

Abstract
Phosphorylation of the C-terminal end of histone H2A.X is the most characterized histone post-translational modification in DNA double-stranded breaks (DSB). DNA-dependent protein kinase (DNA-PK) is one of the three phosphatidylinositol 3 kinase-like family of kinase members that is known to phosphorylate histone H2A.X during DNA DSB repair. There is a growing body of evidence supporting a role for histone acetylation in DNA DSB repair, but the mechanism or the causative relation remains largely unknown. Using bacterially expressed recombinant mutants and stably and transiently transfected cell lines, we find that DNA-PK can phosphorylate Thr-136 in addition to Ser-139 both in vitro and in vivo. Furthermore, the phosphorylation reaction is not inhibited by the presence of H1, which in itself is a substrate of the reaction. We also show that, in contrast to previous reports, the ability of the enzyme to phosphorylate these residues is not affected by the extent of acetylation of the core histones. In vitro assembled nucleosomes and HeLa S3 native oligonucleosomes consisting of non-acetylated and acetylated histones are equally phosphorylated by DNA-PK. We demonstrate that the apparent differences in the extent of phosphorylation previously observed can be accounted for by the differential chromatin solubility under the MgCl2 concentrations required for the phosphorylation reaction in vitro. Finally, we show that although H2A.X does not affect nucleosome conformation, it has a de-stabilizing effect that is enhanced by the DNA-PK-mediated phosphorylation and results in an impaired histone H1 binding.
Keywords: Chromatin, Chromatin Histone Modification, Chromatin Regulation, DNA Damage, Histone Modification, Histones, DNA-PK
Introduction
DNA double-stranded break (DSB)3 repair is an important cellular process that is critical for the maintenance of genome integrity. If improperly repaired, DSBs can have detrimental consequences leading to cancer via chromosomal aberrations such as chromosomal breaks, translocation, and aneuploidy (1).
In the eukaryotic cell the repair mechanism requires the participation of chromatin components. One of the chromatin hallmarks involved is the phosphorylation of histone H2A.X at its highly conserved C-terminal SQE motif (Fig. 1A) (2–4). This phosphorylated form of H2A.X is commonly denoted as γ-H2A.X (2). It is well known that histone H2A.X is phosphorylated ubiquitously at DNA DSB regardless of the source of the damage and of the pathway used in its repair: homologous recombination or non-homologous-end joining (NHEJ). The enzymes responsible for the DSB-induced H2A.X phosphorylation are all members of the PIKK family, such as the DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia mutated (ATM) and ATM and Rad3-related (ATR) (4, 5). The DNA-PK catalytic subunit has been recently shown to play a dominant role in regulating the DNA damage-mediated phosphorylation of H2A.X (6).
FIGURE 1.

Phosphorylation of human H2A.X-Thr-136 by DNA-PK in vitro. A, logos representation of the alignment of the last C-terminal 12 amino acids of histone H2A.X from different vertebrates is shown (Homo sapiens (NP_002096.1), Macaca mulatta (XP_001101949.1), Canis familiaris (XP_853256.1), Mus musculus (NP_034566.1), Gallus gallus (NP_002096.1), Anolis carolinensis (AAWZ01044401), Xenopus tropicalis (NP_001015968.1), Rana catesbeiana (ACO51989.1), Danio rerio (NP_957367.1), and Salmo salar (ACI66032.1)). B, DNA-PK phosphorylates both Ser-139 and Thr-136 of human recombinant H2A.X in vitro. The left panel corresponds to a radioactive DNA-PK reaction, carried out with purified H2A.X mutants H2A.X (lane 1), S139A (lane 2), and T136A/S139E (lane 3) as substrates. Artemis (Ar) was used as a substrate for DNA-PK to serve as a positive control. Proteins were resolved in SDS-PAGE followed by autoradiographic analysis. The right panel shows an SDS-PAGE analysis of H2A.X (lane 1), S139A (lane 2), and T136A/S139E (lane 3). CM is a chicken erythrocyte histone marker. The bottom panel is a schematic representation of the different H2A.X mutants used in the radioactive DNA-PK reaction with the mutations highlighted at the C-terminal tail. C, shown is a deconvoluted nanospray mass spectrum of human recombinant H2A.X phosphorylated with DNA-PK.
In the yeast H2A.X ortholog, Thr-126 and Ser-129 can be both phosphorylated during DSB in a way that appears to depend on the repair pathway. It has been shown that Thr-126 is important for homologous recombination but dispensable for NHEJ (7). The early identification of γ-H2A.X in vertebrates (2) hinted to the possibility that Ser/Thr-136 could also be phosphorylated. However, experimental evidence for this suggestion has been lacking. Determining whether or not this residue is also phosphorylated is important as, in contrast to yeast where homologous recombination is the preferred mechanism of DSB repair, mammalians preferentially use NHEJ (8) where DNA-PK plays a critical role (9).
In addition to the phosphorylation of histone H2A.X, there is a growing body of evidence supporting a role for histone acetylation in DNA DSB repair. Cells with DNA breaks have a transient increase in histone H3 and H4 acetylation (10). Defects in the acetylation of K56 in histone H3 result in sensitivity to genotoxic agents that cause DNA DSBs during replication (11). It has been found that mutations of multiple acetylate-able lysine residues in the H4 tail (12) or mutations of the TIP60 (human histone acetyltransferase complex that mediates H4 acetylation) (13, 14) and NuA4 complexes (yeast homologue of TIP60) (15) confer sensitivity to agents that create DSBs. Although the acetylation of histones is one of the most studied histone modifications and evidence has been provided that histone acetylation enhances phosphorylation of H2A.X by DNA-PK (16), the concerted structural effect, if any, of this histone post-translational modification with that of γ-H2A.X remains to be elucidated.
The structural and functional involvement of this highly characteristic H2A.X phosphorylation is not clearly understood. It is yet unclear whether it elicits a change in chromatin structure allowing for access of DNA repair machineries or if it merely acts as part of the “histone code” that recruits such repair machineries (4, 17). These two possibilities need not necessarily be mutually exclusive. As an example of the latter, it has been shown that γ-H2A.X promotes rapid Rad9 (radiation-sensitive 9) recruitment to DSBs in yeast (18). A direct role on chromatin conformation was suggested by the decrease exerted in its compaction by the serine/glutamic phosphorylation mimics of yeast H2A.X at Ser-129 (19, 20). Arguing against this and against a general role for the C-terminal tail of H2A.X, it was recently shown that yeast Ser/Glu mutants had no effect on chromatin stability and supercoiling or on nucleosome positioning (21).
Here, we show that mammalian Ser/Thr-136 and Ser-139 of mammalian H2A.X are both substrates of DNA-PK and are phosphorylated within the cell context. Acetylation of core histones does not influence the ability of the phosphorylating enzymes to phosphorylate H2A.X and neither does the presence or absence of linker histones. Finally, we observe that H2A.X slightly de-stabilizes the nucleosome and its DNA-PK- mediated phosphorylation impairs linker histone binding.
EXPERIMENTAL PROCEDURES
Histone Plasmid Constructs
A plasmid containing human H2A.X prepared as in Siino et al. (22) was kindly provided to us by Joe Siino, and it was subcloned into a pET11a bacterial expression plasmid. Similar expression vectors consisting of the mutant forms H2A.X-S139A, H2A.X-T136A/S139E, and H2A.X-A138E/S139E were prepared using the appropriate 3′ end primers encompassing these mutations. Constructs containing GFP at the N-terminal end of H2A.X and its mutants were prepared by inserting the coding regions of the previous plasmids into a pEGFP-C1 mammalian expression vector (BD Biosciences Clontech, San Jose, CA).
Histone Expression and Purification
Recombinant human H2A.X and all mutants were expressed in Escherichia coli BL21 (DE3) cells. The bacterial pellet was homogenized with a Dounce homogenizer 6 m guanidinium hydrochloride, 1 mm EDTA, 1 mm dithiothreitol, and 50 mm Tris-HCl (pH 7.5) buffer. The cell lysate was dialyzed against 2 liters of 0.1 m NaCl, 50 mm Tris-HCl (pH 7.5), and 1 mm EDTA buffer for 2 h at 4 °C. The recombinant histone was extracted by 0.5 n HCl. The solubilized histones were precipitated overnight at −20 °C with 6 volumes of cold acetone, pelleted, dried, and resuspended in water. The HCl-extracted histones were further purified using a Macro-Prep CM cation exchange resin (Bio-Rad) with a 0–1 m NaCl gradient in 7 m urea, 10 mm NaOAc (pH 5.2), 5 mm β-mercaptoethanol, and 1 mm EDTA buffer. The eluted histone was dialyzed, lyophilized, and further purified by reverse phase high performance liquid chromatography (HPLC) using a Vydac C4 column as described in Ausió and Moore (23). Native histone H2A-H2B dimmers, H3-H4 tetramers, and histone H1 were purified by a 0–2.5 m NaCl gradient hydroxylapatite fractionation of HeLa S3 (24). Histone H2B was isolated from histone H2A by Vydac C4 reverse phase HPLC as below.
DNA Fragments
A 146-bp random sequence DNA was obtained from chicken erythrocyte nucleosome core particles prepared as described elsewhere (25). A sequence-defined 208-bp DNA fragment was obtained by RsaI digestion of a 208-12 DNA construct consisting of 12 identical copies of a 208-bp fragment of the 5 S rRNA gene of the sea urchin Lytechinus variegatus (26). After digestion with RsaI, the 208-bp DNA was HPLC-purified using a 75 × 7.5-mm Bio-Gel DEAE-5-PW (Bio-Rad) in 0–1 m NaCl gradient in 100 mm Tris-HCl (pH 7.5) buffer.
Antibodies
An antibody against γ-H2A.X was prepared in house (SCL). H2A.X antibody was purchased from Applied Biological Materials (abm, Richmond, BC). GFP and FLAG antibodies were purchased from Abcam (Cambridge, MA) and from Sigma, respectively.
Transient and Stable Transfections
The H2A.X/T136A/T136A-S139E GFP constructs were transfected into HeLa cells using Polyfect Transfection Reagent (Qiagen, Mississauga, ON). Transient transfected HeLa cells were cultured at 37 °C, 5% CO2 and collected after incubating for 24 h. H2A.X/T136A/T136A-S139E with an N-terminal FLAG tag was cloned into the pcDNA 3.1 (−) mammalian expression vector. The H2A.X/T136A/T136A-S139E FLAG constructs were transfected into H2A.X−/− mouse embryonic fibroblasts (MEFs) (kindly provided to us by Andre Nussenzweig) (27). Transfection was carried out using Effectene transfection reagent (Qiagen). The transfected H2A.X−/− MEFs were cultured at 37 °C, 5% CO2, and after 24 h of incubation media were replaced with fresh media containing 800 μg/ml Geneticin. The media were replaced again after 1 week with fresh media containing 400 μg/ml Geneticin. After 1 week of incubation, stably transfected MEFs were then collected, and expression of the protein constructs were confirmed by Western blot analysis using GFP and H2A.X antibodies. Proper incorporation into nucleosomes of the histone constructs expressed in transiently and stably transfected cells was carried out as described elsewhere (28).
Cell Irradiation
HeLa cells or stably transfected MEF cells were subjected to 10 gray irradiation at room temperature using a Varian 6EX medical linear accelerator (Varian Medical Systems, Palo Alto, CA). A calibrated 6 mV x-ray photon beam was used. The beam was directed upwards through a 5-cm-thick slab of water-equivalent material and then through the cell culture dishes to provide uniform irradiation of the cells. A field size of 28 × 28 cm2 was used, and the cells were at a distance of 100 cm from the x-ray target. Maximum uncertainty in the radiation dose was estimated to be ±5%.
Chromatin Preparation from Butyrate-treated and Non-treated HeLa S3 Cells
HeLa S3 cells grown in the presence or the absence of 5 mm sodium butyrate, and nuclei and chromatin fractions were prepared after the protocol previously described (29). In brief, nuclei from butyrate-treated and non-treated HeLa S3 cells were digested with micrococcal nuclease (Worthington Biochemical Corp., Lakewood, NJ). The digestion was carried out at 30 units/mg of DNA for 7 min at 37 °C in 50 mm NaCl, 10 mm Pipes (pH 6.8), 5 mm MgCl2, and 1 mm CaCl2 buffer (with or without 5 mm sodium butyrate). The supernatant obtained after centrifugation for 10 min at 10,000 × g (4 °C) was designated as fraction S1. The pellet was then resuspended and lysed in 0.25 mm EDTA (pH 7.5) to produce, after subsequent centrifugation, a supernatant and a pellet fraction. Stripping of histone H1 from the supernatant fraction was accomplished by treatment with CM-Sephadex C-25 resin in the presence of 0.35 m NaCl as described elsewhere (25). All the buffers used for the chromatin preparation of butyrate-treated HeLa S3 cells contained 5 mm sodium butyrate.
Nucleosome Reconstitution
Recombinant human H2A.X/A138E-S139E or H2A.X phosphorylated with DNA-PK (P-H2A.X) were mixed with HeLa S3 H2B, H3, and H4 histones and reconstituted onto nucleosomes as described previously (23). Histones in stoichiometric amounts were lyophilized and resuspended at 2 mg/ml in a 6 m guanidinium-hydrochloride, 20 mm β-mercaptoethanol, and 50 mm Tris-HCl (pH 7.5) buffer. After incubation for 30 min at room temperature, the samples were dialyzed against distilled and sterilized H2O for 2 h at 4 °C. The dialysis bag was then transferred to another container containing 2 m NaCl, 50 mm Tris-HCl (pH 7.5), 1 mm EDTA, and 1 mm dithiothreitol buffer and dialyzed overnight at 4 °C. For the reconstitution of nucleosomes consisting of cold DNA, the histone mixture prepared was mixed with either 146- or 208-bp DNA at a histone to DNA ratio of (1.12:1 w:w) or (0.79:1 w:w), respectively, in 2 m NaCl, 50 mm Tris-HCl (pH 7.5), and 0.1 mm EDTA buffer. For the [γ-32P]ATP-labeled nucleosome reconstitution, [γ-32P]ATP-labeled 208-bp DNA was mixed with an ∼10-fold amount of cold 208-bp DNA and mixed with histones at a (0.79:1 w:w) histone:DNA ratio in the same 2 m NaCl buffer (30). In all instances nucleosome reconstitution was achieved by a 2 to 0 m NaCl stepwise salt gradient dialysis in 10 mm Tris-HCl (pH 7.5) and 0.1 mm EDTA buffer as described (23). Nucleosomes reconstituted in this way were purified as described below and used for subsequent experiments: H1 binding assay, DNA-PK reaction, and analytical ultracentrifuge analysis.
Sucrose Gradient Purification of Reconstituted Nucleosomes and Native Nucleosomes Isolated from HeLa S3 Cells
Nucleosomes in 10 mm Tris-HCl (pH 7.5) and 0.1 mm EDTA were loaded onto a 5–20% sucrose gradient in 25 mm NaCl, 10 mm Tris-HCl (pH 7.5), and 0.1 mm EDTA buffer and centrifuged at 111,081 × g for 19.6 h in a Beckman SW41Ti rotor at 4 °C. Fractions (0.5 ml) were collected and analyzed by 4% native PAGE.
Gel Electrophoresis and Western Blots
SDS-PAGE was performed as described (31). Native 4% acrylamide PAGE was carried out according to Yager and van Holde (32). Acetic acid/urea gel electrophoresis was performed as previously described (33). Western blots were carried out as described previously (34, 35). H2A.X, γ-H2A.X, GFP, and FLAG antibodies were used at 1:5000; 1:3000; 1:1000, and 1:5000 dilutions, respectively.
Histone H1 Binding
Purified H2A.X/A138E-S139E or H2A.X phosphorylated with DNA-PK (P-H2A.X) nucleosomes (300 ng) were titrated with increasing amounts of HeLa S3 histone H1. The H1 to mononucleosomes molar ratios used were: 0, 0.25, 0.5, 1.0, 1.5, and 2.0. The H1 binding reaction was performed in 50 mm NaCl, 2 mm EDTA, and 20 mm Tris-HCl (pH 7.5) buffer. The mixture was then incubated at room temperature for 30 min. The electrophoretic mobility shift resulting from histone H1 binding was analyzed by agarose gel electrophoresis in 45 mm Tris-HCl (pH 7.5), 45 mm boric acid, and 1 mm EDTA (0.5 × TBE).
Analytical Ultracentrifuge
Nucleosomes reconstituted with 208- or 146-bp DNA and histone octamers consisting of H2A.X or γ-H2A.X were dialyzed against buffers of varying ionic strength (0, 0.1, 0.2, 0.4, and 0.6 m NaCl) and analyzed by sedimentation velocity using a Beckman XL-I analytical ultracentrifuge (Beckman-Coulter, Fullerton, CA). Samples were loaded in double-sector cells with aluminum-filled Epon centerpieces, and runs were performed at 20 °C on an An-55 Al aluminum rotor at 40,000 rpm. UV scans were collected at 260 nm and analyzed by the van Holde and Weischet method (36) using the XL-A Ultra Scan Version 9.3 (37) sedimentation data software (Borries Demeler, Missoula, MT). A value of 0.650 cm3/g was used for the partial specific volume of the nucleosome (25).
DNA-PK Assay
The DNA-PK catalytic subunit and Ku70/80 subunits of DNA-PK were purified from HeLa cells as described previously (38). Purified DNA-PK catalytic subunit (38.1 ng) and Ku (11.9 ng) were incubated in 35 μl of reaction mixture containing 50 mm Tris-HCl (pH 8.0), 4 or 10 mm MgCl2 (buffer), 10 μg/ml sonicated calf thymus DNA, and 0.25 mm ATP containing ∼10 μCi of [γ-32P]ATP. The substrates (histones/nucleosomes) (0.5 μg or 1 μg) were added as indicated. The reaction mixture was incubated for 15 min at 30 °C. Reactions were terminated by the addition of SDS loading buffer for SDS-PAGE or by loading immediately after the reaction for 4% native gel. To prepare γ-H2A.X on a large scale, the reaction was performed in the same way as above with the exception of scaling up substrates and enzymes proportionally and using cold ATP instead of radioactive [γ-32P]ATP. After the reaction, P-H2A.X was filtered through a 0.45-μm Nanosep centrifugal device (Pall, Ann Arbor, MI) and purified with a 4.6 × 250-mm Vydac C18 reverse phase HPLC column (Grace Vydac, St. Hesperia, CA) using a gradient from 0.1% trifluoroacetic acid to 0% acetonitrile (ACN) to 60% ACN in 60 min.
Mass Spectrometry
DNA-PK-phosphorylated H2A.X was analyzed by static manual nanospray using an Applied Biosystems/MDS Sciex QStar Pulsar I fitted with a Protana/Proxeon Nanospray source. Two microliters of DNA-PK-phosphorylated H2A.X were mixed with 2 μl of 60% methanol, 3% formic acid and pipetted into a fused silica gold/palladium-coated nanoES spray tip (Proxeon). Spray was established by applying a tip voltage of 1200 V and collecting a time-of-flight mass spectrometry survey scan 400–1500 m/z consisting of 300 accumulated 1-s scans. Mass spectrometer parameters used were as follows: declustering potential setting of 65, focusing potential setting of 265, and curtain gas setting of 25. The time-of-flight mass spectrometry spectrum was deconvoluted using Analyst QS 1.1 (MDS Sciex) using the Biotools Bayesian reconstruct software. Processing settings were as follows: start mass of 10,000 Da, stop mass of 20,000 Da, step mass 1 Da, signal/noise threshold of 20, minimum intensity of 3%, hydrogen adduct selected, and 20 iterations were used to generate the parent mass of the charged protein envelope.
Magnesium Chloride Titration
Chromatin solubility analysis in the presence of MgCl2 (39) was carried out in 50 mm Tris-HCl (pH 7.5) buffer. The chromatin samples had an approximate A260 = 0.8. The different MgCl2 concentrations were achieved by mixing equal volumes of the chromatin sample in the 50 mm Tris-HCl (pH 7.5) buffer with an equal volume of 2 × MgCl2 in the same buffer while vortexing. The sample mixtures were incubated for 1 h at 4 °C. The chromatin aggregates were centrifuged at 16,000 × g. The absorbance of the supernatant at 260 nm was measured and related to the absorbance of the starting sample in the absence of MgCl2.
RESULTS
Mammalian H2A.X Thr-136 and Ser-139 Are Both Phosphorylated in Vitro and in Vivo
Phosphorylation of mammalian H2A.X of the SQE motif at the C-terminal end of vertebrate H2A.X (Ser-139 in mammals) represents one of the best characterized post-translational modification landmarks associated with double-strand DNA repair. However, other phosphorylatable residues (Ser/Thr) are present in close proximity of the SQE motif targeted by the ATM/ATR and DNA-PK enzymes in many different species. In many vertebrates the SQE sequence is preceded by another potential DNA-PK target motif ((S/T)Q) that is highly conserved (Fig. 1A). Although the potential for phosphorylation of this secondary residue within γ-H2A.X was hinted in the early paper from Bonner and co-workers (2), the demonstration of its existence has remained elusive.
To test whether this site is amenable to DNA-PK phosphorylation in vitro, several human recombinant H2A.X histone versions were created in which the phosphorylatable residues of this histone C-terminal end (TQASQEY) were individually or globally mutated to non-phosphorylatable amino acids and subjected to DNA-PK phosphorylation (Fig. 1B). As can be seen, the TQA motif preceding SQE can be equally phosphorylated by the enzyme, albeit with lower activity. From here on the in vitro double-phosphorylated form of H2A.X will be referred to as P-H2A.X to distinguish it from the in vivo phosphorylated form referred to as γ-H2A.X (2).
The ability of the TQA motif to be phosphorylated in vivo was assessed using HeLa cells transfected with GFP versions of the same constructs used for the in vitro characterization. As shown in Fig. 2A, TQA is phosphorylated in non-irradiated transiently transfected HeLa cells, although to a lesser extent than the SQE counterpart. Fig. 2B shows the reactivity of the γ-H2A.X antibody for DNA-PK-phosphorylated versions of the different constructs. The slightly different reactivity of the antibody toward H2A.X selectively phosphorylated at Thr-136 can in part account for the lower phosphorylation observed with the corresponding transfected version. Irradiation of these transiently transfected cells resulted in the complete disappearance of the H2A.X-GFP constructs (results not shown), most likely as a result of DNA damage impinged by the radiation on the expressing plasmids.
FIGURE 2.

Phosphorylation of human H2A.X-Thr-136 in vivo. A, HeLa cells were transiently transfected with GFP-H2A.X constructs, H2A.X (lane 1), S139A (lane 2), and T136A/S139E (lane 3). The presence of phosphorylated H2A.X and transfected GFP-H2A.X were identified by Western blot using antibody against γ-H2A.X (upper panel) and GFP (lower panel), respectively. Non-transfected HeLa cells (lane H) were used as a marker for endogenous H2A.X. B, H2A.X (lane 1), T136A (lane 2), S139A (lane 3), and T136A/S139E (lane 4) phosphorylated by DNA-PK in vitro were analyzed by Western blot using antibody against γ-H2A.X. The top panel corresponds to SDS-PAGE, and the bottom panel corresponds to the Western blot showing that the γ-H2A.X antibody is able to detect both phosphorylated Thr-136 and phosphorylated Ser-139. CM is chicken erythrocyte histones, used as a marker. C, Western blot analysis of different chromatin fractions (S1, supernatant (SE), and pellet (P)) obtained from HeLa S3 cells using antibodies against γ-H2A.X, H2A.X, and H4 (as a loading control) before and after ionizing irradiation is shown. D, MEF H2A.X−/− cells were stably transfected with different H2A.X mutant constructs and subjected to 10 gray ionizing irradiation. The presence of phosphorylated H2A.X mutants and transfected FLAG-tagged H2A.X mutants in different chromatin fractions (S1, SE, and P) were detected by Western blot using γ-H2A.X (upper panel) and FLAG (lower panel) antibodies. The middle panel shows an SDS-PAGE analysis of the protein loadings for each lane.
Given our inability to recover the transiently expressed GFP-versions of H2A.X after irradiation, stable versions of the different FLAG-H2A.X mutants were produced in MEF H2A.X−/− cells (27), and the chromatin from these cells was digested with micrococcal nuclease and fractionated as described elsewhere (40). Interestingly, native canonical H2A.X in HeLa cells exhibit an uneven distribution in this fractionation that is more pronounced in the case of γ-H2A.X in irradiated cells and that fractionates with the highly insoluble P fraction in a way that is highly dependent on irradiation (see Fig. 2C). Such a fraction may consist of a reversible association of the many large assemblies (including chromatin) that participates in the repair process. This is probably due to the rearrangement of γ-H2A.X from small microfoci into larger foci that takes place in the nucleus of cultured mammalian cells upon irradiation (41, 42).
After irradiation, γ-H2A.X of the MEF H2A.X−/− cells stably transfected with the canonical version of H2A.X exhibits the same distribution. Interestingly, a phosphorylated version of the H2A.X-139A mutant could be observed in the same cells when transfected with this construct. Albeit the signal was, not unexpectedly, low (due in part to the lesser efficiency of the antibody, see above) it was detectable only in the chromatin fraction S.E., which has been ascribed to facultative heterochromatin (40, 43). These results are in good agreement with a recent phospho-proteomic analysis that provided evidence for ionizing irradiation-induced phosphorylation of histone H2A.X on Thr-136 (44).
The DNA-PK-dependent Phosphorylation of H2A.X Is Not Affected by Histone Acetylation
A trinucleosome fraction was prepared from HeLa cells that had been grown in the presence or in the absence of sodium butyrate to increase the extent of histone acetylation. Native and acetylated trinucleosomes containing or lacking histone H1 were prepared by sucrose gradient fractionation before or after extraction of the histone H1 complement. The DNA and histone composition of these fractions is shown in Fig. 3, A and B. The samples, thus, obtained were phosphorylated by DNA-PK in the presence of either 4 or 10 mm MgCl2 (Fig. 3, C and D). As can be seen in Fig. 3C, native and acetylated nucleosomes in the absence of H1 (N and A) exhibit an almost identical extent of phosphorylation that increases slightly when the magnesium concentration was increased from 4 to 10 mm. However, in the presence of histone H1 (NH, AH), acetylated nucleosomes are more phosphorylated than their native counterpart in both 4 and 10 mm MgCl2.
FIGURE 3.

Histone acetylation and H2A.X phosphorylation. Oligonucleosome DNA and proteins were resolved in 1% agarose (A) and in SDS-PAGE (B). Radioactive DNA-PK reaction was carried out with oligonucleosomes as substrates. Labeled nucleosomes and proteins were resolved in 4% native PAGE (C) and in SDS-PAGE (D). Artemis was used as a substrate to serve as a positive control. E, solubility of native and acetylated oligonucleosomes with histone H1 in different concentrations of MgCl2 is shown. N, native oligonucleosome; A, acetylated oligonucleosome; NH, native oligonucleosome with H1; AH, acetylated nucleosomes with H1; SE, supernatant; T, tri-nucleosome; D, di-nucleosome; M, mono-nucleosome.
This enhanced DNA-PK phosphorylation of H1-containing acetylated oligonucleosomes contrasts with the results observed when H1 is not present and is in agreement with previously published results (16). However, when a MgCl2 solubility analysis was carried out with these samples, the results shown in Fig. 3E suggest that the increase in phosphorylation observed in the presence of H1 may simply reflect an increase in MgCl2 solubility as the H1-containing native trinucleosomes are almost completely insoluble at 4 and 10 mm. DNA-PK activity is impaired when the reaction mixture contains less than 4 mm MgCl2. Therefore, levels of H2A.X phosphorylation were assessed at this magnesium concentration for the highest amount of soluble chromatin possible while maintaining DNA-PK activity. The phosphorylation experiment was also performed at 10 mm MgCl2 to better compare our data with the result obtained by Park et al. (16).
DNA-PK Can Phosphorylate Histone H1 in Vitro and in a Chromatin Template
In the course of the phosphorylation assays carried out with oligonucleosomes containing H1, we noticed that a band with an electrophoretic mobility corresponding to that of histone H1 was phosphorylated in addition to H2A.X (Fig. 3D). The same result was obtained when a mononucleosome fraction was used (Fig. 4, lanes 1 and 2). To corroborate that indeed H1 was being phosphorylated, purified HeLa H1 and H3-H4 as well as recombinant H2A.X were phosphorylated in vitro using DNA-PK. The results shown in Fig. 4, lanes 3–5, demonstrate that although the phosphorylation reaction is highly specific for H2A.X, histone H1, albeit to a lower extent, is also a substrate for DNA-PK.
FIGURE 4.
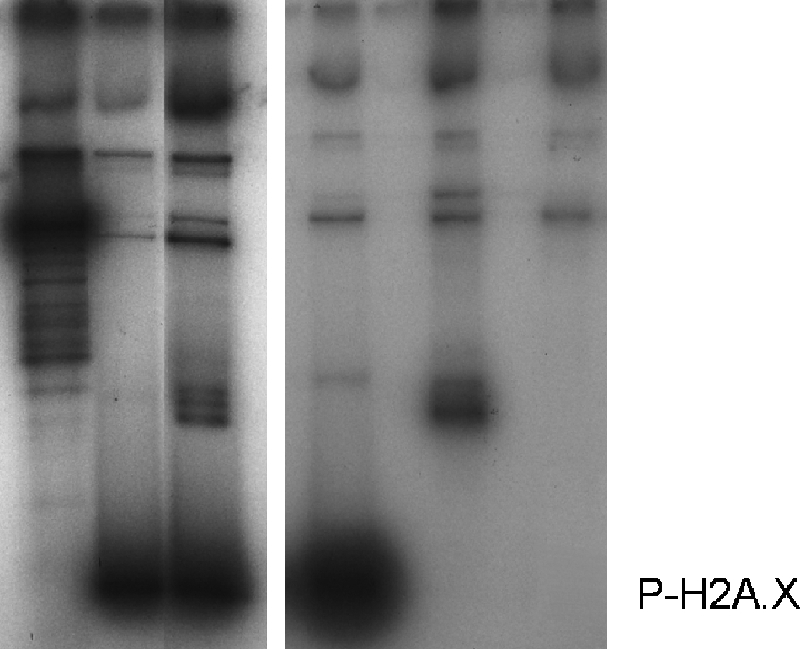
DNA-PK phosphorylates linker histones in vitro and in a nucleosome setting. Lanes 1 and 2 show an autoradiograph of an SDS-PAGE of HeLa mononucleosomes lacking (lane 1) or containing histone H1 (lane 2) upon phosphorylation by DNA-PK using [γ-32P]ATP. Lanes 3–5 show an autoradiograph of an SDS-PAGE of purified HeLa histone H2A.X, histone H1, and H3-H4 tetramers phosphorylated by DNA-PK using [γ-32P]ATP. Artemis (Ar) was used as a substrate to serve as a positive control for these reactions. The high molecular weight bands correspond to autophosphorylated DNA-PK subunits, the DNA-PK catalytic subunit, and Ku 70/80.
Histone H2A.X Decreases the Stability of the Nucleosome and Alters Histone H1 Binding
Next we wanted to look at the potential structural effects of the DNA-PK-mediated phosphorylation of H2A.X on nucleosomes. Fig. 5A shows a native PAGE of native NCPs and NCPs reconstituted with native core histones and recombinant H2A.X and DNA-PK-phosphorylated H2A.X (P-H2A.X) and 146-bp DNA with a random sequence. These particles were then analyzed in the analytical ultracentrifuge using sedimentation velocity (45). The results are shown in Fig. 5, B and C. Native NCPs exhibit a homogeneous sedimentation coefficient distribution with the main peak centered at about 12 S at low salt and at around 10.5 S at 0.6 m NaCl as it has been previously described (25). At NaCl concentrations equal to or above 0.4 m NaCl, minor peaks with smaller sedimentation coefficients are observed that correspond to NCPs with different extents of dissociation. In addition, a peak sedimenting at ∼6 S corresponds to naked (fully dissociated) DNA. In contrast, H2A.X NCPs exhibit a much more unstable pattern where a substantially large part of the NCPs is fully dissociated by 0.6 m NaCl. A higher dissociation was observed when H2A.X was fully phosphorylated. Furthermore, the s20,w values of the undissociated NCP fraction at each NaCl concentration analyzed were slightly lower than those of the native NCPs. This suggests that H2A.X and P-H2A.X NCPs adopt a different conformation, which will be discussed later.
FIGURE 5.

DNA-PK P-H2A.X nucleosome (nuc.) core particles are less stable compared with the H2A.X or H2A nucleosome core particles. A, shown is native 4% PAGE analysis of NCPs, in comparison to NCPs, reconstituted with H2A.X or with γ-H2A.X and a 146-bp random sequence DNA. CFO is a CfoI-digested pBR322 plasmid DNA used as a marker. B, NaCl dependence of the sedimentation coefficient (s20,w) of native NCPs and NCPs reconstituted with H2A.X or with P-H2A.X is shown. Sedimentation velocity experiments were carried out at 40,000 rpm at 20 °C in different NaCl concentrations in 10 mm Tris-HCl and 0.1 mm EDTA (pH 7.5) buffer. D, area of the scan corresponding to material sedimenting as free DNA; N, peak in the scan corresponding to NCPs. C, shown is the percentage of non-dissociating H2A.X/P-H2A.X-containing nucleosomes at different NaCl concentrations calculated from the relative area under peak N of the sedimentation velocity profile shown in B.
We wondered if a potential change in NCP conformation of the H2A.X nucleosomes could affect the binding of histone H1. To this end we reconstituted H2A.X and P-H2A.X-containing nucleosomes using a longer 208-bp DNA template (26). Increasing amounts of H1 were then added to these reconstituted nucleosomes, and the gel shift thus obtained is shown in Fig. 6A. As can be seen, the H2A.X nucleosomes bind more poorly to H1, an effect that is enhanced by Thr-136/Ser-139 phosphorylation. A similar result was obtained when the Ser-139 of H2A.X was replaced by two glutamic acids to mimic the double negative charge introduced by a phosphate at this position. As seen in Fig. 6B, this produced impairment in H1 binding similar to that observed with P-H2A.X in Fig. 6A.
FIGURE 6.

Impaired binding of linker histones to nucleosomes by H2A.X and γ-H2A.X. A, nucleosomes reconstituted with H2A, H2A.X, and P-H2A.X and [γ-32P]ATP-labeled 208-bp DNA were titrated with increasing amounts of histone H1 and analyzed by 1% agarose gel electrophoresis. The histone H1 to nucleosome molar ratios used were 0, 0.5, 1.0, 1.5, and 2.0. The image shows an autoradiograph of the gel. B, shown is an ethidium bromide-stained image of a similar H1-dependent gel shift carried out in a 1% agarose gel using 208-bp reconstituted nucleosomes consisting of H2A.X and H2A.X-A138E/S139E. C, shown is sodium chloride concentration dependence of the sedimentation coefficient (s20,w) of 208-bp reconstituted nucleosomes consisting of native H2A (black circles), H2A.X (black squares), and P-H2A.X (black triangles).
To gain some insight in the potential mechanism involved in the impaired H1 binding, we analyzed the 208-bp reconstituted nucleosomes using the analytical ultracentrifuge. Similar with what is seen in Fig. 5B, a decrease in s20,w of the undissociated nucleosome fraction was observed at any given salt concentration (Fig. 6C). This effect was reminiscent of the decrease in the sedimentation coefficient of the NCPs when core histones become acetylated (46), and it is enhanced by the presence of 208-bp DNA.
Such a decrease in the sedimentation coefficient suggests an unfolding of the particle likely resulting from a release of the DNA ends at entry and exit sites into the nucleosome core particle. Such an alteration in the trajectory of DNA would be consistent with the impaired binding ability of linker histones observed in Fig. 6, A and B.
DISCUSSION
Presence of Several Acetylation-independent Damage-response Phosphorylatable Sites at the C Terminus of H2A.X
Vertebrate H2A.X histone variants contain two PIKK phosphorylatable target sites at their C-terminal end in addition, in many instances, to a tyrosine at the very end of the molecule (Fig. 1A). The C-terminal tyrosine has been recently shown to be phosphorylated by the Williams-Beuren syndrome transcription factor (WSTF), which is also involved in DNA repair (47). It has been speculated that one of the putative roles for such phosphorylation may be needed to adjust chromatin structure for further maintenance of Ser-139 phosphorylation (47). Interestingly, a set of H2A.X having phenylalanine instead of tyrosine at their very C termini (H2A.X-F) has been identified in Xenopus laevis eggs (48). In this instance, an additional SQE motif has been shown to be present (48). Also, yeast H2A.X contains a leucine at its N terminus (49). Yet, its C-terminal end has a sequence (122SAKATKASQEL129) where DNA damage-dependent phosphorylation of Ser-122, Thr-126, and Ser-129 have been reported (7, 50). Therefore, the presence of several phosphorylatable sites at the very end of the C-terminal tail of H2A.X appears to be a general feature of the molecule.
Our results show that, as suspected earlier (2), vertebrate Ser/Thr-136 is amenable to phosphorylation by DNA-PK in vitro and in vivo (Fig. 1, B and C, and Fig. 2, A–D). Of note, Thr-126 in yeast is important for homologous but indispensable for NHEJ (7). Because NHEJ is the main repair pathway in vertebrates, it is then not surprising that Thr/Ser-136 phosphorylation is also important in these organisms (4).
In agreement with what has been hypothesized in (47), it appears that phosphorylation of more than one phosphorylatable amino acid at the C-terminal end of H2A.X maybe important for the manifold chromatin transactions involved in the DNA repair process. In this regard phosphorylation of Ser/Thr-136 could work as a reinforcing structural factor as will be discussed in the following section.
In contrast to what was earlier reported, our results show that the phosphorylation process is unaffected by histone acetylation (Fig. 3). This result is not surprising, as it has been shown in yeast and other organisms that, although H2A.X phosphorylation happens relatively quickly upon DSB damage, histone acetylation is a downstream event that occurs later on (13) upon recruitment of the chromatin remodeling complexes INO8, SWRI (Swi/Snf2-related adenosine triphosphate complex), and NuA4 (15, 52, 53).
Histone H2A.X Partitions with Insoluble Chromatin Fractions in HeLa Cells
In HeLa cells and in other mammalian tissues (results not shown), H2A.X is preferentially distributed in the chromatin-insoluble fraction P (Fig. 2C), which under the experimental digestion conditions used here amounts to ∼65% of the overall chromatin. As mentioned earlier, this may consist of a reversible associating set of large assemblies involved in DNA DSB repair. A smaller amount is detected in the supernatant, and none was detected in supernatant I. Supernatant I is mainly comprised of euchromatin. These two fractions respectively correspond to 25 and 10% of the starting chromatin before digestion. The SI fraction corresponds to nucleosomes from transcriptionally active chromatin (43) while the S.E. fraction is enriched in facultative heterochromatin (40). The P fraction that corresponds to insoluble material which is resistant to micrococcal nuclease digestion, is not associated with facultative heterochromatin markers (40), but contain large macromolecular complexes including transcriptionally engaged chromatin and constitutive heterochromatin (40, 43), and as shown here, it includes the micro- and macro-foci that are cytologically visualized in cultured mammalian cells before and after ionizing irradiation, respectively (41, 54).
This observation has important implications for the yet unresolved issue of chromatin distribution of histone H2A.X in somatic tissues (3, 55). It has long been proposed that H2A.X distributes itself throughout the genome in a rather homogeneous way with approximately one molecule every five nucleosomes assuming that only heteromorphic (H2A.X-H2A) nucleosomes are present (3). Our data (Fig. 2C) are more consistent with a non-homogeneous H2A.X chromatin distribution in which either small “islands” of highly H2A.X-enriched nucleosomes with a less stable conformation (see Fig. 7) are interspersed within more extensive regions with a lower density of the variant or a clustering of the H2A.X nucleosome is present, as has been recently suggested (55). Furthermore, the phosphorylation resulting from irradiation mainly affects the P fraction (compare the distribution of γH2A.X in irradiated versus non-irradiated cells in Fig. 2C). An uneven distribution of the γ-radiation induced γ-H2A.X formation toward preferentially transcribing chromatin has been recently well documented (Refs. 56 and 57; for review, see Ref. 58).
FIGURE 7.

Model proposed to account for the structural implications of histone H2A replacement by H2A.X and its C-terminal phosphorylation. As indicated in the left-hand side of this figure, isolated nucleosomes in solution exhibit a reversible dissociation into its constitutive histone and DNA components that depends on temperature, concentration, and ionic strength (51). Histone H2A.X decreases the stability of the nucleosome, displacing this equilibrium toward the dissociated state (Fig. 5). In addition and as shown in the lower part of this figure, histone H2A.X alters the nucleosome conformation (Fig. 6C) in a way that impairs the binding of linker histones (Fig. 6, A and B).
Finally, the differential chromatin distribution of the phosphorylated forms of the S139A H2A.X mutant (supernatant (SE) fraction) and H2A.X wild type (pellet (P) fraction) in transfected H2A.X−/− MEF cells (Figs. 2, C and D) come in support of their reinforcing structural nature. In the latter instance it is possible that recruitment of DNA repair complexes to phosphorylated Ser-139 site could render the phosphorylated H2A.X-containing nucleosomes insoluble. This would suggest that, in the absence of Ser-139 phosphorylation, Ser/Thr-136 phosphorylation is not sufficient to recruit such complexes to the site of DNA damage. Therefore, phosphorylation of H2A.X at Thr-136 is likely to act as an enhancer to the structural contribution by phosphorylated H2A.X during DNA DSB repair (see below) rather than a signaling factor. As has been earlier demonstrated, trans-histone phosphorylation such as that of histone H1 (Fig. 4) may also have an important contribution to these processes (59). Indeed, DNA-PK-mediated phosphorylation of H1 has been shown to decrease its affinity within the DNA repair context (59).
H2A.X Deposition Has Important Structural Consequences for Chromatin
The issue on whether the histone H2A phosphorylation resulting from DNA damage PIKK enzymes has any effect on chromatin structure has been elusive. An early study carried out in Saccharomyces cerevisiae demonstrated that substitution of serine by glutamic acid (a mutation that is aimed to mimic phosphorylation) at position 129 of yeast H2A.X resulted in chromatin relaxation of the yeast minichromosome. This was not an unexpected result as the C-terminal tail of H2A exits the nucleosome at a position near the pseudo-dyad axis of symmetry where the DNA enters and exits the particle. Therefore, the introduction of negative charges (two per phosphate) at such a location has the potential for an alteration of the path followed by DNA (20). However, a recent publication has challenged these results and shown that S129E does not have any effect on the DNA supercoiling or in nucleosome stability as determined by nucleosome positioning and nuclease accessibility assays (21).
In high contrast to these results we have observed here that the presence of histone H2A.X itself had a very significant ionic strength-dependent destabilization effect on the nucleosome that was enhanced by the presence of DNA-PK-mediated phosphorylation (Fig. 6B). This latter result is in excellent agreement with recently published data that showed a γH2A.X-mediated structural destabilization of the nucleosome that appears to primarily affect the ends of the nucleosomal DNA (60).
The salt-dependent instability of the H2A.X-containing nucleosome was quite unexpected. However, as pointed out in (55), several important amino acid differences in the histone fold of H2A.X, in addition to its very sequence diverging C-terminal tail, could account for such destabilization. In particular the change from asparagine 38 to histidine and from lysine 99 to glycine between human H2A.1 to H2A.X could be potentially involved. The first substitution, which is located in the loop connecting the α1 and α2 helices of the H2A histone fold, deserves special attention as this amino acid is in the H2A region Asn-38—Glu-41 of the H2A-H2B dimer that makes direct contact with the equivalent region of the second H2A.H2B dimer within the protein core of the nucleosome (61, 62). Furthermore, histidine has long been shown to play an important role in the stability of the histone octamer (63). Of note, the ionic strength-dependent instability of the H2A.X nucleosome is very similar to that of the yeast nucleosome, where a histone H2A.X ortholog is present that encompasses 95% of the overall H2A complement (3). Yeast nucleosomes have been shown to be less stable than those consisting of canonical H2A and dissociate completely by 0.5 m NaCl (64). The instability of such nucleosomes would be consistent with a highly dynamic chromatin designed to accommodate the high levels of transcriptional activity of an organism where most of its genes are expressed at a given time.
A similar dynamic chromatin structure is likely to be involved in DNA repair. In terms of the potential physiological implications of the model proposed in Fig. 7 for the H2A.X containing nucleosome, it is tempting to speculate that γ-H2AX together with histone H1 phosphorylation provide a very dynamic nucleosome environment that prevents histone H1 from binding back, thus maintaining an open chromatin organization.
Acknowledgment
We thank Wayne Beckham from the British Columbia Cancer Agency of the Vancouver Island Centre for assistance with the cell irradiation.
This work was supported by Canadian Institute of Health Research Grants MOP-97878 (to S. P. L.-M.) and MOP-57718 (to J. A.).
- DSB
- double-strand break
- ATM
- ataxia telangiectasia mutated
- ATR
- ATM and Rad3-related
- DNA-PK
- DNA-dependent protein kinase
- MEF
- mouse embryonic fibroblast
- NCP
- nucleosome core particle
- NHEJ
- non-homologous end joining
- NuA4
- nucleosome acetyltransferase of H4
- PIKK
- phosphatidylinositol-3 kinase-like family of kinases
- GFP
- green fluorescent protein
- HPLC
- high performance liquid chromatography
- Pipes
- 1,4-piperazinediethanesulfonic acid.
REFERENCES
- 1.Fernandez-Capetillo O., Lee A., Nussenzweig M., Nussenzweig A. (2004) DNA Repair 3, 959–967 [DOI] [PubMed] [Google Scholar]
- 2.Rogakou E. P., Pilch D. R., Orr A. H., Ivanova V. S., Bonner W. M. (1998) J. Biol. Chem. 273, 5858–5868 [DOI] [PubMed] [Google Scholar]
- 3.Pilch D. R., Sedelnikova O. A., Redon C., Celeste A., Nussenzweig A., Bonner W. M. (2003) Biochem. Cell Biol. 81, 123–129 [DOI] [PubMed] [Google Scholar]
- 4.Li A., Eirín-López J. M., Ausió J. (2005) Biochem. Cell Biol. 83, 505–515 [DOI] [PubMed] [Google Scholar]
- 5.Stiff T., O'Driscoll M., Rief N., Iwabuchi K., Löbrich M., Jeggo P. A. (2004) Cancer Res. 64, 2390–2396 [DOI] [PubMed] [Google Scholar]
- 6.An J., Huang Y. C., Xu Q. Z., Zhou L. J., Shang Z. F., Huang B., Wang Y., Liu X. D., Wu D. C., Zhou P. K. (2010) BMC Mol. Biol. 11, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore J. D., Yazgan O., Ataian Y., Krebs J. E. (2007) Genetics 176, 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahaney B. L., Meek K., Lees-Miller S. P. (2009) Biochem. J. 417, 639–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meek K., Dang V., Lees-Miller S. P. (2008) Adv. Immunol. 99, 33–58 [DOI] [PubMed] [Google Scholar]
- 10.Tamburini B. A., Tyler J. K. (2005) Mol. Cell. Biol. 25, 4903–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masumoto H., Hawke D., Kobayashi R., Verreault A. (2005) Nature 436, 294–298 [DOI] [PubMed] [Google Scholar]
- 12.Bird A. W., Yu D. Y., Pray-Grant M. G., Qiu Q., Harmon K. E., Megee P. C., Grant P. A., Smith M. M., Christman M. F. (2002) Nature 419, 411–415 [DOI] [PubMed] [Google Scholar]
- 13.Murr R., Loizou J. I., Yang Y. G., Cuenin C., Li H., Wang Z. Q., Herceg Z. (2006) Nat. Cell Biol. 8, 91–99 [DOI] [PubMed] [Google Scholar]
- 14.Ikura T., Ogryzko V. V., Grigoriev M., Groisman R., Wang J., Horikoshi M., Scully R., Qin J., Nakatani Y. (2000) Cell 102, 463–473 [DOI] [PubMed] [Google Scholar]
- 15.Downs J. A., Allard S., Jobin-Robitaille O., Javaheri A., Auger A., Bouchard N., Kron S. J., Jackson S. P., Côté J. (2004) Mol. Cell 16, 979–990 [DOI] [PubMed] [Google Scholar]
- 16.Park E. J., Chan D. W., Park J. H., Oettinger M. A., Kwon J. (2003) Nucleic Acids Res. 31, 6819–6827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrison A. J., Shen X. (2005) Cell Cycle 4, 568–571 [PubMed] [Google Scholar]
- 18.Javaheri A., Wysocki R., Jobin-Robitaille O., Altaf M., Côté J., Kron S. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 13771–13776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Downs J. A., Lowndes N. F., Jackson S. P. (2000) Nature 408, 1001–1004 [DOI] [PubMed] [Google Scholar]
- 20.Ausió J., Abbott D. W., Wang X., Moore S. C. (2001) Biochem. Cell Biol. 79, 693–708 [PubMed] [Google Scholar]
- 21.Fink M., Imholz D., Thoma F. (2007) Mol. Cell. Biol. 27, 3589–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siino J. S., Nazarov I. B., Svetlova M. P., Solovjeva L. V., Adamson R. H., Zalenskaya I. A., Yau P. M., Bradbury E. M., Tomilin N. V. (2002) Biochem. Biophys. Res. Commun. 297, 1318–1323 [DOI] [PubMed] [Google Scholar]
- 23.Ausió J., Moore S. C. (1998) Methods 15, 333–342 [DOI] [PubMed] [Google Scholar]
- 24.Simon R. H., Felsenfeld G. (1979) Nucleic Acids Res. 6, 689–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ausio J., Dong F., van Holde K. E. (1989) J. Mol. Biol. 206, 451–463 [DOI] [PubMed] [Google Scholar]
- 26.Simpson R. T., Thoma F., Brubaker J. M. (1985) Cell 42, 799–808 [DOI] [PubMed] [Google Scholar]
- 27.Celeste A., Petersen S., Romanienko P. J., Fernandez-Capetillo O., Chen H. T., Sedelnikova O. A., Reina-San-Martin B., Coppola V., Meffre E., Difilippantonio M. J., Redon C., Pilch D. R., Olaru A., Eckhaus M., Camerini-Otero R. D., Tessarollo L., Livak F., Manova K., Bonner W. M., Nussenzweig M. C., Nussenzweig A. (2002) Science 296, 922–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gautier T., Abbott D. W., Molla A., Verdel A., Ausio J., Dimitrov S. (2004) EMBO Rep. 5, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X., Moore S. C., Laszckzak M., Ausió J. (2000) J. Biol. Chem. 275, 35013–35020 [DOI] [PubMed] [Google Scholar]
- 30.Maffey A. H., Ishibashi T., He C., Wang X., White A. R., Hendy S. C., Nelson C. C., Rennie P. S., Ausió J. (2007) Mol. Cell. Endocrinol. 268, 10–19 [DOI] [PubMed] [Google Scholar]
- 31.Laemmli U. K. (1970) Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 32.Yager T. D., van Holde K. E. (1984) J. Biol. Chem. 259, 4212–4222 [PubMed] [Google Scholar]
- 33.Saperas N., Chiva M., Casas M. T., Campos J. L., Eirín-López J. M., Frehlick L. J., Prieto C., Subirana J. A., Ausió J. (2006) FEBS J. 273, 4548–4561 [DOI] [PubMed] [Google Scholar]
- 34.Abbott D. W., Chadwick B. P., Thambirajah A. A., Ausió J. (2005) J. Biol. Chem. 280, 16437–16445 [DOI] [PubMed] [Google Scholar]
- 35.Abbott D. W., Laszczak M., Lewis J. D., Su H., Moore S. C., Hills M., Dimitrov S., Ausió J. (2004) Biochemistry 43, 1352–1359 [DOI] [PubMed] [Google Scholar]
- 36.van Holde K. E., Weischet W. O. (1978) Biopolymers 17, 1387–1403s [Google Scholar]
- 37.Demeler B. (2005) in UltrScan A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments. Modern Analytical Ultracentrifugation: Techniques and Methods (Scott D. J., Hardfing S. E., Rowe A. J. eds) Royal Society of Chemistry, UK [Google Scholar]
- 38.Goodarzi A. A., Lees-Miller S. P. (2004) DNA Repair 3, 753–767 [DOI] [PubMed] [Google Scholar]
- 39.Perry M., Chalkley R. (1982) J. Biol. Chem. 257, 7336–7347 [PubMed] [Google Scholar]
- 40.Ishibashi T., Dryhurst D., Rose K. L., Shabanowitz J., Hunt D. F., Ausió J. (2009) Biochemistry 48, 5007–5017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McManus K. J., Hendzel M. J. (2005) Mol. Biol. Cell 16, 5013–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ismail I. H., Hendzel M. J. (2008) Environ. Mol. Mutagen. 49, 73–82 [DOI] [PubMed] [Google Scholar]
- 43.Henikoff S., Henikoff J. G., Sakai A., Loeb G. B., Ahmad K. (2009) Genome Res. 19, 460–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bennetzen M. V., Larsen D. H., Bunkenborg J., Bartek J., Lukas J., Andersen J. S. (2010) Mol. Cell. Proteomics, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ausió J. (2000) Biophys. Chem. 86, 141–153 [DOI] [PubMed] [Google Scholar]
- 46.Ausio J., van Holde K. E. (1986) Biochemistry 25, 1421–1428 [DOI] [PubMed] [Google Scholar]
- 47.Xiao A., Li H., Shechter D., Ahn S. H., Fabrizio L. A., Erdjument-Bromage H., Ishibe-Murakami S., Wang B., Tempst P., Hofmann K., Patel D. J., Elledge S. J., Allis C. D. (2009) Nature 457, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shechter D., Chitta R. K., Xiao A., Shabanowitz J., Hunt D. F., Allis C. D. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 749–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krebs J. E. (2007) Mol. Biosyst. 3, 590–597 [DOI] [PubMed] [Google Scholar]
- 50.Harvey A. C., Jackson S. P., Downs J. A. (2005) Genetics 170, 543–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ausio J., Seger D., Eisenberg H. (1984) J. Mol. Biol. 176, 77–104 [DOI] [PubMed] [Google Scholar]
- 52.Morrison A. J., Highland J., Krogan N. J., Arbel-Eden A., Greenblatt J. F., Haber J. E., Shen X. (2004) Cell 119, 767–775 [DOI] [PubMed] [Google Scholar]
- 53.van Attikum H., Fritsch O., Hohn B., Gasser S. M. (2004) Cell 119, 777–788 [DOI] [PubMed] [Google Scholar]
- 54.Bewersdorf J., Bennett B. T., Knight K. L. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 18137–18142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinto D. M., Flaus A. (2010) Subcell. Biochem. 50, 55–78 [DOI] [PubMed] [Google Scholar]
- 56.Cowell I. G., Sunter N. J., Singh P. B., Austin C. A., Durkacz B. W., Tilby M. J. (2007) PloS one 2, e1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vasireddy R. S., Karagiannis T. C., El-Osta A. (2010) Cell. Mol. Life Sci. 67, 291–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Costes S. V., Chiolo I., Pluth J. M., Barcellos-Hoff M. H., Jakob B. (2010) Mutat. Res., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kysela B., Chovanec M., Jeggo P. A. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 1877–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heo K., Kim H., Choi S. H., Choi J., Kim K., Gu J., Lieber M. R., Yang A. S., An W. (2008) Mol. Cell 30, 86–97 [DOI] [PubMed] [Google Scholar]
- 61.Mariño-Ramírez L., Kann M. G., Shoemaker B. A., Landsman D. (2005) Expert Rev. Proteomics. 2, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 63.Eickbush T. H., Moudrianakis E. N. (1978) Biochemistry 17, 4955–4964 [DOI] [PubMed] [Google Scholar]
- 64.Piñeiro M., Puerta C., Palacián E. (1991) Biochemistry 30, 5805–5810 [DOI] [PubMed] [Google Scholar]