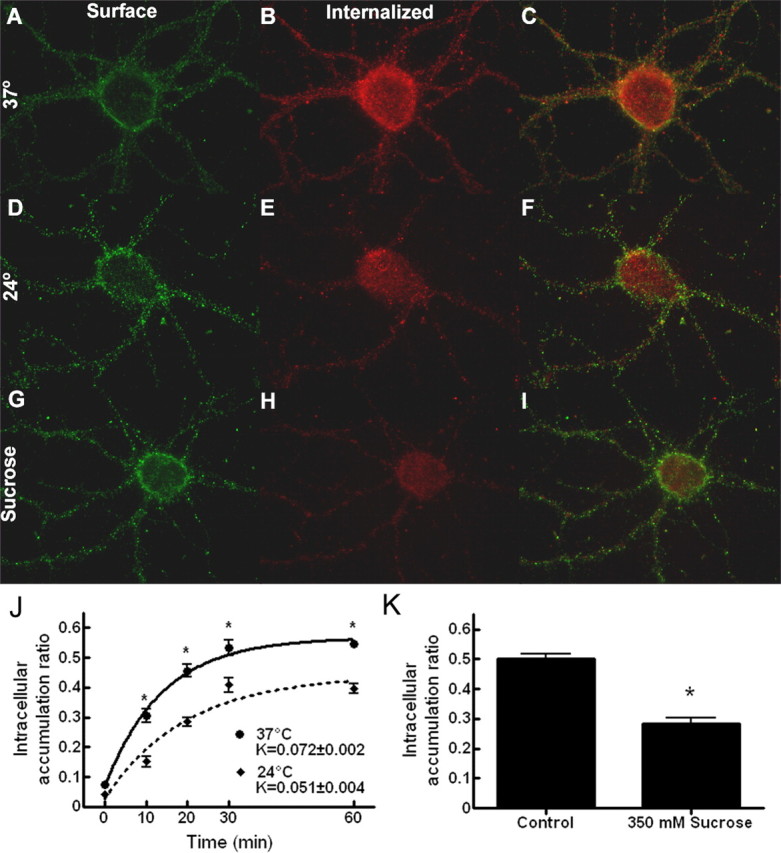
To test the hypothesis that the redistribution of GABARs to the intracellular compartment is modulated by seizure activity, the rate at which GABARs accumulate within the intracellular compartment was examined during bursting and compared with that obtained for control neurons (basal activity). After antibody feeding for 1 h at 4°C, the bursting-treated neurons were incubated at 37°C in 0 [Mg2+]o, promoting recurrent epileptiform bursting. Color-combined images of control and bursting-treated neurons incubated for 10 and 30 min at 37°C after completion of antibody feeding are shown in Figure 5A-D. At both 10 and 30 min, GABARs have redistributed to the intracellular compartment under both conditions, as demonstrated by the intracellular immunoreactivity (red). However, the surface immunoreactivity (green) at 10 and 30 min is greater for the control neurons than the bursting-treated neurons, suggesting that a greater percentage of the antibody-tagged receptors of the bursting-treated neurons have redistributed to the intracellular compartment.
Figure 5.
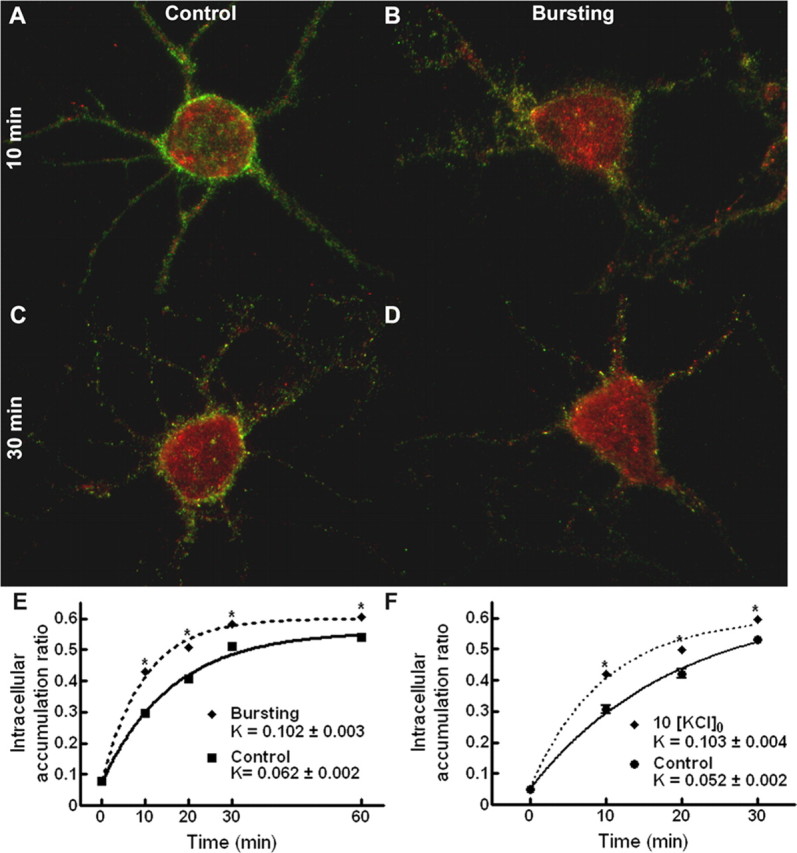
Epileptiform bursting accelerated intracellular accumulation of GABAR. A-D, Color-merged images of antibody-tagged surface (green) and internalized (red) receptors for neurons incubated in standard, control external medium (A, C) or with neuronal activity increased in 0 [Mg2+]o (B, D) at 37°C for 10 min (A, B) and 30 min (C, D) after antibody tagging of the GABARs. E, Quantification of the GABAR intracellular accumulation ratio for neurons incubated in either standard, control external medium or in 0 [Mg2+]o at 37°C for 0, 10, 20, 30, and 60 min after antibody tagging. Solid line, Time course of GABAR intracellular accumulation in standard, control external medium (r2 = 0.97). Dotted line, Time course of GABAR intracellular accumulation in 0 [Mg2+]o (r2 = 0.98). Compared with control neurons, the intracellular accumulation ratio was increased at 10, 20, 30, and 60 min, and there was a nearly 65% increase in the intracellular accumulation rate constant. *p < 0.01. F, Quantification of the GABAR intracellular accumulation ratio for neurons incubated in either standard, control external medium or in 10 [K+]o at 37°C for 0, 10, 20, 30, and 60 min after antibody tagging of the GABARs. Solid line, Time course of GABAR intracellular accumulation in standard, control external medium (r2 = 0.94). Dotted line, Time course of GABAR intracellular accumulation in 10 [KCl]o (r2 = 0.97). Bursting induced by a high external potassium concentration also increased the intracellular accumulation at 10, 20, and 30 min and the intracellular accumulation rate constant. *p < 0.01.
The GABAR intracellular accumulation ratio for bursting-treated neurons was compared with that for control neurons (Fig. 5E), and it was increased at 10, 20, 30, and 60 min (n = 15 from 3 replicates; ANOVA, p < 0.01). At 30 and 60 min, the GABAR intracellular accumulation ratio measured for the bursting neurons was ∼14% greater than controls. Bursting resulted in a nearly 65% increase in the intracellular accumulation rate constant (Kbursting = 0.102 ± 0.003 min-1 vs Kcontrol = 0.062 ± 0.002 min-1).
To confirm that the increase in the rate of GABAR intracellular accumulation was an effect of increased neuronal activity, GABAR redistribution to the intracellular compartment was also assessed by inducing bursting by increasing the extracellular potassium concentration. Concentrations of KCl above 6.5 mm have been used previously to increase neuronal activity and induce recurrent epileptiform bursting (Rutecki et al., 1985; Traynelis and Dingledine, 1988; Jensen and Yaari, 1997). After antibody feeding, the neurons were incubated in an external medium with a potassium concentration of 10 mm (10 [K+]o). The GABAR intracellular accumulation ratio for neurons incubated in 10 [K+]o at 37°C was measured at 10, 20, and 30 min after antibody feeding and compared with controls (Fig. 5F). Incubation in 10 [K+]o resulted in an increase of the GABAR intracellular accumulation ratio at each time point (n = 15 from 3 replicates; ANOVA, p < 0.01) and an increase of the rate of GABAR redistribution to the intracellular compartment (KKCl = 0.103 ± 0.004 min-1 vs Kcontrol = 0.052 ± 0.004 min-1).
We tested whether inhibition of bursting would block the effect of bursting on the intracellular accumulation of GABARs. Inhibition of bursting in 0 [Mg2+]o requires the presence of both the NMDA receptor antagonist d-APV and the AMPA receptor antagonist DNQX (Mangan and Kapur, 2004). After antibody feeding, neurons were incubated in either 0 [Mg2+] (bursting) or 0 [Mg2+]o and 50 μm d-APV and 20 μm DNQX (bursting + d-APV/DNQX). The GABAR intracellular accumulation ratio for the bursting and bursting + d-APV/DNQX neurons at 37°C was measured at 30 min after antibody feeding. Compared with the bursting neurons (intracellular accumulation ratio, 0.574 ± 0.006; n = 15 from 3 replicates), the GABAR intracellular accumulation ratio for bursting + d-APV/DNQX neurons was smaller (intracellular accumulation ratio of 0.470 ± 0.06; n = 15 from 3 replicates; t test, p < 0.01) (Fig. 6) and similar to that observed previously at 30 min for control neurons incubated in the standard, control external medium. The results of this experiment and the experiment performed in 10 [K+]o strongly suggest that the increase in GABAR intracellular accumulation observed in the nominally magnesium-free environment is likely the result of the recurrent bursting and not a nonspecific result of incubation in 0 [Mg2+]o.
Figure 6.

Inhibition of excitatory neurotransmission reduced the intracellular accumulation of GABARs in the 0 [Mg2+]o condition. A, B, Color-merged images of antibody-tagged surface (green) and internalized (red) receptors for neurons incubated in either 0 [Mg]o (A, Bursting) or 0 [Mg]o with excitation blocked (B, Bursting + d-APV/DNQX) at 37°C for 30 min after antibody tagging of the GABARs. Compared with bursting neurons, the excitatory block resulted in a reduction in the intracellular accumulation.
Endocytosis can be inhibited in a hyperosmolar medium; therefore, GABAR intracellular accumulation in 0 [Mg2+]o supplemented with 350 mm sucrose was measured. GABAR internalization occurs predominantly via clathrin-dependent endocytosis, and this process can be inhibited by incubation in a hyperosmolar medium (Hansen et al., 1993; Kittler et al., 2000). After antibody feeding, neurons were incubated in either 0 [Mg2+] (bursting) or 0 [Mg2+]o and 350 mm sucrose (bursting + sucrose). The GABAR intracellular accumulation ratio for the bursting and bursting + sucrose neurons at 37°C was measured at 30 min after antibody feeding. Compared with the bursting neurons (intracellular accumulation ratio, 0.577 ± 0.006; n = 15 from 3 replicates), the GABAR intracellular accumulation ratio for bursting + sucrose neurons was smaller (0.424 ± 0.007; n = 15 from 3 replicates; t test, p < 0.01) (Fig. 7). Although incubating in the hyperosmolar medium reduced the intracellular accumulation at 30 min in 0 [Mg2+]o, GABAR intracellular accumulation was still observed, suggesting that a subset of GABAR internalization may occur via a clathrin-independent mechanism (Cinar and Barnes, 2001).
Figure 7.

Hyperosmolar solutions reduced intracellular accumulation of GABARs during bursting. A, B, Color-merged images of antibody-tagged surface (green) and internalized (red) receptors for neurons incubated in either 0 [Mg]o (A, Bursting) or 0 [Mg]o supplemented with 350 mm sucrose (B, Bursting + Sucrose) at 37°C for 30 min after antibody tagging of the GABARs. Compared with bursting neurons, the addition of sucrose resulted in a reduction of the intracellular accumulation.
An increase of the rate of GABAR redistribution under conditions of prolonged bursting raised the question of whether a reduction in the rate of intracellular accumulation would occur during periods of decreased neuronal activity. Such activity modulation of receptor trafficking has been demonstrated previously for the AMPA receptor (Ehlers, 2000). Neuronal activity was inhibited by the addition of the fast sodium channel blocker tetrodotoxin (2 μm TTX) to the extracellular environment. The GABAR intracellular accumulation ratio for neurons in the presence of TTX was measured at 10, 20, 30, and 60 min (Fig. 8). Compared with control neurons, the GABAR intracellular accumulation ratio was decreased at these time points (n = 15 neurons from 3 replicates; ANOVA, p < 0.01). At 30 and 60 min, the GABAR intracellular accumulation ratio measured for neurons in the presence of TTX was ∼30% less than that of controls. Blocking action potential-dependent activity resulted in a 22% decrease in the intracellular accumulation rate constant (KTTX = 0.056 ± 0.003 min-1 vs K37°C = 0.072 ± 0.002 min-1). In total, these findings demonstrate that the rate at which GABARs are redistributed to the intracellular compartment is, in part, affected by the level of neuronal activity.
Figure 8.

Inhibition of neuronal activity reduced GABAR internalization. A-D, Color-merged images of antibody-tagged surface (green) and antibody-tagged internalized (red) receptors for neurons incubated in standard, control external medium (A, C) or with neuronal activity inhibited through the addition of 2 μm TTX to the standard, control external medium (B, D) at 37°C for 10 min (A, B) and 30 min (C, D) after antibody tagging of the GABARs. E, Quantification of the GABAR intracellular accumulation ratio for neurons incubated in either standard, control external medium or with neuronal activity inhibited by the addition of 2 μm TTX to the standard, control external medium at 37°C for 0, 10, 20, 30, and 60 min after antibody tagging of the GABARs. Solid line, Time course of GABAR intracellular accumulation in standard, control external medium (r2 = 0.85). Dotted line, Time course of GABAR intracellular accumulation in TTX (r2 = 0.75). Compared with control neurons, the intracellular accumulation ratio for TTX neurons was decreased at 10, 20, 30, and 60 min, and there was a 22% decrease in the intracellular accumulation rate constant. *p < 0.01.
Discussion
The major findings of this study were (1) that GABA-mediated synaptic inhibition in a network of cultured hippocampal neurons was diminished after a period of prolonged epileptiform bursting, and (2) that the intracellular accumulation of GABARs was modulated by neuronal activity.
Plasticity of GABAergic synaptic transmission during SE
Although previous studies have implied that the reduction of GABA-mediated inhibition that occurs during SE is the result of rapid modification of GABARs (Kapur and Coulter, 1995; Gibbs et al., 1997; Kapur and Macdonald, 1997), the effect of SE on GABAergic synaptic transmission has not been studied in the past. This study directly demonstrates that prolonged epileptiform bursting results in a modification of GABAergic synaptic transmission. There were profound changes in inhibitory synaptic transmission in neurons subjected to prolonged bursting: amplitude and charge transfer were reduced, decay was prolonged, and frequency was decreased.
These findings help to partially explain the rapid loss of efficacy of drugs during SE. Current treatment of SE uses drugs such as the benzodiazepines and barbiturates that act on GABARs (Lowenstein and Alldredge, 1998; Kapur, 2002). Although benzodiazepines are quite effective during the early phase of SE, they fail to control prolonged self-sustained, subtle SE (Treiman et al., 1998; Mayer et al., 2002). The development of resistance to benzodiazepines during SE has also been observed in experimental models of SE in which the benzodiazepine diazepam controlled brief seizures but failed after prolonged SE (Walton and Treiman, 1988). In these animal models, the development of resistance to benzodiazepine was quite rapid (Kapur and Macdonald, 1997; Jones et al., 2002; Goodkin et al., 2003). The development of drug resistance during SE in both clinical SE and in animal models of SE could result from the rapid modification of inhibitory synaptic transmission during SE demonstrated in this study.
Plasticity of GABAR intracellular accumulation during SE
The basal rate of intracellular accumulation of GABARs was rapid (τ1/2 = 9.6 min) and reached a plateau of ∼50% by 30 min. Increasing synaptic activity in the form of epileptiform bursting resulted in both faster kinetics and an increase in the percentage of receptors that accumulated in the intracellular space. Reducing synaptic activity had the converse effect: the kinetics were slower and there was a decrease in the percentage of internalized receptors. Whereas the kinetics of surface GABARs has not been measured directly in the past, studies have suggested that the redistribution of GABAA receptors between surface and intracellular compartments can be rapid. For example, both BDNF (Brunig et al., 2001) and insulin (Wan et al., 1997) have been shown to rapidly alter synaptic transmission by redistributing surface and internalized GABARs.
Rapid basal intracellular accumulation of AMPA receptors and NMDA receptors has been measured previously using biotinylation assays and immunofluorescent techniques (Ehlers, 2000; Lin et al., 2000). The τ1/2 of AMPA receptors was found to be similar to that measured for GABARs in this study, ranging between ∼7 and 10 min. However, in the basal condition, only between 16 and 25% of surface AMPA receptors were removed from the surface when the internalization reached a plateau (Ehlers, 2000; Lin et al., 2000). Similarly, NMDA receptors also have a short basal τ1/2 of 7 min, but the extent of internalization is limited to 6% (Ehlers, 2000). In contrast to NMDA and AMPA receptors, a much larger fraction of GABARs, 50% of the receptors, have internalized at the time of plateau, suggesting that a larger fraction of surface GABAA receptors are subjected to trafficking.
The internalization of GABARs was modulated by neuronal activity; recurrent bursting enhanced it, and activity blockade reduced it. The effect of activity on GABAR internalization has not been studied in the past; however, neuronal activity is known to enhance internalization of AMPA receptors but not NMDA receptors.
Neuronal activity could modulate the endocytosis of GABARs by either ligand-dependent or -independent mechanisms (Barnes, 2001). GABAR internalization is known to occur directly in response to the administration of GABA. Tehrani and Barnes (1991) demonstrated that the percentage of intracellular GABARs increased in response to incubation of cultured cortical neurons in 1 μm GABA for 1-2 h. Calkin and Barnes (1994) have demonstrated that incubation of cortical chick neurons for 2 h in 200 μm GABA also resulted in internalization of GABARs.
GABAR ligand-independent mechanisms could be activated by stimulation of NMDA receptors and the subsequent influx of calcium. NMDA receptor stimulation is known to be an important feature of SE (Borris et al., 2000). Several studies have demonstrated that stimulation of NMDA receptors can reduce GABA-mediated inhibition (Stelzer et al., 1987; Stelzer, 1990; Lu et al., 2000), and blocking NMDA receptors during SE prevents the loss of inhibition (Kapur and Lothman, 1990). Although the link between NMDA activation and the reduction of GABA-mediated inhibition has not been fully characterized, Stelzer and Shi (1994), Chen and Wong (1995), and Lu et al. (2000) have provided evidence that the activation of phosphatase 2B (calcineurin) is involved with this pathway. Whereas Jones and Westbrook (1997) demonstrated that calcineurin had a direct effect on the functional properties of GABARs, Wang et al. (2003) have suggested that the induction of long-term depression at CA1 inhibitory synapses resulted in a reduction in the number of synaptic GABARs and that this reduction was dependent on calcineurin. This link between NMDA activation and internalization of GABARs may not be limited to activation of calcineurin because other second-messenger systems are capable of modulating GABAR trafficking (Chapell et al., 1998; Connolly et al., 1999; Filippova et al., 2000; Cheng and Yeh, 2003; Jovanovic et al., 2004).
A seizure-induced increase in internalization of GABARs is the likely explanation for the diminished mIPSC amplitude observed in bursting-treated neurons
It has been proposed that each vesicle of GABA released by the presynaptic terminal contains enough GABA to result in complete occupancy of the synaptic GABAR population (De Koninck and Mody, 1994; Poncer et al., 1996). Under this saturating condition, the number and type of postsynaptic GABARs will influence the mIPSC amplitude. An increase in the percentage of internalized receptors and a concomitant decrease in the number of postsynaptic GABARs would explain the reduction in mIPSC amplitude observed in the bursting-treated neurons. The conclusion that an increase in the rate of GABAR intracellular accumulation during SE contributes to diminished synaptic strength is supported by previous studies that have demonstrated that the postsynaptic GABAR population is modified during SE (Kapur and Coulter, 1995; Gibbs et al., 1997; Kapur and Macdonald, 1997). However, the saturation theory is debated and no consensus has thus far emerged (Frerking et al., 1995; Frerking and Wilson, 1996), and a reduction of presynaptic vesicular content could also contribute to the reduction of mIPSC amplitude (Hill et al., 1998).
In addition to diminished amplitude and charge transfer, the mIPSC decay was prolonged in the bursting-treated neurons. Because the mIPSC decay is shaped in part by GABAR desensitization and thus GABAR subunit composition (Jones and Westbrook, 1995; Haas and Macdonald, 1999), the prolonged mIPSC decay in the bursting treated-neurons suggests that the GABARs occupying the synapse after bursting were different from those in control neurons. A similar change in the properties of the synaptic GABAR population has been postulated to occur after chronic drug exposure (Hsu et al., 2003; Liang et al., 2004). GABARs with distinct subunit composition and properties are present in synaptic and extrasynaptic membrane (Brunig et al., 2001; Christie and de Blas, 2002; Christie et al., 2002; Caraiscos et al., 2004; Mangan et al., 2005), and it is possible that extrasynaptic receptors move into synapses during bursting, thus altering the decay properties of mIPSCs. A similar mechanism is described during long-term potentiation, in which extrasynaptic AMPA receptors are incorporated into silent synapses (for review, see Poncer, 2003).
The finding that mIPSC frequency was reduced in bursting-treated neurons could potentially be explained by the reduction in mIPSC amplitude, resulting in a higher frequency of mIPSCs falling below the level of detection. Alternatively, the reduction in frequency could support a presynaptic modification in vesicular release. Additional studies designed to study the effect of SE on the probability of GABA vesicle release and content are required.
In summary, we found that the rate of GABAR intracellular accumulation was modulated by activity. An increase in the rate of GABAR intracellular accumulation may be one of the mechanisms by which GABA inhibition is reduced during the prolonged seizures of SE. Future experiments are required to determine the mechanism by which neuronal activity modulates GABAR trafficking, as well as to explain the other changes in GABAergic synaptic transmission that occur as a result of prolonged paroxysmal bursting.
Footnotes
This work was supported by the National Institutes of Health Grants NS-040337 (J.K.), NS-44370 (J.K.), NS-048413 (H.P.G.), and HD-001421 (H.P.G.). We thank Ashley Renick for the preparation of the hippocampal cultures, Chengsan Sun for his assistance in the fluorescent internalization assay, and Kevin Kelly, Catherine Croft Swanwick, and Stacey Trotter for their very careful reading of this manuscript and thoughtful comments.
Correspondence should be addressed to Dr. Howard P. Goodkin, Department of Neurology, University of Virginia, P.O. Box 800394, Charlottesville, VA 22908. E-mail: hpg9v@virginia.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255511-10$15.00/0
References
- Barnes Jr EM (2001) Assembly and intracellular trafficking of GABAA receptors. Int Rev Neurobiol 48: 1-29. [DOI] [PubMed] [Google Scholar]
- Borden LA, Czajkowski C, Chan CY, Farb DH (1984) Benzodiazepine receptor synthesis and degradation by neurons in culture. Science 226: 857-860. [DOI] [PubMed] [Google Scholar]
- Borris DJ, Bertram EH, Kapur J (2000) Ketamine controls prolonged status epilepticus. Epilepsy Res 42: 117-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunig I, Penschuck S, Berninger B, Benson J, Fritschy JM (2001) BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABAA receptor surface expression. Eur J Neurosci 13: 1320-1328. [DOI] [PubMed] [Google Scholar]
- Calkin PA, Barnes Jr EM (1994) Gamma-aminobutyric acid-A (GABAA) agonists down-regulate GABAA/benzodiazepine receptor polypeptides from the surface of chick cortical neurons. J Biol Chem 269: 1548-1553. [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA (2004) Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci USA 1010: 3662-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapell R, Bueno OF, Alvarez-Hernandez X, Robinson LC, Leidenheimer NJ (1998) Activation of protein kinase C induces gamma-aminobutyric acid type A receptor internalization in Xenopus oocytes. J Biol Chem 273: 32595-32601. [DOI] [PubMed] [Google Scholar]
- Chen QX, Wong RK (1995) Suppression of GABAA receptor responses by NMDA application in hippocampal neurones acutely isolated from the adult guinea-pig. J Physiol (Lond) 482: 353-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Yeh HH (2003) Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABA(A) receptor-mediated responses via postsynaptic mechanisms. J Physiol (Lond) 548: 711-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie SB, de Blas AL (2002) α5 Subunit-containing GABAA receptors form clusters at GABAergic synapses in hippocampal cultures. Neuro Report 13: 2355-2358. [DOI] [PubMed] [Google Scholar]
- Christie SB, Miralles CP, de Blas AL (2002) GABAergic innervation organizes synaptic and extrasynaptic GABAA receptor clustering in cultured hippocampal neurons. J Neurosci 22: 684-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinar H, Barnes Jr EM (2001) Clathrin-independent endocytosis of GABAA receptors in HEK 293 cells. Biochemistry 40: 14030-14036. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Kittler JT, Thomas P, Uren JM, Brandon NJ, Smart TG, Moss SJ (1999) Cell surface stability of gamma-aminobutyric acid type A receptors. Dependence on protein kinase C activity and subunit composition. J Biol Chem 274: 36565-36572. [DOI] [PubMed] [Google Scholar]
- de Blas AL, Vitorica J, Friedrich P (1988) Localization of the GABAA receptor in the rat brain with a monoclonal antibody to the 57,000 Mr peptide of the GABAA receptor/benzodiazepine receptor/Cl- channel complex. J Neurosci 8: 602-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koninck Y, Mody I (1994) Noise analysis of miniature IPSCs in adult rat brain slices: properties and modulation of synaptic GABAA receptor channels. J Neurophysiol 71: 1318-1335. [DOI] [PubMed] [Google Scholar]
- DeLorenzo RJ, Pellock JM, Towne AR, Boggs JG (1995) Epidemiology of status epilepticus. J Clin Neurophysiol 12: 316-325. [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B (1990) Quantal analysis of inhibitory synaptic transmission in the dentate gyrus of rat hippocampal slices: a patch-clamp study. J Physiol (Lond) 430: 213-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511-525. [DOI] [PubMed] [Google Scholar]
- Engel D, Endermann U, Frahm C, Heinemann U, Draguhn A (2000) Acute effects of gamma-vinyl-GABA on low-magnesium evoked epileptiform activity in vitro. Epilepsy Res 40: 99-107. [DOI] [PubMed] [Google Scholar]
- Filippova N, Sedelnikova A, Zong Y, Fortinberry H, Weiss DS (2000) Regulation of recombinant γ-aminobutyric acid (GABA)A and GABAC receptors by protein kinase C. Mol Pharmacol 57: 847-856. [PubMed] [Google Scholar]
- Frerking M, Wilson M (1996) Saturation of postsynaptic receptors at central synapses? Curr Opin Neurobiol 10: 395-403. [DOI] [PubMed] [Google Scholar]
- Frerking M, Borges S, Wilson M (1995) Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron 15: 885-895. [DOI] [PubMed] [Google Scholar]
- Gibbs III JW, Sombati S, DeLorenzo RJ, Coulter DA (1997) Physiological and pharmacological alterations in postsynaptic GABAA receptor function in a hippocampal culture model of chronic spontaneous seizures. J Neurophysiol 77: 2139-2152. [DOI] [PubMed] [Google Scholar]
- Goodkin HP, Liu X, Holmes GL (2003) Diazepam terminates brief but not prolonged seizures in young, naive rats. Epilepsia 44: 1109-1112. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G (1998) Rat hippocampal neurons in low-density culture. In: Culturing nerve cells, pp 339-370. Cambridge, MA: MIT.
- Haas KF, Macdonald RL (1999) GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol (Lond) 514: 27-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 39: 85-100. [DOI] [PubMed] [Google Scholar]
- Hansen SH, Sandvig K, van Deurs B (1993) Clathrin and HA2 adaptors: effects of potassium depletion, hypertonic medium, and cytosol acidification. J Cell Biol 121: 61-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Draguhn A, Meierkord H (1996) The pathophysiology of seizure generation. In: The treatment of epilepsy (Shorvon S, Dreifuss F, Fish D, Thomas D, eds), pp 3-19. Oxford: Blackwell Science.
- Hill MW, Reddy PA, Covey DF, Rothman SM (1998) Contribution of sub-saturating GABA concentrations to IPSCs in cultured hippocampal neurons. J Neurosci 18: 5103-5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F-C, Waldeck R, Faber DS, Smith SS (2003) Neurosteroid effects on GABAergic synaptic plasticity in hippocampus. J Neurophysiol 89: 1929-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson GD, McIntosh AM, Briellmann RS, Berkovic SF (1998) Hippocampal sclerosis studied in identical twins. Neurology 51: 78-84. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Yaari Y (1997) Role of intrinsic burst firing, potassium accumulation, and electrical coupling in the elevated potassium model of hippocampal epilepsy. J Neurophysiol 77: 1224-1233. [DOI] [PubMed] [Google Scholar]
- Jones DM, Esmaeil N, Maren S, Macdonald RL (2002) Characterization of pharmacoresistance to benzodiazepines in the rat Li-pilocarpine model of status epilepticus. Epilepsy Res 50: 301-312. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL (1995) Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 15: 181-191. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL (1997) Shaping of IPSCs by endogenous calcineurin activity. J Neurosci 17: 7626-7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ (2004) Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABAA receptor phosphorylation, activity, and cell-surface stability. J Neurosci 24: 522-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J (2002) Prehospital treatment of status epilepticus with benzodiazepines is effective and safe. Epilepsy Curr 4: 1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, Coulter DA (1995) Experimental status epilepticus alters gamma-aminobutyric acid type A receptor function in CA1 pyramidal neurons. Ann Neurol 38: 893-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur J, Lothman EW (1990) NMDA receptor activation mediates the loss of GABAergic inhibition induced by recurrent seizures. Epilepsy Res 5: 103-111. [DOI] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL (1996) Status epilepticus: a proposed pathophysiology. In: The treatment of epilepsy (Shorvon S, Dreifuss F, Fish D, Thomas D, eds), pp 258-268. Oxford: Blackwell Science.
- Kapur J, Macdonald RL (1997) Rapid seizure-induced reduction of benzodiazepine and Zn2+ sensitivity of hippocampal dentate granule cell GABAA receptors. J Neurosci 17: 7532-7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Delmas P, Jovanovic JN, Brown DA, Smart TG, Moss SJ (2000) Constitutive endocytosis of GABAA receptors by an association with the adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J Neurosci 20: 7972-7977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohling R, Vreugdenhil M, Bracci E, Jefferys JG (2000) Ictal epileptiform activity is facilitated by hippocampal GABAA receptor-mediated oscillations. J Neurosci 20: 6820-6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Cagetti E, Olsen RW, Spigelman I (2004) Altered pharmacology of synaptic and extrasynaptic GABAA receptors on CA1 hippocampal neurons is consistent with subunit changes in a model of alcohol withdrawal and dependence. J Pharmacol Exp Ther 310: 1234-1245. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M (2000) Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci 3: 1282-1290. [DOI] [PubMed] [Google Scholar]
- Lowenstein DH, Alldredge BK (1998) Status epilepticus. N Engl J Med 338: 970-976. [DOI] [PubMed] [Google Scholar]
- Lu YM, Mansuy IM, Kandel ER, Roder J (2000) Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron 26: 197-205. [DOI] [PubMed] [Google Scholar]
- Maconochie DJ, Zempel JM, Steinbach JH (1994) How quickly can GABAA receptors open? Neuron 12: 61-71. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Bertram III EH (1998) Ontogeny of altered synaptic function in a rat model of chronic temporal lobe epilepsy. Brain Res 799: 183-196. [DOI] [PubMed] [Google Scholar]
- Mangan PS, Kapur J (2004) Factors underlying bursting behavior in a network of cultured hippocampal neurons exposed to zero magnesium. J Neurophysiol 91: 946-957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan PS, Sun C, Carpenter M, Goodkin HP, Sieghart W, Kapur J (2005) Cultured hippocampal pyramidal neurons express two kinds of GABAA receptors. Mol Pharmacol 67: 775-788. [DOI] [PubMed] [Google Scholar]
- Mayer SA, Claassen J, Lokin J, Mendelsohn F, Dennis LJ, Fitzsimmons BF (2002) Refractory status epilepticus: frequency, risk factors, and impact on outcome. Arch Neurol 59: 205-210. [DOI] [PubMed] [Google Scholar]
- Mtchedlishvili Z, Kapur J (2003) A presynaptic action of the neurosteroid pregnenolone sulfate on GABAergic synaptic transmission. Mol Pharmacol 64: 857-864. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Cull-Candy S, Farrant M (1997) Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron 19: 697-709. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Hajos N, Somogyi P, Mody I (1998) Increased number of synaptic GABAA receptors underlies potentiation at hippocampal inhibitory synapses. Nature 395: 172-177. [DOI] [PubMed] [Google Scholar]
- Poncer JC (2003) Hippocampal long term potentiation: silent synapses and beyond. J Physiol (Lond) 97: 415-422. [DOI] [PubMed] [Google Scholar]
- Poncer JC, Durr R, Gahwiler BH, Thompson SM (1996) Modulation of synaptic GABAA receptor function by benzodiazepines in area CA3 of rat hippocampal slice cultures. Neuropharmacology 35: 1169-1179. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Lebeda FJ, Johnston D (1985) Epileptiform activity induced by changes in extracellular potassium in hippocampus. J Neurophysiol 54: 1363-1374. [DOI] [PubMed] [Google Scholar]
- Schulz R, Ebner A (2001) Prolonged febrile convulsions and mesial temporal lobe epilepsy in an identical twin. Neurology 57: 318-320. [DOI] [PubMed] [Google Scholar]
- Sombati S, Delorenzo RJ (1995) Recurrent spontaneous seizure activity in hippocampal neuronal networks in culture. J Neurophysiol 73: 1706-1711. [DOI] [PubMed] [Google Scholar]
- Spruston N, Johnston D (1992) Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons. J Neurophysiol 67: 508-529. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jaffe DB, Johnston D (1994) Dendritic attenuation of synaptic potentials and currents: the role of passive membrane properties. Trends Neurosci 17: 161-166. [DOI] [PubMed] [Google Scholar]
- Stelzer A (1990) Regulation of GABAA currents by excitatory amino acids. Adv Exp Med Biol 268: 255-263. [DOI] [PubMed] [Google Scholar]
- Stelzer A, Shi H (1994) Impairment of GABAA receptor function by N-methyl-d-aspartate-mediated calcium influx in isolated CA1 pyramidal cells. Neuroscience 62: 813-828. [DOI] [PubMed] [Google Scholar]
- Stelzer A, Slater NT, ten Bruggencate G (1987) Activation of NMDA receptors blocks GABAergic inhibition in an in vitro model of epilepsy. Nature 326: 698-701. [DOI] [PubMed] [Google Scholar]
- Tehrani MH, Barnes Jr EM (1991) Agonist-dependent internalization of gamma-aminobutyric acidA/benzodiazepine receptors in chick cortical neurons. J Neurochem 57: 1307-1312. [DOI] [PubMed] [Google Scholar]
- Tehrani MH, Barnes Jr EM (1993) Identification of GABAA/benzodiazepine receptors on clathrin-coated vesicles from rat brain. J Neurochem 60: 1755-1761. [DOI] [PubMed] [Google Scholar]
- Tehrani MH, Barnes Jr EM (1997) Sequestration of gamma-aminobutyric acidA receptors on clathrin-coated vesicles during chronic benzodiazepine administration in vivo. J Pharmacol Exp Ther 283: 384-390. [PubMed] [Google Scholar]
- Towne AR, Pellock JM, Ko D, DeLorenzo RJ (1994) Determinants of mortality in status epilepticus. Epilepsia 35: 27-34. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R (1988) Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol 59: 259-276. [DOI] [PubMed] [Google Scholar]
- Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, Handforth A, Faught E, Calabrese VP, Uthman BM, Ramsay RE, Mamdani MB (1998) A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med 339: 792-798. [DOI] [PubMed] [Google Scholar]
- VanLandingham KE, Heinz ER, Cavazos JE, Lewis DV (1998) Magnetic resonance imaging evidence of hippocampal injury after prolonged focal febrile convulsions. Ann Neurol 43: 413-426. [DOI] [PubMed] [Google Scholar]
- Walton NY, Treiman DM (1988) Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol 101: 267-275. [DOI] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT (1997) Recruitment of functional GABA receptors to postsynaptic domains by insulin. Nature 388: 686-690. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Haditsch U, Tu W, Cochrane K, Ahmadian G, Tran L, Paw J, Wang Y, Mansuy I, Salter MM, Lu YM (2003) Interaction of calcineurin and type-A GABA receptor gamma 2 subunits produces long-term depression at CA1 inhibitory synapses. J Neurosci 123: 826-836. [DOI] [PMC free article] [PubMed] [Google Scholar]