

Abstract
FGF2 (fibroblast growth factor 2), but not vascular endothelial growth factor (VEGF), stimulates sustained activation of ERK2/1 for endothelial NOS3 (nitric-oxide synthase 3) protein expression in ovine fetoplacental artery endothelial cells (oFPAEC). We deciphered herein the downstream signaling of ERK2/1 responsible for NOS3 expression by FGF2 in oFPAEC. FGF2, but not VEGF, increased NOS3 mRNA levels without altering its degradation. FGF2, but not VEGF, trans-activated sheep NOS3 promoter, and this was dependent on ERK2/1 activation. FGF2 did not trans-activate NOS3 promoters with deletions upstream of the consensus AP-1 site (TGAGTC A, −678 to −685). Trans-activation of wild-type NOS3 promoter by FGF2 was significantly inhibited when either the AP-1 or the cAMP-response element (CRE)-like sequence (TGCGTCA, −752 to −758) was mutated and was completely blocked when both were mutated. EMSA analyses showed that FGF2, but not VEGF, stimulated AP-1 and CRE DNA-protein complexes primarily composed of JunB and Fra1. Chromatin immunoprecipitation assays confirmed JunB/Fra1 binding to NOS3 promoter AP-1 and CRE elements in intact cells. FGF2, but not VEGF, stimulated JunB and Fra1 expressions; all preceded NOS3 up-regulation and were inhibited by PD98059. Down-regulation of JunB or Fra-1, but not c-Jun, blocked FGF2 stimulation of NOS3 expression and NO production. AP-1 inhibition suppressed FGF2 stimulation of NOS3 expression in human umbilical vein EC and uterine artery endothelial cells. Thus, FGF2 induction of NOS3 expression is mainly mediated by AP-1-dependent transcription involving JunB and Fra1 up-regulation via sustained ERK2/1 activation in endothelial cells.
Keywords: Endothelium, Gene Expression, Growth Factors, MAP Kinases (MAPKs), Transcription, Transcription Factors, Placenta
Introduction
During normal pregnancy at a time when fetal weight and uterine and placental blood flows increase exponentially, there are significantly increased maternal-fetal tissue expressions of angiogenic factors, including FGF2 (fibroblast growth factor 2) and vascular endothelial growth factor (VEGF)3 (1, 2). Concomitantly, production of the potent vasodilator nitric oxide (NO) and expression of endothelial NOS3 (nitric-oxide synthase 3) are markedly increased within the maternal-fetal vascular beds (3, 4). Compelling evidence suggests that complex interplays between locally produced angiogenic factors (FGF2 and VEGF) and the NOS3-NO system play a critical role in regulating angiogenesis and vasodilatation (i.e. two key routes for up-regulating uterine and placental blood flows that directly correlate to fetal growth, survival, and neonatal outcomes) (1, 2, 5). Impaired placental angiogenesis and NO signaling are commonly associated with various pregnancy complications, such as intrauterine fetal growth retardation and preeclampsia (1).
Increased NO production during pregnancy is derived, at least in part, from activation of placental endothelial NOS3 by FGF2 and VEGF (6, 7). However, significant differences exist in the effects of FGF2 and VEGF on the placental endothelial NOS3-NO system. For example, in ovine fetoplacental artery endothelial cells (oFPAEC), we have shown that, although both acutely stimulate NO production, VEGF does so with greater potency than FGF2 (8); surprisingly, FGF2, but not VEGF, stimulates NOS3 protein expression (6, 7). Both activate similar signaling pathways, including phosphatidylinositol 3-kinase/AKT1 (protein kinase B) and mitogen-activated protein kinase (MAPK) members, such as extracellular-signal regulated kinase 2/1 (ERK2/1) and Jun N-terminal kinase 1/2 (JNK1/2); however, differences in intensity and temporal patterns of the activation signals may explain the differential effects of FGF2 and VEGF on NOS3 expression in oFPAEC (7). Of note, sustained ERK2/1 activation apparently plays a key role in differentiating why FGF2, but not VEGF, stimulates NOS3 protein expression (7). The mechanism of such regulation is unknown; however, it is known that activated ERK2/1 translocate into the nucleus, where they activate various transcription factors to regulate gene transcription (9, 10). Although NOS3 was initially identified as a “constitutive” enzyme in endothelial cells (EC), its expression has been shown to be regulated at multiple levels by both physiological and pathological stimuli (11). NOS3 promoter trans-activation-initiated transcription is a major route for de novo NOS3 protein synthesis; however, NOS3 expression is also associated with mRNA stability (12).
NOS3 promoter contains various DNA elements for binding of many transcription factors (13–15). A consensus 12-O-tetradecanoylphorbol-13-acetate response element ([TGA(C/G)TCA], −678 to −685) for binding AP-1 (activator protein-1) is critical for NOS3 expression in EC (13–15). Adjacent to the AP-1 site, there is a conserved cAMP-responsive element (CRE, TGACGTCA)-like sequence (TGCGTCA, −752 to −758) for binding CRE-binding protein (CREB) that is required for prostacyclin-induced NOS3 expression in bovine aortic EC (16). AP-1 represents a family of dimeric complexes of Jun (c-Jun, JunB, and JunD) homodimers and Jun heterodimers with Fos (c-Fos, FosB, Fra-1, and Fra-2) or activation transcription factor (ATF) family proteins (ATF1 to -3, Jun dimerization protein) (17, 18). CREB homo- or heterodimerizes with Fos and ATF members and binds to both CRE and AP-1 elements (19).
AP-1- and CREB-dependent transcriptions are critical for endothelial NOS3 expression in response to prostacyclins, hypoxia, insulin, erythropoietin, and shear stress (16, 20–24); however, their role in endothelial NOS3 expression by angiogenic growth factors has yet to be determined. FGF2 and VEGF are capable of activating both AP-1 and CREB to initiate transcription of angiogenic genes (9, 10, 25, 26). In this study, we hypothesized that FGF2 and VEGF differentially activate AP-1 and/or CREB, thereby leading to differential control of NOS3 expression in placental artery EC. We found that up-regulation of NOS3 mRNA and protein expression by FGF2, but not VEGF, is associated with sustained binding of AP-1 transcription factors to both AP-1 and CRE-like elements in the NOS3 promoter in conjunction with JunB and Fra1 protein up-regulation via an ERK2/1 dependent pathway. JunB and Fra1 down-regulation effectively inhibited the FGF2-induced NOS3 expression and NO production in oFPAEC. Thus, differential activation of AP-1 plays a key role in the differential regulation of NOS3 expression by FGF2 and VEGF.
EXPERIMENTAL PROCEDURES
Cell Culture, Experimental Conditions, and Total Cell Extracts
Three primary oFPAEC lines were isolated by collagenase digestion from second degree placental arteries obtained from late pregnant (day 120–130 of gestation, term ∼145 days) sheep placentas and validated and were cultured and used as described (7). Uterine artery EC (UAEC) were prepared from the same animals as described previously (15). The animal use protocol was approved by the University of California San Diego Animal Subjects Committee, and we followed the National Research Council's Guide for the Care and Use of Laboratory Animals throughout the study. Human umbilical vein EC (HUVEC) were isolated from cords of healthy term placentas using a protocol approved by the Institutional Review Boards of the University of California San Diego. HUVEC were cultured in EBM-2 medium containing BulletKit supplements (Lonza Walkersville, Inc., Walkersville, MD) and used as described previously (27). Following treatment with FGF2 or VEGF, cell lysates were prepared as described previously (28). The protein content was measured by a Bradford procedure using bovine serum albumin as the standard.
SDS-PAGE and Immunoblotting
Protein samples were separated on SDS-PAGE and transferred to polyvinylidene difluoride membranes. Membranes were blocked in 5% nonfat dried milk in 0.05% Tris-buffered saline (TBST) for 1 h and probed in primary antibody overnight at 4 °C. The following antibodies were used: mouse anti-NOS3 monoclonal antibodies (BD Biosciences) at 1:1000; rabbit anti-Fra1 polyclonal antibody and anti-JunB monoclonal antibody at 1:500 (both from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA)); anti-β-actin monoclonal antibody (Ambion, Austin, TX) at 1:10,000; and anti-β-tubulin polyclonal antibody (Cell Signaling, Danvers, MA) at 1:2000. All antibodies were diluted in TBST containing 5% nonfat dry milk. After washing (3 × 10 min) with TBST, the membranes were incubated with peroxidase-conjugated secondary Ab. Blots were visualized using ChemiGlow substrate (Alpha Innotech Corp., San Leandro, CA). Digital images were captured using the Alpha Innotech ChemiImager Imaging System with a CCD camera and quantified using the ChemiImager 4400 software.
RNA Extraction and Real-time Quantitative Reverse Transcription-PCR
Total RNA was extracted with TRIzol (Invitrogen), quantified, and stored at −80 °C until analysis. Total RNA (1 μg) was reverse transcribed for quantifying NOS3 mRNA by real-time PCR exactly as described previously (29). Briefly, all PCRs were run in triplicate with SYBR Green and a master mix containing hot start Taq polymerase (Qiagen, San Diego, CA) and 50 ng of total RNA equivalent/reaction. PCR was run with denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 45 s. The Bio-Rad iCycler equipped with a real-time optical fluorescent detection system was used for SYBR Green detection. Cyclophillin was used as a housekeeping mRNA. Primers used for NOS3 and cyclophillin were exactly the same as described (29), which were designed based on bovine sequences. An artificial 100-base single-stranded DNA standard was used to generate a standard curve for cDNA quantification. Extrapolation of unknowns from the standard curve was performed using Prism 3 (GraphPad Software, San Diego, CA), predicting unknowns from the standard curve Ct values.
To determine the effects of growth factors on NOS3 mRNA stability, actinomycin D chase studies were performed as described previously (30). Briefly, serum-starved subconfluent (∼60%) cells were treated with or without FGF2 (10 ng/ml) for 18 h. Actinomycin D (5 μg/ml) was added to stop transcription. Total RNA samples were harvested at 0, 3, 6, 9, and 12 h for analyzing NOS3 and cyclophillin mRNA levels.
Generation of Sheep NOS3 Promoter-Luciferase Reporter Constructs
We have recently isolated a sheep NOS3 promoter (positioned at −1283 to +22) by PCR and prepared a luciferase reporter construct named 1283pNOS3-pGL3-Luc. This vector was then used to prepare constructs of the NOS3 promoter with various deletions (15). The wild-type sheep NOS3 promoter-driven luciferase reporter construct was used to prepare constructs with mutations of AP-1 and/or CRE elements by site-directed mutagenesis as described previously (15). The mutated AP-1 constructs were with mutations at −680, −681, −682, and −685 (aGAtctA), and the mutated CRE constructs were with mutations at −752, −754, and −758 (cGCGcCg). Double AP-1/CRE-mutated constructs were with same mutations in both AP-1 and CRE elements.
Cell Transfection and Luciferase Assays
Luciferase reporter gene expression assays were performed as described previously (15). Briefly, subconfluent oFPAEC were transfected with vectors using Targefect F-2 reagent (Targeting Systems, San Diego, CA) according to the manufacturer's protocol. Thymidine kinase-Renilla luciferase vector (Promega) was used as the internal control. The transfection was carried out at 37 °C for 4 h. The cells were allowed to recover in MCD131 containing 10% fetal bovine serum for 18–20 h and then treated with starvation medium with or without FGF2 or VEGF (10 ng/ml) for another 24 h. Firefly and Renilla luciferase activities were measured using a dual reporter assay kit (Promega) according to the manufacturer's protocol, and a ratio of the two for each sample was calculated. Each treatment was tested in quadruplicates and repeated three times using different cell preparations.
Nuclear Extract and Electrophoretic Mobility Shift Assay (EMSA)
Serum-starved subconfluent (∼60%) oFPAEC were treated with FGF2 or VEGF for various times. Nuclear extracts were prepared as described previously (15) and kept at −70 °C until use. The sense and antisense sheep NOS3 AP-1 (5′-CCCCAACTTGAGTCACAGGGG-3′, −678 to −685, AP-1 underlined) and “CRE-like” (5′-TGGGGAAGCATGCGTCACTGGATGACA-3′, −752 to −758, CRE underlined) oligonucleotides were biotinylated using a biotin 3′-end DNA labeling kit (Pierce). The biotinylated oligonucleotides were annealed and used as the probe. EMSA was performed with the Pierce LightShift Chemiluminescent EMSA kit. Briefly, nuclear extracts (10 μg) were incubated with the labeled probe in binding buffer (10 mm Tris, 50 mm potassium chloride, 1 mm dithiothreitol, 1 μg of poly(dI-dC), 0.1 mm EDTA, 2.5% glycerol, 5 mm magnesium chloride, 2 μg of bovine serum albumin, total volume of 20 μl) for 20 min at room temperature. Competition was carried out with unlabeled probe for 20 min prior to the addition of biotinylated probes. For supershift assays, 2 μg of the following antibodies (all from Santa Cruz Biotechnology, Inc.) were added in the reaction and incubated at 4 °C for 2 h: anti-c-Jun (SC-45X), anti-JunB (SC-8051X), anti-JunD (SC-74X), anti-c-Fos (SC-52X), anti-FosB (SC-7203X), anti Fra-1 (SC-605X), and anti-Fra-2 (SC-604X). Anti-CREB (catalog no. 9197) and anti-phospho-Ser133-CREB (catalog no. 9191), both from Cell Signaling, were tested at 1 μg/reaction. The binding complexes were separated on 6% non-denaturing PAGE in 0.5× Tris borate/EDTA buffer, transferred to nylon membrane. The membranes were UV cross-linked and probed with an anti-biotin-horseradish peroxidase antibody. The binding complexes were visualized with ChemiGlow substrate and quantified.
Chromatin Immunoprecipitation (ChIP) Assay
The ChIP assay was performed as described previously (31). Cells (∼107/group) were grown to 70% confluence, serum-starved overnight, and treated with growth factors for 3 h. Formaldehyde (1%) was added for cross-linking at 37 °C for 10 min. The cells were collected and resuspended in 0.5 ml of lysis buffer. Sheared DNA was diluted in dilution buffer. 10% of each sample was stored as control input DNA, and the remaining lysates were cleared with 50 μl of protein G-Sepharose for 4 h at 4 °C. Immunoprecipitation (IP) was done with specific anti-JunB or anti-Fra1 antibodies or anti-mouse and rabbit IgGs overnight at 4 °C, followed by incubation with protein G-agarose beads (50 μl) for 4 h at 4 °C. The beads were washed and extracted as described previously (31). Eluates were reverse cross-linked at 65 °C overnight. DNA fragments were purified and analyzed by real-time PCR using a Roche Applied Science LightCycler 1.5 analyzer. A 108-bp fragment containing ovine NOS3 AP-1 site was amplified by using primers of 5′-CTGAATCGCAGCTTCCTG-3′ (−722 to −705) and 5′-GTATTCCATTCCCTCCGG-3′ (−631 to −614), and a 113-bp fragment containing the ovine NOS3 CRE site was amplified by using primers of 5′-TGACCGCTGGGTCTATCTG (−813 to −794) and 5′-AAGGGCAGGAAGCTGCGAT-3′ (−719 to −700) in sheep NOS3 promoter. Melting curves were used to determine appropriate annealing temperatures for AP-1 (53 °C) and CRE (55 °C) real-time analyses. Input DNA was used as internal control, and IgG samples were used as negative controls. Relative levels of DNA were estimated with respect to input DNA.
Overexpression and Down-regulation of AP-1 Subunits
Recombinant adenoviral vectors (25 multiplicity of infection, plaque-forming units/cell) carrying the coding region of c-Jun or JunB in sense or antisense orientations were used to overexpress or repress JunB or c-Jun proteins overnight as described previously (15). Transient transfections of antisense and sense Fra-1 mammalian cytomegalovirus vectors (kind gifts of Dr. Colburn) were used to repress or overexpress Fra-1 as described (32). The cells were serum-starved and then treated with or without FGF2 (10 ng/ml) for another 24 h. Culture media were stored at −20 °C. Cellular proteins were immunoblotted as above.
Determination of Total Nitric Oxide (NOx) Production
Medium samples (100 μl) were analyzed for total NO production (nitrite and nitrate, NOx) by chemiluminescence using a Sievers NO analyzer as described previously (8). Total NOx was calculated by a standard curve generated with sodium nitrate (0.1–10 μm). Data were presented as nmol/mg total protein/24 h.
Effects of AP-1 Decoy Oligodeoxynucleotides (ODN) on NOS3 Expression
Decoy AP-1 ODN was used to determine if FGF2 up-regulates NOS3 via AP-1 in HUVEC. ODN was designed based on the NOS3 promoter surrounding the consensus AP-1 site. The sense sequence of the dumbbell-shaped, phosphorothioated decoy AP-1 ODN (CDODN) used was as follows: 5′-CCCCACTTGAGTCATGGGGGT-3′ (AP-1 is underlined). A scrambled ODN (sense strain 5′-GGATCCATCTCTGCGAAGACG-3′) was used as a control. ODN was annealed and ligated with T4 DNA ligase to generate a covalently ligated dumbbell-shaped ODN molecule. HUVEC (∼60% confluence) were transfected with 100 nm decoy ODN using Fugene 6 (Roche Applied Science). Six hours post-transfection, medium was replaced with fresh EBM2 medium containing FGF2 (10 ng/ml) or phorbol 12-myristate 13-acetate (PMA) (100 nm). After 24 h treatment, cellular proteins were harvested for immunoblotting.
Statistical Analysis
Each experiment was performed three times with cells derived from different pregnant ewes. Data are presented as means ± S.E. and were analyzed by Student's t test or one-way analysis of variance using SigmaStat (Jandel Scientific). When an F test was significant (p < 0.05), treatments were compared with controls by LDS multiple comparisons. Comparisons between treatments (FGF2 versus VEGF) over time were performed using univariate analysis via two-way analysis of variance. p < 0.05 was regarded as significant.
RESULTS
FGF2, but Not VEGF, Trans-activates NOS3 Promoter; Role of ERK2/1
We have shown that FGF2, but not VEGF, increases NOS3 protein and mRNA levels in oFPAEC in a time- and dose-dependent manner (6, 7). Because both de novo mRNA synthesis via transcription and stabilization of mRNA are involved in NOS3 mRNA expression (12), actinomycin D chase studies were performed to test the effects of FGF2 and VEGF on NOS3 mRNA stability. When actinomycin D was added to block de novo NOS3 mRNA synthesis, the levels of NOS3 mRNA decreased in both FGF2-treated and control cells in a time-dependent manner. The half-life of NOS3 mRNA in FGF2-treated cells (10.9 ± 1.2 h) did not differ from that (9.3 ± 0.7 h) in untreated controls (Fig. 1A).
FIGURE 1.
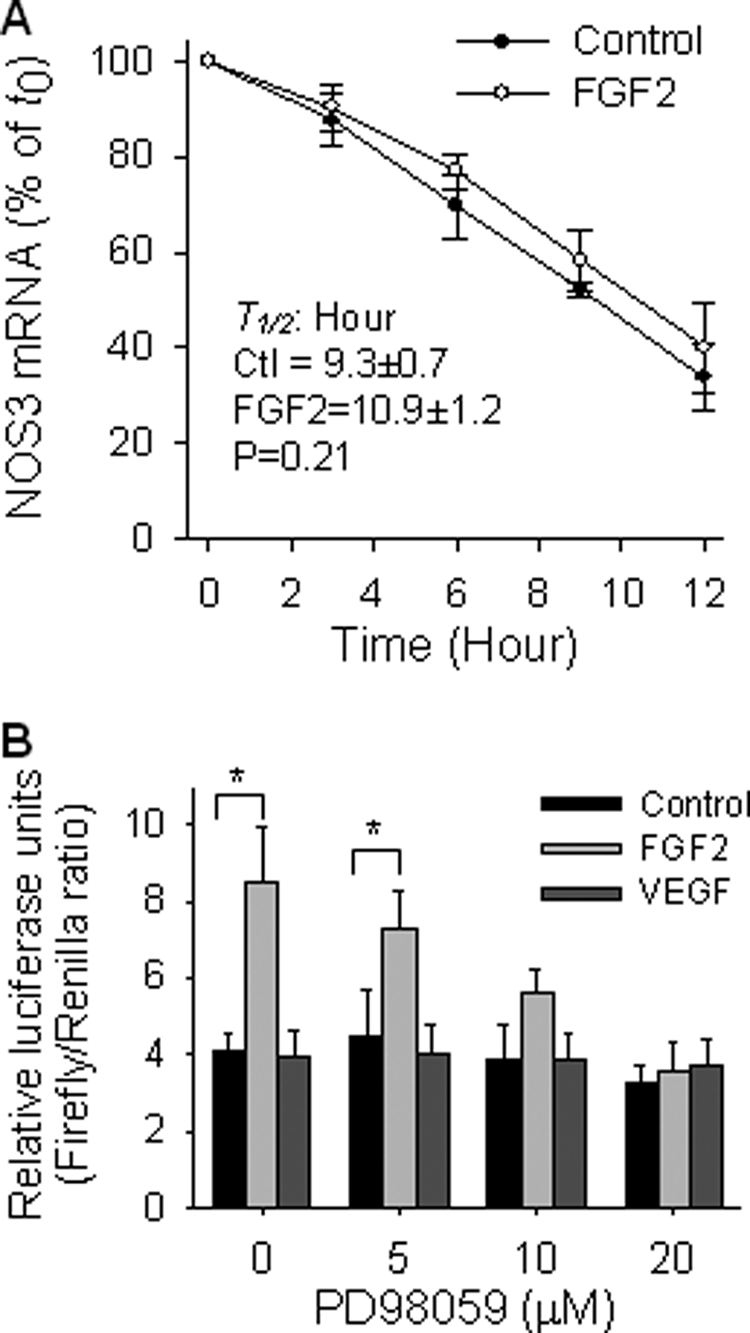
FGF2, but not VEGF, trans-activates NOS3 promoter in oFPAEC. A, quiescent cells were treated with or without FGF2 (10 ng/ml) for 18 h. Actinomycin D (5 μg/ml) was then added into the cultures. Total RNA samples were extracted at 0, 3, 6, 9, and 12 h postactinomycin D. NOS3 and 18 S were analyzed using real time quantitative reverse transcription-PCR. Quantitative data are presented as a percentage of relative NOS3 expression at time 0. Data were summarized as mean ± S.E. from three independent experiments. B, cells were transfected with luciferase construct driven by the wild-type sheep NOS3 promoter (−1283/+22) and co-transfected with the thymidine kinase-Renilla luciferase vector. After treatment with FGF2 or VEGF (10 ng/ml) for an additional 24 h in the presence of various concentrations of PD98059, trans-activation of the NOS3 promoter was measured by luciferase reporter gene expressions as a ratio of firefly/Renilla luciferase activities, as described under “Experimental Procedures.” Quantitative data are expressed as mean ± S.E. from three independent experiments. *, p < 0.05 versus untreated control.
We next determined if FGF2 and/or VEGF trans-activate a luciferase reporter construct driven by a sheep NOS3 promoter (1283 bp) that possesses basal and stimulated NOS3 transcription activities in primary uterine artery EC (15). FGF2, but not VEGF, significantly increased the activity of this reporter construct transfected in oFPAEC. PD98059 dose-dependently inhibited trans-activation of this NOS3 promoter by FGF2; PD98059, at 10–20 μm, which effectively inhibits FGF2- and VEGF-induced ERK2/1 activation (6, 7), completely blocked FGF2-induced NOS3 promoter activity (Fig. 1B).
Both the Consensus AP-1 and the CRE-like Elements of NOS3 Promoter Are Necessary for FGF2 Trans-activation of NOS3 Promoter
We transiently transfected the luciferase reporter constructs driven by the sheep NOS3 1283-bp promoter and its various deletions (15) in oFPAEC to define potential FGF2-inducible transcription factor DNA binding sites. FGF2, but not VEGF, stimulated the 1283- and 757-bp sheep NOS3 promoter activity by ∼2- and 1.71-fold that of controls (p < 0.05; Figs. 1B and 2A). The 757-bp deletion construct resulted in reduced basal NOS3 promoter activity, suggesting that sequence between −757 and −1283 appears to be involved in basal NOS3 expression, consistent with a previous report in human NOS3 promoter (14). FGF2 no longer trans-activated the −636 and −322 bp NOS3 promoter deletion constructs (Fig. 2A). Analyses of sheep NOS3 promoter sequence (−1283 to −636 bp) sensitive to FGF2 stimulation revealed at least two ERK2/1-sensitive sites: a consensus AP-1 site at −685 to −678 (TGAGTCA) and a CRE-like site at −758 to −751 (TGCGTCA). These sites are conserved in other mammalian NOS3 promoters (15, 16).
FIGURE 2.

Effects of FGF2 and VEGF on NOS3 promoter trans-activation potential; role of AP-1 and CRE sites. A, deletion analyses. Cells were transfected with luciferase reporter constructs driven by the wild-type sheep NOS3 promoter (−1283/+22) or its deletions. B, site-directed mutagenesis study. Cells were transfected with the luciferase reporter construct driven by the 1305-bp NOS3 promoter of either wild type (WT) or of mutations in the AP-1, CRE, or both sites. In both panels, cells were also co-transfected with a thymidine kinase-Renilla luciferase vector as internal control. After treatment with FGF2 or VEGF (10 ng/ml) for an additional 24 h, trans-activation of NOS3 promoter was measured. Quantitative data are expressed as mean ± S.E. (error bars) (n = 3) of the ratio of firefly/Renilla luciferase activities from three independent experiments. *, p < 0.05 versus untreated control.
To confirm that trans-activation of NOS3 AP-1- and CRE-like elements is critical for FGF2-induced NOS3 expression, we compared the effects of FGF2 on the trans-activation of the 1283-bp wild-type sheep NOS3 promoter with those of the same promoter but with mutations at the AP-1 and CRE sites. FGF2 increased the activity of the wild-type sheep NOS3 promoter by 90% (p < 0.05). This increase was reduced to 33% when the AP-1 was mutated and to 23% when the CRE was mutated; when both were mutated, FGF2 did not trans-activate the NOS3 promoter (Fig. 2B).
FGF2, but Not VEGF, Increases AP-1 Binding to NOS3 Promoter
We then determined the effects of FGF2 and VEGF on DNA-binding complex formed on the consensus ovine NOS3 promoter AP-1 site. We first confirmed that a well known AP-1 activator PMA induced a strong NOS3 AP-1-DNA complex, which could be blocked with excess unlabeled probe (15). FGF2, but not VEGF, increased AP-1 DNA binding activity in a time-dependent manner. The stimulatory effect of FGF2 on AP-1 binding to the NOS3 AP-1 site maximized at 2 h and was maintained at least up to 6 h post-FGF2 treatment (Fig. 3A).
FIGURE 3.

Effects of FGF2 and VEGF on DNA-protein complex formation at AP-1- and CRE-like sites. Quiescent cells were treated with VEGF or FGF2 (10 ng/ml) for 0, 1, 2, 4, and 6 h, and nuclear extracts were prepared. EMSAs were performed as described under “Experimental Procedures.” Representative EMSA images shown depict one typical experiment for the consensus AP-1 site (A) and the CRE-like site (B). Quantitative data are expressed as the mean ± S.E. (error bars) (n = 3) of -fold untreated (time 0) controls. *, p < 0.05 versus control (time 0). C, quiescent cells were stimulated with forskolin or FGF2 for various times. Total protein samples were analyzed for phosphorylated and total CREB protein levels by immunoblotting.
Treatment with forskolin, presumptively activating protein kinase A, did not increase the CRE binding activity in oFPAEC (Fig. 3B). However, forskolin potently activated CREB within 5–60 min, whereas FGF2 only phosphorylated CREB within 5 min (Fig. 3C). Thus, the inability of forskolin of inducing CRE DNA binding was not due to a deficiency of CREB activation in oFPAEC. In contrast, PMA markedly induced DNA binding on this site. FGF2 and, to a lesser extent, VEGF increased the formation of a CRE-binding complex. FGF2 time-dependently increased the band intensity of a CRE-binding complex, maximizing at 6 h post-FGF2 stimulation in a similar fashion as with the consensus AP-1 site. The maximal response of FGF2-induced CRE binding activity (4.58-fold) was stronger than that of the FGF2-induced AP-1 binding activity. VEGF also significantly increased the CRE binding activity at 1 and 2 h post-stimulation (86 and 98% versus time 0, respectively), which returned to base line at 4–6 h (Fig. 3B).
FGF2, but Not VEGF, Induces JunB and Fra1 Binding to the Consensus AP-1 Site on Sheep NOS3 Promoter
We analyzed the composition of the AP-1-binding complex by EMSA using sheep NOS3 promoter AP-1 probe co-incubated with antibodies against the AP-1 family members of proteins. When specific antibodies against the Jun (c-Jun, JunB, and JunD) or Fos (c-Fos, FosB, Fra-1, and Fra2) family of proteins were added into the EMSA binding reactions of control cells, we observed a “supershift” band co-incubated with either c-Jun, JunB, JunD, or Fra-1, but not with c-Fos, FosB, or Fra-2 antibodies. Treatment with FGF2, but not VEGF, increased the intensity of the supershift band caused by anti-JunB or anti-Fra-1 polyclonal antibodies, indicating that FGF2-induced AP-1 complex contains JunB and Fra-1. More than half of the AP-1-binding complex was supershifted by anti-JunB Ab, indicating JunB to be a major subunit of the AP-1 complex formed under basal conditions and by FGF2 stimulation (Fig. 4A).
FIGURE 4.
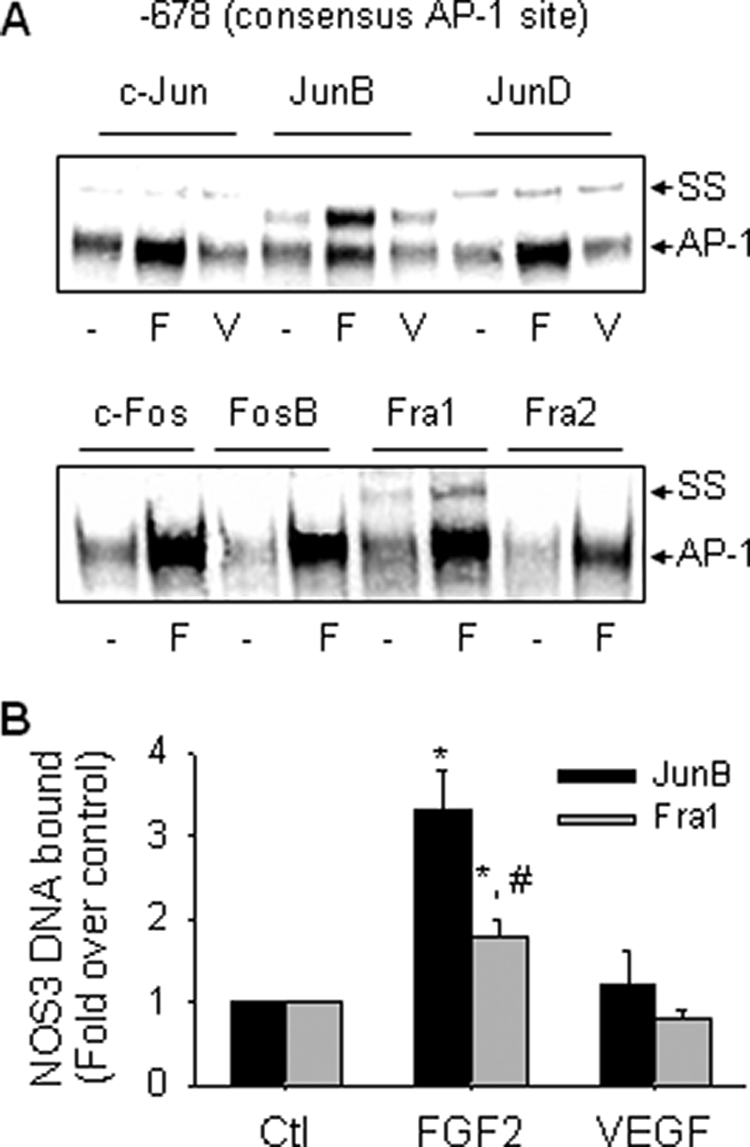
FGF2, but not VEGF, increases JunB/Fra1 binding to the consensus AP-1 site. A, oFPAEC were treated with or without 10 ng/ml FGF2 (F) or VEGF (V) for 3 h, and nuclear extracts were prepared. Supershift assays for AP-1 subunit members using an oligonucleotide probe containing the sheep NOS3 consensus AP-1 element were performed as described under “Experimental Procedures.” Representative “supershift” EMSA images shown depict similar results from three independent experiments. B, oFPAEC were treated with or without 10 ng/ml FGF2 (F) or VEGF (V) for 3 h and then used for ChIP assays. The real-time PCR signals obtained for IP with specific JunB/Fra1 antibodies were estimated with respect to input DNA, and results are expressed as mean ± S.E. (n = 3) of -fold changes over controls. *, p < 0.05 versus control. #, p < 0.05, JunB versus Fra1.
ChIP assays were performed with anti-JunB or anti-Fra-1 antibody to test if FGF2 induced direct AP-1 binding to the proximal sheep NOS3 promoter in intact oFPAEC in situ. In control cells, we observed basal association of JunB and Fra-1 AP-1 with a 108-bp sheep NOS3 promoter PCR product amplified by real-time PCR, which contains the consensus AP-1 site. FGF2, but not VEGF, increased JunB/Fra1 AP-1 binding to the NOS3 promoter region. The PCR product was not amplified in the IgG-immunoprecipitated DNA (Fig. 4B). Of note, NOS3 DNA bound to JunB was significantly greater than that to Fra1.
FGF2, but Not VEGF, Induces JunB and Fra1 Binding to Sheep NOS3 Promoter CRE-like Site
PMA, but not forskolin, trans-activated the sheep NOS3 promoter CRE element, suggesting that it functions more likely as an AP-1 site in oFPAEC (Fig. 3B). We investigated the subunit composition of the transcription factors binding to this element. EMSA with various antibodies against the AP-1 and CREB families of transcription factors was first performed. When specific JunB and Fra-1 Abs were added, we observed a supershift EMSA band in all samples. FGF2, but not VEGF, increased the supershift band caused by co-incubation with antibodies of JunB and Fra-1 but not c-Jun (Fig. 5A). No apparent supershift band was observed with antibodies of other Jun or Fos family proteins (data not shown). These data are consistent with those in Fig. 4A showing that JunB and Fra-1 are the major components of the AP-1 complex induced by FGF2 on the NOS3 promoter AP-1 site. Also of note was that more than half of the complex was supershifted by anti-JunB antibody in all samples, indicating JunB to be a major component of the binding complex formed in oFPAEC under basal conditions and in response to FGF2 stimulation. The addition of either CREB or phospho-CREB antibodies did not result in supershift bands of the CRE-binding complex in EMSA using nuclear proteins from either control or FGF2-, VEGF-, or forskolin-treated oFPAEC (Fig. 5A), consistent with the finding that forskolin did not increase the CRE-binding complex in oFPAEC (Fig. 3B). Thus, the NOS3 promoter CRE-like site preferentially binds JunB and Fra1 in response to FGF2 stimulation.
FIGURE 5.
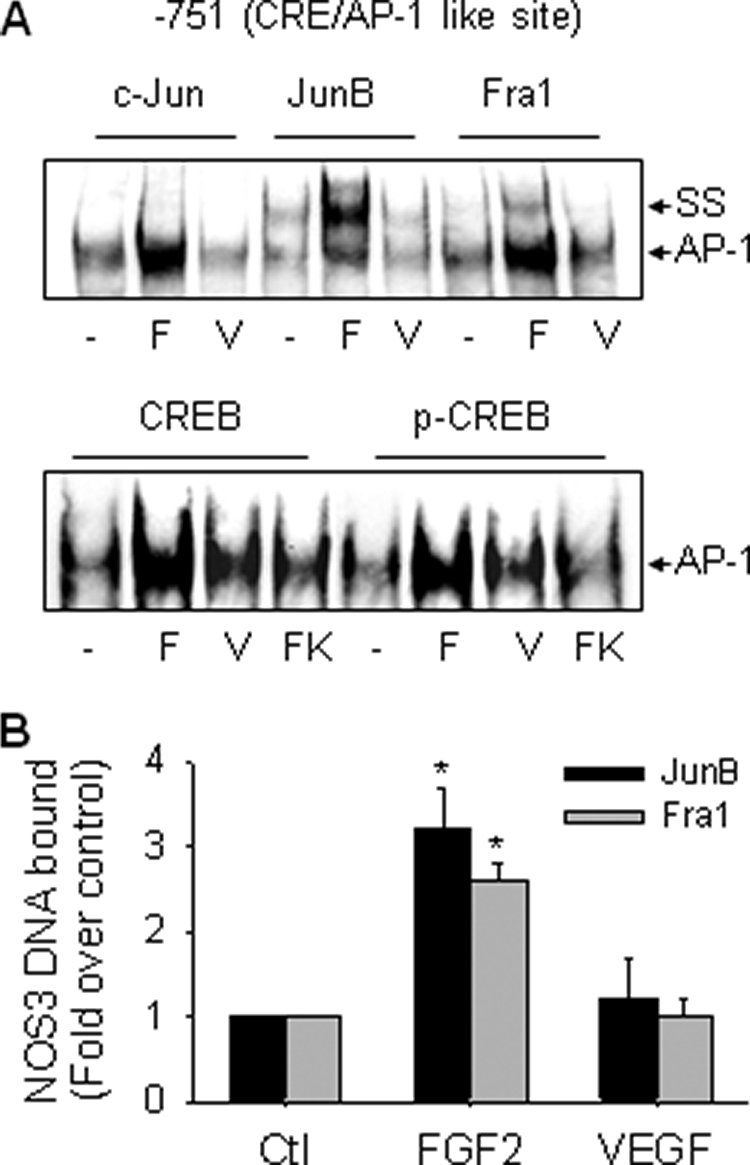
FGF2, but not VEGF, increases JunB/FRA1 binding to the CRE-like site. A, oFPAEC were treated with or without 10 ng/ml FGF2 (F) or VEGF (V) for 3 h. Supershift using an oligonucleotide probe containing the sheep NOS3 promoter CRE-like element was performed as described under “Experimental Procedures.” Representative supershift EMSA images shown depict similar results from three independent experiments for binding of members of the Jun/Fos families and of CREB and phospho-CREB to the CRE-like element. B, oFPAEC were treated with or without 10 ng/ml FGF2 or VEGF for 2 h and then used for ChIP assays. The real-time PCR signals obtained for IP with specific JunB/Fra1 antibodies were estimated with respect to input DNA, and results are expressed as mean ± S.E. (error bars) (n = 3) of -fold changes over controls (Ctl). *, p < 0.05 versus control.
ChIP analyses with anti-JunB and anti-Fra1 antibodies were then performed to solidify this notion. Basal binding of JunB and Fra-1 proteins to a 113-bp real-time PCR product of the proximal sheep NOS3 promoter containing the CRE-like site was found in control cells. FGF2, but not VEGF, increased JunB and Fra1 association with this element (Fig. 5B). CREB or phospho-CREB binding to this site was not found in intact oFPAEC by ChIP assays (data not shown). In contrast to the AP-1 element, FGF2 stimulated the binding of JunB and Fra-1 to the CRE-like element with comparable intensities. Increased Fra-1 binding to CRE-like elements many cause increased AP-1 binding to the NOS3 CRE elements, as shown in Fig. 3B.
FGF2, but Not VEGF, Up-regulates c-Jun, JunB, and Fra1 Expression in oFPAEC; Role of ERK2/1
AP-1 mediated trans-activation of gene expression occurs via de novo synthesis of AP-1 proteins or post-transcriptional modifications of the preexisting AP-1 components (17, 18). We recently showed that FGF2 induces potent and sustained (up to 12 h) activation of ERK2/1 in oFPAEC (7), raising a question as to whether sustained ERK2/1 activation up-regulates AP-1 protein expression, which in turn trans-activates the NOS3 promoter AP-1 site. High levels of basal c-Jun, JunB, and JunD proteins and lower levels of basal c-Fos and Fra-1 proteins, but not Fos-B and Fra-2, were readily detectable in oFPAEC. FGF2, but not VEGF, significantly increased the levels of c-Jun, JunB, and Fra-1 proteins in a time-dependent manner. JunB, c-Jun, and Fra-1 proteins were significantly increased at 2 h and sustained up to 24 h; however, significant differences exist in their time courses. JunB and c-Jun proteins peaked between 4 and 8 h, whereas Fra-1 expression increased constantly over time with the highest at 24 h. JunB protein expression was mostly responsive to FGF2, with a ∼5-fold increase above base line at 4–8 h, maintained at high levels up to 24 h. VEGF only modestly increased c-Jun and JunB proteins at early times, and the effects were not significant (Fig. 6).
FIGURE 6.

Effects of FGF2 and VEGF on c-Jun, JunB, and Fra-1 protein expression in oFPAEC. Cells were treated with each growth factor (10 ng/ml) for 0, 2, 4, 8, 12, and 24 h and then harvested for immunoblotting of c-Jun (A), JunB (B), and Fra-1 (C) proteins. Representative blots shown depict one typical experiment. Quantitative data are expressed as the mean ± S.E. (error bars) (n = 3) of -fold control (untreated) value. *, p < 0.05 versus control.
Activation of MAPK, including ERK2/1 and JNK1/2, plays a key role in up-regulating AP-1 proteins by a variety of extracellular stimuli (17). Because the time courses of FGF2 induction of c-Jun, JunB, and Fra-1 proteins were similar to that of ERK2/1 activation by FGF2 and sustained ERK2/1 activation was required for FGF2-induced NOS3 protein expression (7), we studied the effects of ERK2/1 inhibition on FGF2 induction of JunB, c-Jun, and Fra-1 protein expression. Blockade of ERK2/1 activation by PD98059 inhibited FGF2-induced JunB and Fra-1, but not c-Jun, protein expression (p < 0.05). However, when PD98059 was given 30 min or 1 h after FGF2 stimulation, only JunB induction was attenuated. FGF2 induction of Fra-1 was only partially blocked by PD98059 when given 1 h post-FGF2 stimulation (Fig. 7). Thus, JunB was the FGF2-induced AP-1 component most sensitive to inhibition of sustained ERK2/1 activation. Fra-1 protein expression by FGF2 is possibly mediated by acute ERK2/1 activation. FGF2 induction of c-Jun protein was not affected by inhibition of ERK2/1 activation alone, suggesting that other pathways activated by FGF2, such as JNK1/2 and AKT1 (7), may be involved in this process in oFPAEC.
FIGURE 7.
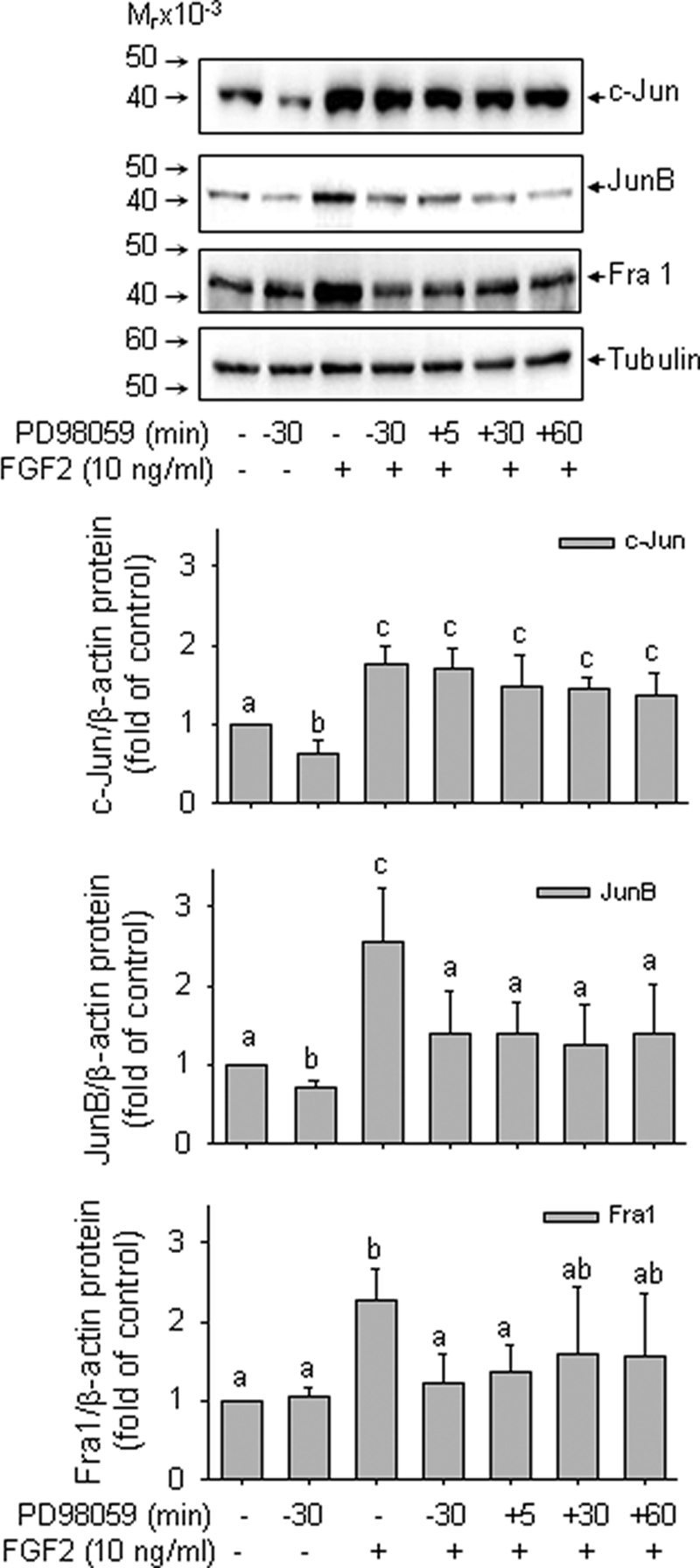
Role of ERK2/1 in FGF2 induction of AP-1 components in oFPAEC. Serum-starved cells were either pretreated with PD98058 (10 μm) for 30 min followed by FGF2 (10 ng/ml) or treated first with FGF2 and then with PD98058 at 5, 30, and 60 min post-FGF2 treatment. Protein samples were harvested 2 h after FGF2 stimulation and analyzed for c-Jun, JunB, and Fra-1 proteins. Representative blots shown depict one typical experiment. Bar graphs summarize data (means ± S.E. (error bars)) of -fold control values from three independent experiments. Bars with different letters (a versus b versus c) differ significantly (p < 0.05), and bars with two letters do not differ from bars with either letter.
FGF2 Stimulation of NOS3 Expression Requires JunB and Fra1 in oFPAEC
Infection with sense JunB adenovirus increased JunB protein levels by >10-fold over base line, whereas infection with the antisense JunB adenovirus decreased JunB protein by ∼50% compared to that of base line and also blocked FGF2-induced increases in JunB protein levels. Infection with the empty adenovirus did not alter JunB expression. Compared with controls, infection with sense JunB adenovirus significantly increased the levels of NOS3 protein. JunB overexpression did not further increase NOS3 expression in FGF2-treated cells. However, infection of antisense JunB adenovirus decreased the FGF2-induced NOS3 protein by 45% (p < 0.05). Concomitant infection with sense and antisense JunB adenoviruses blocked FGF2-induced NOS3 protein expression (Fig. 8A). Infection with antisense c-Jun adenovirus did not inhibit basal or FGF2-induced NOS3 protein expression. Infection with sense c-Jun adenovirus increased basal NOS3 protein expression by 197% compared with controls (p < 0.05). Moreover, FGF2 induced 170% increase in NOS3 protein expression, which was further significantly increased to 373% of basal level by c-Jun overexpression (Fig. 8B).
FIGURE 8.

FGF2-increased NOS3 up-regulation requires JunB and Fra1 but not c-Jun. A, cells were infected with or without empty, antisense (AS), or sense (S) JunB adenoviruses for 18 h. After recovery in complete culture medium, cells were starved in the presence or absence of FGF2 (10 ng/ml) for 24 h. B, cells were treated similarly to A but with sense and/or antisense c-Jun adenoviruses. C, cells were treated similarly to A but with sense and/or antisense Fra1-expressing cytomegalovirus constructs. For all treatments, total protein samples were analyzed for JunB, c-Jun, Fra1, NOS3, and β-actin levels. Representative blots shown depict a typical experiment for each panel. Bar graphs summarize data (means ± S.E. (error bars)) of -fold control values from three independent experiments. Bars with different letters (a versus b versus c) differ significantly (p < 0.05), and bars with two letters do not differ from bars with either letter.
Transfection of antisense Fra1 vector decreased Fra1 expression by 40% of basal levels, whereas sense Fra1 vector increased Fra1 protein by 2-fold. Contrasting to c-Jun/JunB overexpression, increased Fra1 expression did not alter basal NOS3 levels (Fig. 8C). However, transfection of antisense Fra1 without or with sense Fra1 decreased FGF2 induction of NOS3 expression by 20 and 35%, respectively (p < 0.05). Thus, similar to antisense JunB, antisense Fra1 can inhibit NOS3 expression, although the former had a stronger inhibitory effect on FGF2 induction of NOS3 in oFPAEC.
In comparison with empty adenovirus infections, co-infections of sense and antisense Jun adenoviruses led to >80% reduction of Jun overexpression by sense Jun adenoviruses alone; co-infections still resulted in greater Jun protein levels, similar to those of FGF2 stimulation. However, co-infection of sense/antisense Jun adenoviruses did not enhance endothelial nitric-oxide synthase protein expression similarly as did FGF2. These data indicate that additional mechanisms other than Jun/AP-1 expression, possibly linked to protein kinase-dependent Jun phosphorylation/activation by FGF2 (33), might be involved in FGF2 induction of endothelial nitric-oxide synthase protein expression.
FGF2 Increases NO Production in oFPAEC; Role of JunB, Fra1, and ERK2/1
We tested whether changes observed in NOS3 expression by FGF2 affect NO production in oFPAEC. Treatment with FGF2 for 24 h stimulated a 2-fold increase in NOx production compared with controls (Fig. 9). JunB down-regulation inhibited FGF2 stimulation of NOx production by 95% (p < 0.05), and JunB overexpression increased NO production by 2.07-fold (p < 0.05). However, JunB overexpression did not further increase FGF2-stimulated NO production. Fra1 down-regulation decreased FGF2-induced NOx production by 30% (p < 0.05). Fra1 overexpression did not alter basal or FGF2-induced NOx production. PD98059 inhibited basal and FGF2-induced NOx production by 46% (p < 0.05) and 94% (p < 0.05), respectively (Fig. 9).
FIGURE 9.

FGF2 increases total NOx levels in oFPAEC via JunB and ERK. Subconfluent and quiescent oFPAEC were pretreated with or without JunB adenoviruses, as shown in Fig. 8A, and Fra-1 vectors, as in Fig. 8C, or in the presence or absence of PD98059 (10 μm). The cells were then treated with FGF2 (10 ng/ml) for 24 h. Culture media were sampled for analyses of NOx production by chemiluminescence using an NO Sievers analyzer. Data were calculated by a standard curve generated by sodium nitrate. Bars with different letters (a versus b versus c versus d) differ significantly (p < 0.05), and bars with two letters do not differ from bars with either letter. Error bars, S.E.
Effects of AP-1 Inhibition on FGF2 Induction of NOS3 in HUVEC and UAEC
We investigated if FGF2 stimulates NOS3 protein expression and if AP-1 has a role in this process in other EC. Similarly to oFPAEC, FGF2 and the AP-1 activator PMA stimulated NOS3 protein expression in HUVEC (Fig. 10) and UAEC (15). When transfected with the AP-1 CDODN, but not the scrambled ODN, presumptively “trapping” the AP-1 proteins (34), FGF2 and PMA did not stimulate NOS3 protein expression in HUVEC (Fig. 10). FGF2 also stimulated NOS3 protein expression in UAEC. Treatment with a selective pharmacological AP-1 inhibitor curcumin dose-dependently inhibited FGF2-induction of NOS3 protein expression in oFPAEC, HUVEC, and UAEC (supplemental Fig. S1).
FIGURE 10.
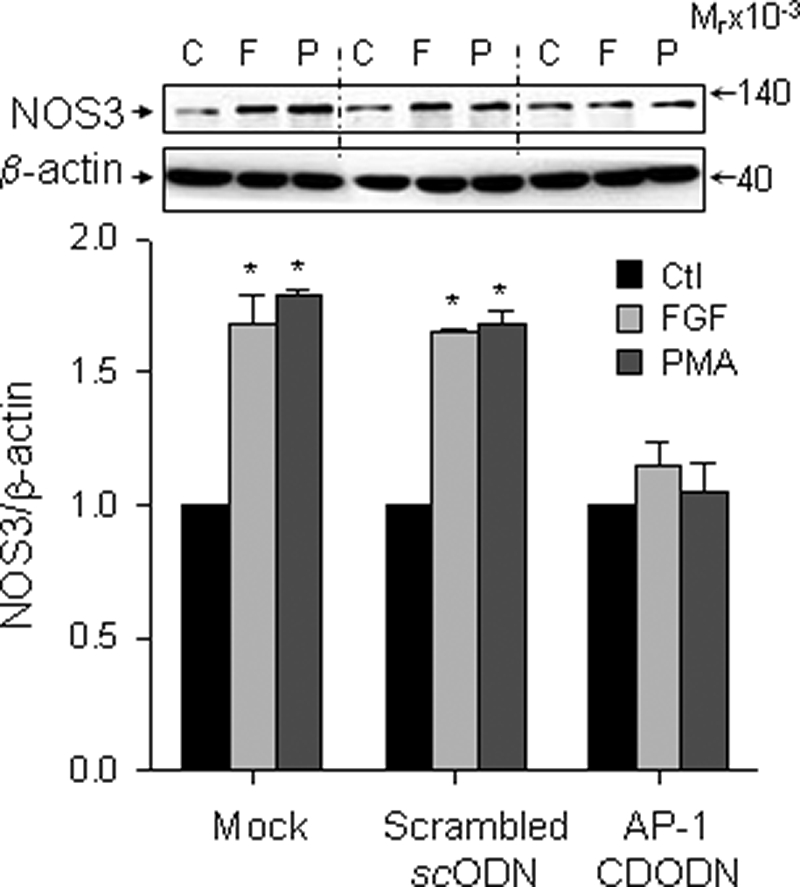
FGF2 up-regulates NOS3 expression in HUVEC via AP-1. Subconfluent HUVEC were transfected with a decoy AP-1 CDODN or scrambled ODN and then treated without (C) or with FGF2 (10 ng/ml; F) or PMA (100 nm; P) for 24 h. Cellular proteins were harvested for immunoblotting of NOS3 and β-actin. Representative blots of one typical experiment shown depict three independent experiments using HUVEC from different placentas. Quantitative data are expressed as the mean ± S.E. (n = 3) of -fold control (Ctl) (untreated) value. *, p < 0.05 versus controls. Error bars, S.E.
DISCUSSION
We have shown that FGF2 stimulation of NOS3 mRNA/protein expression is associated with potent trans-activation of the NOS3 promoter without significant mRNA degradation in oFPAEC. These data suggest that de novo mRNA synthesis via activation of transcription largely accounts for FGF2-induced NOS3 expression. Expression analyses of luciferase reporter constructs driven by sheep 1283-bp NOS3 promoter and its various deletions reveal that the proximal AP-1 and CRE-like elements are the primary cis-elements responsible for NOS3 expression by FGF2 in oFPAEC. This notion is strongly supported by the findings that trans-activation of the wild-type NOS3 promoter-driven luciferase construct by FGF2 was lost when both sites were deleted or mutated.
Our current study has established a critical role of AP-1 in mediating FGF2 induction of NOS3 expression in EC as evidenced by the following: 1) FGF2 stimulates endothelial nitric-oxide synthase expression in various types of EC, which is attenuated by AP-1 inhibition; 2) FGF2 stimulates time-dependent increases in NOS3 promoter AP-1 DNA binding activity; 3) FGF2-induced AP-1-DNA complex consists of JunB and Fra1; and 4) FGF2 stimulates JunB/AP-1 binding to NOS3 promoter in intact oFPAEC in situ. Other AP-1 subunits, such as c-Jun, JunD, c-Fos, FosB, and Fra2, seem not to play a major role. These data further solidify a critical role of AP-1 in regulating NOS3 expression, as reported in uterine artery EC expressing c-Jun (15) and in other EC stimulated by various extracellular stimuli (21–23).
Adjacent to the AP-1, a CRE-like element (-TGCGTCA-, −752 to −758) in the NOS3 promoter is identified as a CREB binding site for mediating NOS3 expression by prostacyclin (16) and hypoxia (24) in bovine aortic EC. This site is also involved in FGF2 induction of NOS3 expression in oFPAEC; however, it seems not to function as a classical CREB but rather an AP-1 site in mediating FGF2 induction of NOS3 transcription due to the following reasons. First, the CRE differs by only one base pair from AP-1, providing a structural base for it as AP-1. Second, forskolin dramatically activates CREB; however, it does not increase the NOS3 CRE DNA binding. Third, PMA markedly increases the CRE DNA binding. Fourth, FGF2 increases the CRE DNA binding; however, the binding complex does not contain CREB but instead contains JunB and Fra1. Interestingly, VEGF stimulates the CRE DNA binding; however, the response is relatively short (<1 h). This does not suffice to initiate NOS3 transcription in intact cells because of the following. 1) VEGF does not stimulate the direct association of JunB and/or Fra1 at the CRE site as measured by ChIP assays. 2) VEGF does not stimulate the DNA binding on the consensus AP-1, nor does it increase NOS3 mRNA and protein expression in oFPAEC.
The role of ERK2/1 in NOS3 expression seems to vary among the stimuli and EC surveyed. In oFPAEC, FGF2 and angiotensin II up-regulate NOS3 via ERK2/1 activation (6, 35); however, shear stress-induced NOS3 does require ERK2/1 activation (36). Of specific interest to angiogenic growth factors, we have shown previously that FGF2 and VEGF activate ERK2/1; however, different signal intensities and duration of FGF2 and VEGF-induced ERK2/1 activation may differentiate the effects of the two growth factors on NOS3 expression in oFPAEC (6–8). FGF2 and VEGF rapidly stimulate ERK2/1 nuclear translocation (6). FGF2, but not VEGF, trans-activates the NOS3 promoter via an AP-1-dependent mechanism, which is attenuated by PD98059. Collectively, these data have shown a critical role of the ERK2/1-AP-1 pathway in the FGF2-induced NOS3 transcription in placental EC. This concept is well in line with previous studies showing a critical role of AP-1 in NOS3 up-regulation by insulin in bovine aortic EC (22) and by shear stress in fetal pulmonary artery EC (23) and that in both cases NOS3 up-regulation is sensitive to ERK2/1 inhibition. Our conclusion is further solidified by the following findings. First, FGF2 increased JunB/Fra1 AP-1-binding complexes to NOS3 promoter AP-1- and CRE-like sites. Second, FGF2 stimulates JunB and Fra1 expressions, which are sensitive to PD98059. Third, JunB or c-Jun overexpression increases NOS3 expression. Fourth, down-regulation of JunB and Fra1 significantly lowers FGF2-induced NOS3 expression.
In keeping with our recent findings that FGF2 induces sustained activation of ERK2/1 in oFPAEC (17, 18, 33), FGF2 rapidly initiates and induces sustained up-regulation of AP-1 proteins. Different AP-1 subunits undergo similar but not identical regulation by angiogenic growth factors. JunB expression is most sensitive to FGF2 stimulation, followed by c-Jun, and JunD does not respond to FGF2. The expression of the Fos family members is also differentially modulated by FGF2. FGF2 rapidly increases Fra1 expression but does not stimulate c-Fos expression. Fra1 expression by FGF2 lasts for at least 24 h and is sensitive to ERK2/1 inhibition. These data agree with previous studies showing that Fra-1 is highly inducible by ERK2/1 activation as compared with c-Fos (32, 37) and that insulin stimulates sustained (up to 24 h) activation of AP-1 factors, which contain mainly Fra-1 in CHO cells (32, 38). In addition to increased expression of AP-1 proteins, post-transcriptional regulation of preexisting AP-1 proteins, such as phosphorylation by ERK2/1, JNK1/2, and phosphatidylinositol 3-kinase/Akt (32, 33), also plays a critical role in AP-1 dependent transcription (17, 18). In addition to ERK2/1, activation of JNK1/2 and phosphatidylinositol 3-kinase/Akt is also required for NOS3 expression by FGF2 (7). Further studies are needed to delineate whether these pathways are involved in NOS3 expression in EC.
Up-regulation of multiple AP-1 subunits by FGF2 in oFPAEC also raises the following critical question. What is the specific role that each AP-1 component plays in FGF2-induced placental endothelial NOS3 expression? FGF2 stimulation of JunB seems necessary and sufficient for subsequent NOS3 up-regulation and NO production in oFPAEC because they are reversed by JunB down-regulation. The inability of c-Jun down-regulation to inhibit FGF2-induced NOS3 expression may also support the importance and specificity of JunB in FGF2-induced NOS3 expression. However, c-Jun overexpression results in greater NOS3 protein level than that of JunB overexpression. Only overexpression of c-Jun, and not of Jun-B or Fra1, enhanced FGF2-induced NOS3 protein expression. Thus, c-Jun and JunB may both up-regulate basal NOS3 expression, but only c-Jun mediates FGF2 effects. These findings confirm both redundancy and specific activities of c-Jun and JunB as described in other systems (17, 36). For example, c-Jun deletion leads to liver and heart defects in mice; “knock-in” of JunB alone rescues c-Jun-null mice from embryonic death and liver failure (39). It is speculated that embryonic lethality of c-Jun-null mice is due to the inability of c-Jun target tissues to express sufficient quantities of other AP-1 members capable of regulating similar sets of genes (39). Similarly, it is possible that a clear role of JunB and a minor role of Fra1 in FGF2 up-regulation of NOS3 probably originated from JunB and Fra1 being the most highly inducible AP-1 subunits by FGF2 in oFPAEC.
JunB functions as either trans-activator or trans-repressor, depending on the promoter of a given target gene (18, 33). JunB antagonizes c-Jun trans-activation in promoters that express one single AP-1 element like cyclin D1, whereas it trans-activates promoters that possess multiple AP-1 sites (40, 41). In oFPAEC, we have shown that JunB binds to both AP-1 and CRE-like elements, consistent with the fact that JunB trans-activates promoters with more than one element for binding JunB. The trans-activation potential of c-Jun and JunB also depends on the AP-1 and CRE flanking sequences and its dimerization partner (33, 42). For instance, c-Jun/ATF2 dimers strongly activate the c-Jun promoter containing two CRE sites while weakly activating cyclin D promoter that contains an AP-1 and a CRE site, and the contrary is true for a tethered c-Jun/c-Fos dimer (43).
Fra1 seems to play a supportive role in FGF2 up-regulation of oFPAE cell NOS3. FGF2 induced only Fra1, and not c-Fos, in oFPAEC. Fra1 and Fra2 do not possess a trans-activation domain, and they presumptively repress the transcriptional activity of c-Fos and Fos-B (37). However, they bind to Jun proteins and stabilize the heterodimer to prevent the degradation of Jun members (37). Fra1 overexpression did not increase basal levels of NOS3, as did JunB and c-Jun overexpressions in oFPAEC. Fra1 requires stronger ERK2/1 activation to activate gene expression (32). Fra1 overexpression does not increase basal NOS3 expression, potentially because basal ERK2/1 activation does not suffice to activate Fra1.
In sum, JunB and Fra1/AP-1 play a major role in FGF2 up-regulation of NOS3 in oFPAEC, and sustained ERK activation is a key upstream route in the differential up-regulation of NOS3 by FGF2 and VEGF. We conclude that differential activation of AP-1 plays a key role in the differential regulation of NOS3 expression by FGF2 and VEGF in endothelial cells.
Supplementary Material
Acknowledgments
We thank Drs. Arlin Blood and Shannon Bragg from Loma Linda University for assistance in the use of the Sievers NO Analyzer. We also thank Dr. Nancy Colburn (NCI, National Institutes of Health) for providing the sense and antisense cytomegalovirus-Fra1 vectors.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 HL64703 (to J. Z.) and RO1 HL74947 and HL70562 (to D.-b. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- VEGF
- vascular endothelial growth factor
- oFPAEC
- ovine fetal placental artery endothelial cell(s)
- NO
- nitric oxide
- CRE
- cyclic AMP-response element
- CREB
- CRE-binding protein
- ATF
- activation transcription factor
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated protein kinase
- JNK
- Jun N-terminal kinase
- EMSA
- electrophoretic mobility shift assay
- ChIP
- chromatin immunoprecipitation
- ODN
- oligodeoxynucleotide
- EC
- endothelial cell(s)
- UAEC
- uterine artery endothelial cell(s)
- HUVEC
- human umbilical vein endothelial cell(s)
- Ab
- antibody
- IP
- immunoprecipitation
- NOx
- total nitric oxide
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1.Redman C. W., Sargent I. L. (2005) Science 308, 1592–1594 [DOI] [PubMed] [Google Scholar]
- 2.Borowicz P. P., Arnold D. R., Johnson M. L., Grazul-Bilska A. T., Redmer D. A., Reynolds L. P. (2007) Biol. Reprod. 76, 259–267 [DOI] [PubMed] [Google Scholar]
- 3.Zheng J., Bird I. M., Chen D. B., Magness R. R. (2005) J. Physiol. 565, 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson S. H., Steinsland O. S., Wang Y., Yallampalli C., Dong Y. L., Sanchez J. M. (2000) Circ. Res. 87, 406–411 [DOI] [PubMed] [Google Scholar]
- 5.Reynolds L. P., Caton J. S., Redmer D. A., Grazul-Bilska A. T., Vonnahme K. A., Borowicz P. P., Luther J. S., Wallace J. M., Wu G., Spencer T. E. (2006) J. Physiol. 572, 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zheng J., Bird I. M., Melsaether A. N., Magness R. R. (1999) Endocrinology 140, 1399–1407 [DOI] [PubMed] [Google Scholar]
- 7.Mata-Greenwood E., Liao W. X., Zheng J., Chen D. B. (2008) Placenta 29, 708–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng J., Wen Y., Song Y., Wang K., Chen D. B., Magness R. R. (2008) Biol. Reprod. 78, 143–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powers C. J., McLeskey S. W., Wellstein A. (2000) Endocr. Relat. Cancer 7, 165–197 [DOI] [PubMed] [Google Scholar]
- 10.Chang F., Steelman L. S., Lee J. T., Shelton J. G., Navolanic P. M., Blalock W. L., Franklin R. A., McCubrey J. A. (2003) Leukemia 17, 1263–1293 [DOI] [PubMed] [Google Scholar]
- 11.Fish J. E., Marsden P. A. (2006) Cell Mol. Life Sci. 63, 144–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Searles C. D. (2006) Am. J. Physiol. Cell Physiol. 291, C803–C816 [DOI] [PubMed] [Google Scholar]
- 13.Zhang R., Min W., Sessa W. C. (1995) J. Biol. Chem. 270, 15320–15326 [DOI] [PubMed] [Google Scholar]
- 14.Karantzoulis-Fegaras F., Antoniou H., Lai S. L., Kulkarni G., D'Abreo C., Wong G. K., Miller T. L., Chan Y., Atkins J., Wang Y., Marsden P. A. (1999) J. Biol. Chem. 274, 3076–3093 [DOI] [PubMed] [Google Scholar]
- 15.Qian X. X., Mata-Greenwood E., Liao W. X., Zhang H., Zheng J., Chen D. B. (2007) Mol. Cell. Endocrinol. 279, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niwano K., Arai M., Tomaru K., Uchiyama T., Ohyama Y., Kurabayashi M. (2003) Circ. Res. 93, 523–530 [DOI] [PubMed] [Google Scholar]
- 17.Shaulian E., Karin M. (2002) Nat. Cell Biol. 4, E131–E136 [DOI] [PubMed] [Google Scholar]
- 18.Hess J., Angel P., Schorpp-Kistner M. (2004) J. Cell Sci. 117, 5965–5973 [DOI] [PubMed] [Google Scholar]
- 19.Sands W. A., Palmer T. M. (2008) Cell. Signal. 20, 460–466 [DOI] [PubMed] [Google Scholar]
- 20.Hoffmann A., Gloe T., Pohl U. (2001) J. Cell. Physiol. 188, 33–44 [DOI] [PubMed] [Google Scholar]
- 21.Rui T., Feng Q., Lei M., Peng T., Zhang J., Xu M., Abel E. D., Xenocostas A., Kvietys P. R. (2005) Cardiovasc. Res. 65, 719–727 [DOI] [PubMed] [Google Scholar]
- 22.Fisslthaler B., Benzing T., Busse R., Fleming I. (2003) Nitric Oxide 8, 253–261 [DOI] [PubMed] [Google Scholar]
- 23.Wedgwood S., Mitchell C. J., Fineman J. R., Black S. M. (2003) Am. J. Physiol. Lung Cell Mol. Physiol. 284, L650–L662 [DOI] [PubMed] [Google Scholar]
- 24.Min J., Jin Y. M., Moon J. S., Sung M. S., Jo S. A., Jo I. (2006) Hypertension 47, 1189–1196 [DOI] [PubMed] [Google Scholar]
- 25.Varghese S., Rydziel S., Canalis E. (2000) Endocrinology 141, 2185–2191 [DOI] [PubMed] [Google Scholar]
- 26.Mohan R., Sivak J., Ashton P., Russo L. A., Pham B. Q., Kasahara N., Raizman M. B., Fini M. E. (2000) J. Biol. Chem. 275, 10405–10412 [DOI] [PubMed] [Google Scholar]
- 27.Liao W. X., Feng L., Zhang H., Zheng J., Moore T. R., Chen D. B. (2009) Mol. Endocrinol. 23, 1428–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao W. X., Magness R. R., Chen D. B. (2005) Biol. Reprod. 72, 530–537 [DOI] [PubMed] [Google Scholar]
- 29.Ducsay C. A., Hyatt K., Mlynarczyk M., Kaushal K. M., Myers D. A. (2006) Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R1406–R1413 [DOI] [PubMed] [Google Scholar]
- 30.Yoshizumi M., Perrella M. A., Burnett J. C., Jr., Lee M. E. (1993) Circ. Res. 73, 205–209 [DOI] [PubMed] [Google Scholar]
- 31.Shang Y., Myers M., Brown M. (2002) Mol. Cell 9, 601–610 [DOI] [PubMed] [Google Scholar]
- 32.Young M. R., Nair R., Bucheimer N., Tulsian P., Brown N., Chapp C., Hsu T. C., Colburn N. H. (2002) Mol. Cell. Biol. 22, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mechta-Grigoriou F., Gerald D., Yaniv M. (2001) Oncogene 20, 2378–2389 [DOI] [PubMed] [Google Scholar]
- 34.Bielinska A., Shivdasani R. A., Zhang L. Q., Nabel G. J. (1990) Science 250, 997–1000 [DOI] [PubMed] [Google Scholar]
- 35.Zheng J., Wen Y., Chen D. B., Bird I. M., Magness R. R. (2005) Biol. Reprod. 72, 1421–1428 [DOI] [PubMed] [Google Scholar]
- 36.Li Y., Zheng J., Bird I. M., Magness R. R. (2004) Biol. Reprod. 70, 785–796 [DOI] [PubMed] [Google Scholar]
- 37.Milde-Langosch K. (2005) Eur. J. Cancer 41, 2449–2461 [DOI] [PubMed] [Google Scholar]
- 38.Hurd T. W., Culbert A. A., Webster K. J., Tavaré J. M. (2002) Biochem. J. 368, 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Passegué E., Jochum W., Behrens A., Ricci R., Wagner E. F. (2002) Nat. Genet. 30, 158–166 [DOI] [PubMed] [Google Scholar]
- 40.Bakiri L., Lallemand D., Bossy-Wetzel E., Yaniv M. (2000) EMBO J. 19, 2056–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu J. C., Cressman D. E., Taub R. (1993) Cancer Res. 53, 3789–3794 [PubMed] [Google Scholar]
- 42.Deng T., Karin M. (1993) Genes Dev. 7, 479–490 [DOI] [PubMed] [Google Scholar]
- 43.Bakiri L., Matsuo K., Wisniewska M., Wagner E. F., Yaniv M. (2002) Mol. Cell. Biol. 22, 4952–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.