

Abstract
hnRNP K, a member of the family of heterogeneous ribonucleoproteins, is known to exert various functional roles in the nucleus, cytoplasm, and mitochondria to affect different cellular processes including chromatin remodeling, transcription, splicing, and translation. Here we report, for the first time, that hnRNP K is specifically involved in human LDL receptor (LDLR) gene transcription in HepG2 cells. We show that depletion of hnRNP K by siRNA transfection reduces the expression of LDLR mRNA and protein by more than 50% as measured by quantitative real-time PCR and Western blot analysis. Importantly, we show that the decay rate of LDLR mRNA is not affected by hnRNP K siRNA transfection, whereas the LDLR promoter activity is significantly decreased. Furthermore, overexpression of hnRNP K increased the LDLR promoter activity by the luciferase reporter assay. By utilizing a series of mutational and deletional constructs of LDLR promoter luciferase reporters, we mapped the K-responsive element to the repeat 3 (R3) sequence of the LDLR promoter. Electrophoretic mobility shift assays show that the K protein binds to a single-stranded DNA probe containing the CT-rich element of R3, which is in contrast to the requirement of double-stranded DNA for Sp1 to bind to R3. Finally, chromatin immunoprecipitation assays reveal a direct interaction of hnRNP K with the LDLR promoter in intact HepG2 cells. These new findings provide strong evidence demonstrating that hnRNP K is an important transactivator for human LDLR gene transcription. This work sheds new light on our current understanding of how LDLR gene expression is controlled at the transcriptional level.
Keywords: Gene Regulation, Gene Transcription, General Transcription Factors, Lipoprotein Metabolism, Lipoprotein Receptor, CT-rich Element, LDL Receptor, hnRNP K, Transactivator
Introduction
Elevated levels of the low density lipoprotein (LDL)2 in the blood are strongly correlated with an increased risk of coronary artery disease and myocardial infarction (1, 2). The LDL is removed from the bloodstream through the LDL receptor (LDLR)-mediated endocytosis pathway in the liver (2). This discovery of the role of LDLR revolutionized thoughts on familial hypercholesterolemia (FH) and ways to treat hyperlipidemia (3). Although it has been shown that LDLR expression can be regulated at different levels, it is mainly regulated at the transcriptional level by sterol through the sterol response element (SRE) residing within the promoter of the LDLR gene (4). In addition to the sterol-dependent pathway that controls the expression of LDLR, sterol-independent pathways that regulate LDLR transcription by cytokine oncostatin M have also been reported by our laboratory (5, 6). With the study on proprotein convertase subtilisin/kexin type 9 (PCSK9), it has been well established that PCSK9 negatively regulates LDLR protein expression on the liver cell surface through its direct interaction with the receptor (7–10). Moreover, our laboratory recently identified several mRNA-binding proteins, which appear to be involved in the post-transcriptional regulation of LDLR (11). Clearly, final expression levels of LDLR are subject to tight regulation by a complex network at different processing levels.
The hnRNP K protein was originally identified as a member of heterogeneous nuclear ribonucleoproteins, which is involved in the metabolism of pre-mRNAs that contain cytidine-rich sequences (12). hnRNP K belongs to poly(C)-binding proteins (PCBPs) that contain three conserved K homology (KH) domains and are characterized by high affinity for poly(C) (13, 14). It has been shown that hnRNP K binds RNA (15, 16), which is involved in pre-mRNA splicing (17), transport of mRNA from the nucleus to the cytosol (18), and translational silencing of 15-lipoxygenase (LOX) mRNA (19); The K protein also binds single- and double-stranded DNA (20, 21) and functions as either a transcription factor (21–23) or a repressor (24, 25). Specifically, hnRNP K is able to activate transcription from the single-stranded CT-element (26). In addition, hnRNP K acts as a docking platform for protein-protein interaction (27, 28). Furthermore, it has been reported that the K protein, as a cofactor for p53, plays key roles in coordinating transcriptional responses to DNA damage (29). Thus, hnRNP K is a multifunctional protein that is implicated in chromatin remodeling, transcription, splicing, and translation processes through its interaction with diverse molecules including proteins, DNAs, and RNAs (30, 31).
Here, through an array of experiments with siRNA-mediated depletion and plasmid-mediated overexpression, we present strong evidence showing that hnRNP K is directly involved in LDLR gene expression at the transcriptional level. Interestingly, we demonstrate that as a transactivator, hnRNP K interacts with a CT-rich stretch residing within the repeat 3 sequence of the LDLR promoter in a manner that requires a single-stranded DNA, which is different from the action of a well known repeat 3 binding factor Sp1, as our data showed that Sp1 only binds the double-stranded repeat 3 sequence.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
The hepatoma-derived cell line HepG2 was purchased from the American Type Culture Collection (ATCC) and was maintained in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (Omega Scientific, Inc., Tarzana, CA) and 1% penicillin/streptomycin solution (Mediatech, Inc., Herndon, VA). FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN) was used to transfect plasmids into HepG2 cells according to the manufacturer's instructions.
siRNA Knockdown
siRNA against human hnRNP K (Cat. AM16708, ID: 11245) and Silencer negative siRNA control (Cat. AM4635) were purchased from Applied Biosystems. siPORT™ NeoFXTM transfection reagent (Ambion Inc, Austin, TX) was used to transfect siRNA into HepG2 cells in siRNA knockdown assays according to the manufacturer's instructions with minor modifications. 0.5 μl/well of 2 μm siRNA in 96-well plates (1.5 × 104 cells/well) or 12.5 μl/well of 2 μm siRNA in 6-well plates (0.5 × 106 cells/well) was used for each transfection. The knockdown effects were analyzed 48 h after siRNA transfection.
RNA Isolation and Real-time RT-PCR
Total RNA was extracted from HepG2 cells using UltraSpecTM total RNA isolation reagent (Biotecx Laboratories). 2 μg of total RNA was reverse transcribed with a high-capacity cDNA reverse transcription kit using random primers according to the manufacturer's instructions. Real-time PCR was performed on the cDNA using an ABI Prism 7900-HT Sequence Detection System (Applied Biosystems). Human LDLR and GAPDH Pre-Developed TaqMan Assay Reagents from Applied Biosystems were used to assess mRNA expression in HepG2 cells. Each cDNA sample was run in triplicate. All values are reported as means ± S.D. of the triple measurement of each cDNA sample.
Analysis of DiI-LDL Uptake after hnRNP K Knockdown
0.5 × 106 HepG2 cells/well were seeded into a 6-well cell culture plate. siRNA knockdown procedure was described above. After 2 days transfection with siRNA, DiI-LDL (5 μg/ml) was incubated with cells for 4 h, and cells were washed three times with cold phosphate-buffered saline. DiI-LDL uptake was examined with a fluorescent microscope and single-cell fluorescence intensities were subsequently measured by a FACScan instrument (Becton Dickinson Immunocytometry Systems). The mean fluorescence value (MFV) of scrambled siRNA-treated samples is expressed as 100%.
Cell Viability Assay
To assess possible effects of K depletion on HepG2 cell proliferation, HepG2 cells seeded in a 96-well culture plate were transfected with the specific siRNA or the scrambled control siRNA. After 48 h, the number of viable cells in culture was determined by using the CellTiter-Glo Luminescent Cell Viability Assay kit (Promega). Triplicate wells were assayed for each siRNA condition. The luminescent signal was recorded by SpectraMax® Microplate Luminometer (Molecular Device).
Northern Blotting and Actinomycin D Treatment
Northern blot analysis of LDLR and GAPDH mRNA using 32P-labeled DNA probe was conducted as previously described (32). Two days after transfection with scrambled or hnRNP K siRNA into HepG2 cells, actinomycin D (5 μg/ml) was added to inhibit the de novo mRNA synthesis and incubated for the indicated periods of time. For Northern blot analysis, 10 μg of total RNA per sample were loaded onto a 1.2% denatured agarose gel and run for 2 h at a voltage of 120 V. The separated RNAs were then transferred to BrightStar-Plus® positively charged nylon membrane (Ambion, Cat. No. AM10104). The targeted RNAs were hybridized and exposed to Storage Phosphor Screen (Amersham Biosciences) overnight. The bands were visualized by Typhone 2000. Actinomycin D was purchased from Sigma Chemicals Co. (Cat. No. A-9415).
Western Blotting
Western blotting was performed according to standard procedures as described previously (11) on whole cell lysates from HepG2 cells. Briefly, 50 μg of protein extract for each sample was loaded into PAGEr® Gold Precast gels (Lonza Rockland, Inc.). Separated proteins were transferred onto nitrocellulose membrane and incubated with indicated antibodies. The membranes were developed and visualized using ECL Plus Western blotting reagent (GE Healthcare, Piscataway, NJ) and Kodak Image Station 4000R (Kodak Molecular Imaging Systems, New Haven). Goat anti-hnRNP K polyclonal antibody (sc16554, 1:500) was purchased from Santa Cruz Biotechnologies, Inc., and chicken polyclonal to human LDLR (ab14056, 1:2500) antibody was purchased from Abcam, Inc.
Luciferase Reporter Assay
Firefly luciferase reporter constructs containing either wild-type or mutant LDLR proximal promoter were individually transfected into HepG2 cells along with Renilla luciferase (pRL-TK) reporter as an internal transfection efficiency control. Briefly, 1 × 104 HepG2 cells/well were seeded onto 96-well plates (Corning Inc., Corning, NY) the day before transfection, 100 ng of firefly luciferase plasmid plus 5 ng of Renilla luciferase per well were cotransfected using FuGENE 6 transfection reagent (Roche Diagnostics). Two days after transfection, cells were lysed with 1× passive lysis buffer, and luciferase activity was analyzed using the Dual-Luciferase® reporter Assay System (Promega Corporation, Madison, WI) on a 96-well plate reader (SpectraMax® L microplate luminometer, Molecular Devices).
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation (ChIP) assays were performed according to a previously published protocol (33). Briefly, the cultured HepG2 cells were cross-linked by the addition of formaldehyde to a final concentration of 1.42% for 15 min at room temperature. Cross-linking was quenched with 125 mm glycine for 5 min at room temperature, and cells were washed twice with cold phosphate-buffered saline before scraping and centrifugation. Then, cell pellets were resuspended and lysed with immunoprecipitation (IP) buffer-containing protease inhibitors. The cell lysates were sonicated to shear chromatin DNA with an average length of about 500–1000 bp. 2 μg of polyclonal goat anti-hnRNP K antibody (sc16554, Santa Cruz Biotechnologies, Inc.) were incubated with 500 μl of IP buffer containing 250 μg of sheared chromatin in an ultrasonic water bath for 30 min at 4 °C. The same amount of normal goat IgG was used as mock IP. For detecting the acetylated status of acetyl-histone H3 and H4, 1 μg of anti-acetyl-Histone H3 (Cat. No. 06–599, Millipore) or anti-acetyl-Histone H4 (Cat. No. 06–866, Millipore) were incubated with 25 μg of sheared chromatin in a total volume of 500 μl IP buffer. 20 μl of protein G-agarose beads (Cat. No.16–266, Millipore) were used to precipitate the potential DNA-hnRNP K complex; the precipitated DNAs were isolated by using Chelex 100 slurry. DNA precipitated from 20 μg of sheared chromatin was used as input control. The isolated DNAs were analyzed by PCR with 35 cycles using the following primers: forward 5′-cgatgtcacatcggccgttcg-3′ and reverse 5′-cacgacctgctgtgtcctagctggaa-3′, the PCR amplicon is 180 base pairs in length (6).
Electrophoretic Mobility Shift Assay
5′-biotin end-labeled single-stranded sense oligonucleotides were synthesized by Elim Biopharmaceuticals, Inc. The double-stranded biotin-labeled oligonucleotides were made by annealing the biotin end-labeled sense oligonucleotide with unlabeled antisense complementary oligonucleotide at a 1:1 molar ratio in the annealing buffer (10 mm Tris (pH 8.0), 1 mm EDTA (pH 8.0), and 50 mm NaCl) at 95 °C for 5 min. Afterward, the reaction was slowly cooled down to room temperature. The biotin-labeled single-stranded and double-stranded oligonucleotides were stored at −20 °C before use. EMSA was performed using a LightShift® Chemiluminescent EMSA kit purchased from Pierce according to the manufacturer's instructions.
Briefly, 50 fmol of 5′-biotin end-labeled sense DNA probes were incubated with 0.2 μg of purified recombinant hnRNP K protein in 1× binding buffer containing 10 mm Tris (pH 7.5), 50 mm KCl, 1 mm dithiothreitol, 2.5% glycerol, 5 mm MgCl2, 50 ng/μl of poly(dI-dC), and 0.05% Nonidet P-40 in a total of 20 μl of reaction mixture for 20 min at room temperature. For the observation of a supershift complex, 1 μg of corresponding antibody was added, and the mixture was incubated for another 10 min at room temperature before loading onto a 5% native polyacrylamide gel. The binding reactions were electrophoretically transferred to a positively charged Nylon membrane (Ambion, Cat. No. AM10104). The transferred DNA was cross-linked to the nylon membrane in a Bio-Rad GS GENE LINKER at a setting of 150 mJoules. The biotin-labeled DNAs were detected by chemiluminescence, and signals were recorded by the Kodak Image Station 4000R. The recombinant human hnRNP K was purchased from Abnova (Cat. No. H00003190-P01) and OriGene Technologies (Cat. No. TP301843), respectively; and human recombinant transcription factor Sp1 was purchased from Promega (Cat. No. E6391). The sense sequences of biotinylated probes and competitors used in the EMSA assays were as follows: LDLR wild-type: 5′-GGTGAAGACATTTGAAAATCACCCCACTGCAAACTCCTCCCCCTGCTAGAA-3′; LDLR mutant R3D: 5′-GGTGAAGACATTTGAAAATCACCCCACTGCAAACTCCTCttCCTGCTAGAA-3′; c-Myc promoter: 5′-TCCTCCCCACCTTCCCCACCCTCCCCACCCTCCCCATAAGCGCCCTCCCGG-3′; LDLR intron 1: 5′-GAGAATCGCTTGAACCTGGGAGCAGGATGTTGCAGTGAACCGATATCACAC-3′. The small bold letters are mutated nucleotides, and the sequence located within intron 1 of the LDLR gene was used as a nonspecific competitor.
Statistical Analysis
For luciferase reporter assays, each experiment is representative of at least three independent experiments with a minimum of triplicates per condition. Real-time RT-PCR results were summarized from at least three independent assays, and data from Dil-LDL uptake were an average from two independent assays. Significant differences between control and treatment groups or between wild-type and mutated vectors were assessed by one-way ANOVA with post test of Bonferroni's multiple comparison test or by two tailed Student's t test. Values of p < 0.05 were considered statistically significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
RESULTS
Knockdown of hnRNP K Down-regulates LDLR mRNA and Protein Levels
To explore the role of hnRNP K in the regulation of LDLR expression, we used siRNA against human hnRNP K to deplete the transcript of hnRNP K in HepG2 cells. After transfection with specific siRNA, hnRNP K expression decreased significantly in both mRNA and protein levels compared with scrambled siRNA transfection as shown in Fig. 1, A and B. As a consequence, depletion of hnRNP K by siRNA lowered the amount of LDLR mRNA by 55% of control as evidenced by real-time RT-PCR (Fig. 1C). Similarly, Western blotting showed that the LDLR protein level dramatically decreased in specific hnRNP K siRNA-treated HepG2 cells compared with scrambled siRNA-transfected cells (Fig. 1D). Furthermore, LDLR-mediated DiI-LDL uptake assays showed that the DiI-LDL intracellular fluorescence intensity decreased upon hnRNP K depletion by siRNA knockdown compared with scrambled siRNA transfection (Fig. 1E). Single cell fluorescence intensities measured by FACS analysis indicated a 36% reduction of endocytosed DiI-LDL in hnRNP K-depleted cells compared with those in scrambled knockdown samples (Fig. 1F).
FIGURE 1.

hnRNP K knockdown attenuates LDLR expression. A, 0.5 × 106 HepG2 cells/well were plated into a 6-well cell culture plate, and 12.5 μl/well of 2 μm scrambled or hnRNP K siRNAs were simultaneously transfected into HepG2 cells for 2 days before total RNA or protein extraction. RT-PCR was performed to detect hnRNP K and GAPDH mRNA, respectively. B, total protein was extracted from HepG2 cells and hnRNP K and β-actin were detected by Western blotting, respectively. C, real-time PCR was performed to measure the LDLR mRNA levels from cDNA samples obtained from A. LDLR mRNA levels were normalized by GAPDH mRNA expression levels. ***, p < 0.001. The data shown are average of three separate transfections. D, Western blotting was performed to detect the LDLR and β-actin protein levels in both scrambled or hnRNP K siRNA-transfected samples. E, HepG2 cells were seeded onto 6-well plates and transfected with siRNAs for 48 h; receptor-mediated uptake of DiI-LDL was examined with a fluorescent microscope after incubation with DiI-LDL (5 μg/ml) for 4 h. F, the uptake of DiI-LDL uptake in HepG2 cells was further analyzed by FACS. The mean fluorescence value (MFV) of control cells transfected with scrambled siRNA was defined as 100%, and the MFV in hnRNP K siRNA-transfected cells were normalized by that value. ***, p < 0.001. The data shown are the average of two independent experiments. G, real-time PCR analysis of mRNA expression of K target genes and nontargeted genes using cDNA samples obtained from A. ***, p < 0.001. H, cell proliferation rates were determined using the CellTiter-Glo Luminescent Cell Viability Assay kit as described under “Experimental Procedures.”
We further assessed the specificity of siRNA on K-target genes and off-target genes. The promoters of c-myc and eukaryotic translation initiation factor 4E (eIF4E) contain the K-recognition sequence and are transcriptionally regulated by K (34). Real-time RT-PCR analysis showed that the c-Myc and eIF4E mRNA levels along with LDLR were significantly reduced compared with that of the scrambled siRNA-transfected cells. In contrast, the mRNA expression of peroxisome proliferator-activated receptor α (PPARα), HMG CoA reductase (HMGCR), and CCAAT enhancer-binding protein β (C/EBPβ) that are not regulated by K was not affected by the cellular depletion of hnRNP K. These data demonstrated that K siRNA transfection did not generate an off-target nonspecific effect on gene expression. Moreover, cell proliferation assays (Fig. 1H) showed that the cell growth rate of HepG2 cells was not affected by the K siRNA transfection within 48–72 h. In addition, the morphology of HepG2 cells was not obviously altered by cellular depletion of K. Altogether these data consistently demonstrated that hnRNP K is required for LDLR expression.
hnRNP K Transcriptionally Regulates LDLR
hnRNP K is a multifunctional protein that has been shown to regulate its target genes at different levels. We next asked at which level hnRNP K may participate in the regulation of LDLR gene expression. hnRNP K is known as an RNA-binding protein because of its RNA binding property and was reported to stabilize rennin mRNA (35). We first examined if hnRNP K can stabilize LDLR mRNA. We used specific siRNA to knockdown hnRNP K in HepG2 cells together with actinomycin D treatment to block de novo mRNA synthesis, while scrambled siRNA was used as control. Northern blotting was performed to examine the remaining LDLR mRNA after actinomycin D treatment (Fig. 2A). The results first demonstrated that actinomycin D treatment decreased LDLR mRNA levels in both scrambled and hnRNP K siRNA-treated samples along the time course and that knockdown of hnRNP K significantly decreased the LDLR mRNA levels compared with control samples (Fig. 2A). However, the turnover ratio remained unchanged between scrambled and hnRNP K siRNA-transfected groups, indicating that hnRNP K may not participate in the control of LDLR expression at the post-transcriptional level (Fig. 2B). We postulated that K protein may regulate LDLR gene transcription as a transcription activator. To this end, we used the previously characterized LDLR promoter luciferase reporter construct pLDLR234Luc (5) to transfect HepG2 cells combined with either hnRNP K overexpression or siRNA knockdown in transient transfection assays. The Renilla luciferase (pRL-TK) reporter was cotransfected with pLDLR234Luc as an internal transfection efficiency control. Results showed that siRNA knockdown of hnRNP K decreased luciferase activity (Fig. 2C), while overexpression of the K protein increased luciferase activity (Fig. 2D) of the LDLR promoter reporter, indicating that hnRNP K transcriptionally regulates LDLR gene expression through the proximal region of the LDLR promoter.
FIGURE 2.

Transcriptional regulation of LDLR expression by hnRNP K. A, HepG2 cells were seeded onto 6-well cell culture plates, and total RNA was extracted as described in the legend to Fig. 1 except that actinomycin D (5 μg/ml) was added and incubated for the indicated periods of time before collecting cells. 10 μg/sample of total RNA was loaded onto a 1.2% denatured agarose gel. LDLR and GAPDH transcripts were detected by 32P-labeled DNA probes, respectively. B, the density of LDLR and GAPDH mRNA bands was calculated and LDLR mRNA levels were normalized by corresponding GAPDH values. Linear degradation curves were obtained by plotting the relative LDLR mRNA values against time. The data represent the average of two independent experiments. C, HepG2 cells were transfected by scrambled or hnRNP K siRNA on the day when cells were seeded. pLDLR234Luc and pRL-TK were cotransfected at a ratio of 20:1 the next day and incubated for two more days before lysis. D, pLDLR234Luc together with pRL-TK at a ratio of 20:1 were transiently transfected into HepG2 cells by FuGene 6 along with pcDNA3-myc-hnRNP K or mock vector for 2 days before examining luciferase activity. Results are presented as a ratio of firefly luciferase activity to Renilla. *, p < 0.05 and **, p < 0.01 compared with control. The data shown are representative of three separate transfections with similar results.
Repeat 3 of LDLR Promoter Is Required for LDLR Gene Transactivation by hnRNP K
To identify the corresponding sequence on the LDLR promoter that is involved in hnRNP K-mediated transactivation, a series of LDLR promoter luciferase reporters (Fig. 3A) were further employed upon siRNA knockdown of hnRNP K. HepG2 cells were transfected by either scrambled siRNA or hnRNP K siRNA for 1 day before transfection of the luciferase reporters. Fig. 3B showed the normalized promoter activities, indicating that all mutations on LDLR promoter greatly reduced the reporters' luciferase activity; and Fig. 3C showed the promoter activities in specific siRNA-transfected cells relative to that of scrambled control siRNA. These data demonstrated that that the R1, R2, and R3D mutation reduced promoter activity by 95, 85, and 98%, respectively. Whereas the activities of reporters containing mutations in repeat 1 (LDLR234-R1) and repeat 2 (LDLR234-R2) were further reduced by K-depletion, the promoter activity of LDLR234-R3D, that was lowest among all the mutated promoters, was not further reduced by K-depletion. These results suggest that hnRNP K interacts with sequences within R3 to activate the LDLR gene transcription. We further tested the effect of hnRNP K siRNA on luciferase activity of pLDLR-R3 that contains the LDLR promoter sequence +13 to −52 including the R3 while excluding R1 and R2 sequences (5). Depletion of hnRNP K also reduced luciferase activity of pLDLR-R3; thus providing additional evidence to localize the K-responsive element to the repeat 3 region of the LDLR promoter.
FIGURE 3.

The repeat 3 of the LDLR promoter is responsive to hnRNP K knockdown. A, schematic representation of the wild type, 3 mutants, and 1 deletion of pLDLR234Luc reporters. To perform siRNA knockdown and dual-luciferase assay on 96-well plates, 0.5 μl/well of 2 μm small RNA were mixed with 1.5 × 104 HepG2 cells/well. The next day, 100 ng/well of pLDLR234Luc reporter plus 5 ng of pRL-TK reporter as transfection control were cotransfected into these cells. Two days after transfection with different reporters, cells were lysed. The normalized luciferase activities of various reporters are presented in B, and in C the relative luciferase activity in scrambled siRNA-transfected cells is expressed as 1. The data shown are representative of three separate transfections with similar results.
Cholesterol negatively mediates LDLR expression through the SRE-1 element located in R2 (4). The fact that R2 mutation that disrupted SREBP binding to the SER-1 site did not abolish the knockdown effect of hnRNP K suggests that the interaction of hnRNP K with LDLR promoter is independent of the classical cholesterol negative feedback regulation mediated by the SREBP pathway. To further demonstrate the cholesterol-independent activation of hnRNP K on LDLR transcription, we examined the effects of siRNA transfections on LDLR promoter activities in cells cultured in medium containing lipoprotein cholesterol-depleted serum (LPDS) and in medium containing cholesterol (LPDS + cholesterol). HepG2 cells were transfected with scrambled or hnRNP K siRNA and seeded into 96-well plates. The next day, plasmids pLDLR234Luc or pLDLR1563Luc together with pRL-TK internal control were transfected into these HepG2 cells. The culture medium was changed to MEM containing 10% LPDS or MEM containing 10% LPDS plus cholesterol on the third day and incubated for 24 h before lysis. Results showed that knockdown of hnRNP K significantly reduced the pLDLR234Luc (Fig. 4A) and pLDLR1563Luc (Fig. 4B) reporter activities on both cholesterol-containing or -depleting medium, further confirming that the transcriptional regulation of LDLR by hnRNP K is independent of the SRE-1/SREBP regulatory pathway.
FIGURE 4.
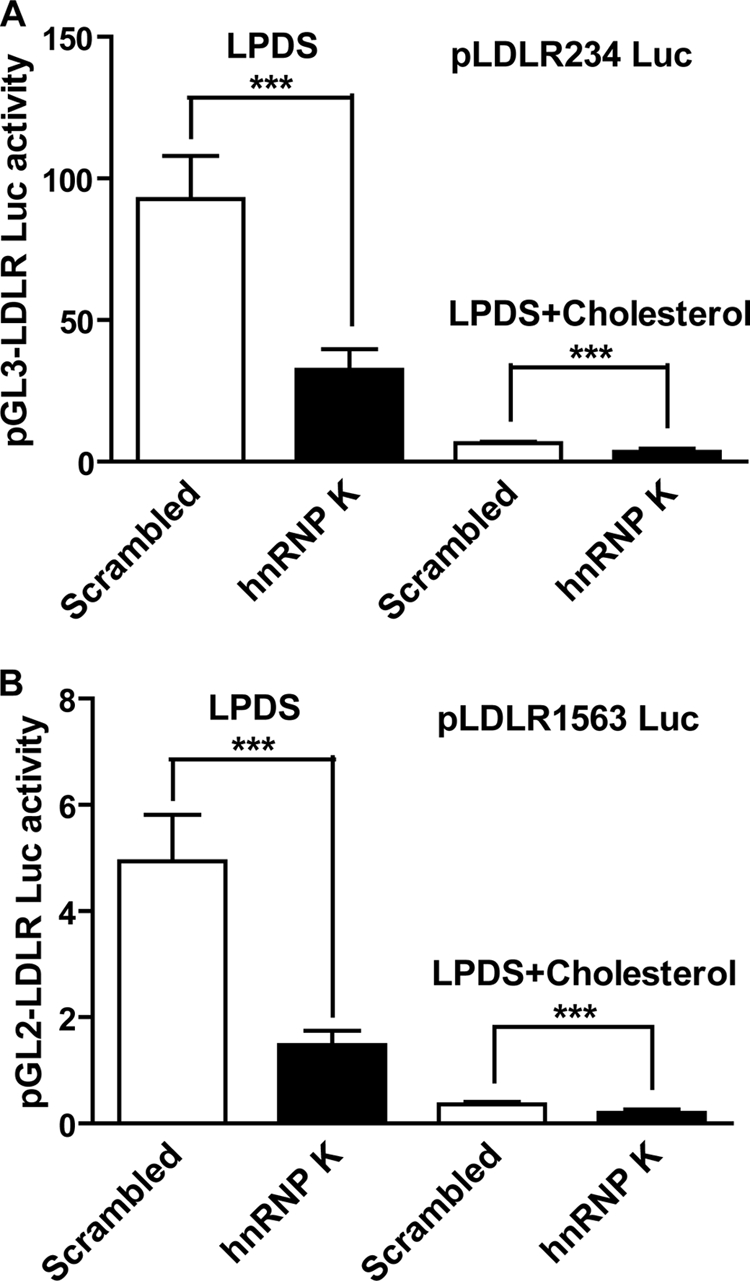
LDLR transactivation by hnRNP K is independent of intracellular cholesterol levels. A, 0.5 μl/well of 2 μm small RNA was mixed with 1.5 × 104 HepG2 cells/well on 96-well cell culture plates. The next day, 100 ng/well of pLDLR234Luc reporter plus 5 ng of pRL-TK reporter as transfection control were transfected into these cells. Two days after reporters' transfection, culture medium was changed to MEM containing 10% LPDS or MEM containing 10% LPDS plus cholesterol (10 μg/ml cholesterol + 1 μg/ml 25-hydroxycholesterol), and cells were incubated for 24 h prior to cell lysis for performing luciferase activity assays. B, dual-luciferase analysis was performed as described in Fig. 4A using pLDLR1563Luc. Each value represents the mean ± S.D. of 6 wells per condition. ***, p < 0.001. The data shown are representative of three separate transfections with similar results.
hnRNP K Binds a Single-stranded CT-rich Element
hnRNP K has been reported to act through a CT-rich element located on promoters of several genes (23). It may act in the same way on the LDLR promoter. After comparison with known hnRNP K binding sites, we found that a CT-rich like element exists within the Sp1 binding site of R3 of the LDLR promoter. Thus, we postulated that hnRNP K positively controls the LDLR gene expression through interaction with the single-stranded CT-rich like element of the LDLR gene. To test this hypothesis, EMSA was performed using biotinylated probes and recombinant hnRNP K protein to examine if hnRNP K can bind the CT-rich element of the LDLR gene. Our results showed that hnRNP K protein binds a single-stranded (ss) probe containing the CT-rich-like element of R3, and excess specific competitor inhibited the formation of the probe-hnRNP K complex, whereas an nonspecific competitor failed to inhibit the biotinylated probe binding to K protein. Furthermore, when an anti-hnRNP K antibody was applied to the probe-protein mixture, the band was further shifted (Fig. 5A). As a positive control, hnRNP K also was shown to bind a c-Myc single-stranded CT element (Fig. 5A). Importantly, the mutation of two nucleotides from CC → TT within this CT-rich stretch on R3D oligonucleotide could not inhibit the binding of K to the biotinylated probe (Fig. 5B). It is interesting to note that unlabeled c-Myc probe was able to completely block the binding of K protein to biotinylated LDLR probe (Fig. 5B). This result provided the evidence that linked the unresponsiveness of the mutated reporter pLDLR234-R3D to K depletion (Fig. 3C) with the loss of K binding to the mutated recognition site on the LDLR promoter.
FIGURE 5.

hnRNP K binds single-stranded LDLR probe containing CT-rich element of LDLR gene promoter. A, EMSA assays were carried out using 5′-biotin end-labeled single-stranded LDLR or c-myc DNA probes with LightShift® Chemiluminescent EMSA kit. Unlabeled single-stranded probes were used as specific competitors, and a DNA fragment from intron 1 of LDLR gene was used as a nonspecific competitor in 100-fold excess. To observe the specific shifted bands, recombinant hnRNP K protein was added to the reaction mixture. An antibody against hnRNP K was added into the DNA-protein reaction mixture to detect supershifted bands. B, EMSA assays were carried out to examine the ability of mutated R3D oligonucleotide to compete the K binding to the labeled probe. C, EMSA assays were carried out using 5′-biotin end-labeled single- or double-stranded LDLR promoter probes. Shifted and supershifted bands were detected by mixing recombinant hnRNP K protein or hnRNP K protein plus anti-hnRNP K antibody, respectively.
To determine if hnRNP K binds double-stranded (ds) probe, similar EMSA assays were performed using biotinylated ds probe instead of ss probe. Results clearly demonstrated that hnRNP K only binds to the ss probe not the ds probe (Fig. 5C). It is known that Sp 1 binds to R3 as well as R1 to maintain the basal transcriptional activity of the LDLR gene. To determine whether Sp1 could bind to single-stranded R3, we performed the EMSA with recombinant Sp1 protein and biotinylated ss R3 probe and the ds R3 probe. Results in Fig. 6 clearly show that recombinant Sp1 only binds to the ds DNA rather than the ss DNA.
FIGURE 6.

Sp1 only binds the double-stranded LDLR DNA probe. EMSA assays were carried out using 5′-biotin end-labeled single- or double-stranded LDLR DNA probes with recombinant purified Sp1 protein. EMSA was conducted as described in legend of Fig. 5.
hnRNP K Locates on the LDLR Promoter Region in Vivo
hnRNP K may regulate LDLR gene transcription in vivo by its binding activity to chromatin. We performed a ChIP assay using samples with or without hnRNP K depletion and antibody to hnRNP K. Results showed that hnRNP K specifically binds the promoter region encompassing the R3 of the LDLR gene, as normal IgG did not precipitate detectable DNA and depletion of hnRNP K by siRNA lost the immunoprecipitation signal (Fig. 7A, upper panel). We validated the specificity of the ChIP assay by examination of the K binding to the irrelevant PCSK9 proximal promoter region (36) or to the DNA fragment of PCSK9 exon 12. These DNA fragments were not present in the immunoprecipitates of control and K-depleted samples (Fig. 7B).
FIGURE 7.

ChIP analyses of hnRNP K and acetylated histone H3 and H4 association with the LDLR promoter in HepG2 cells. A, antibodies to hnRNP K, acetylated histone H3, H4, and HNF1β were used in a ChIP analysis followed by PCR to amplify a 180-bp region surrounding the repeat 3 sequence of LDLR promoter from genomic DNA isolated from HepG2 cells transfected with scrambled siRNA or hnRNP K siRNA. Normal rabbit IgG was included in the assay as negative controls for nonspecific binding. The data shown are representative of three independent assays with similar results. B, ChIP assay was performed to examine the binding of K to the PCSK9 promoter and exon 12 regions as nonspecific controls.
It has been well established that levels of acetylated histone H3 and H4 associated with gene promoters are markers of gene transcriptional status. We further examined acetylated histone H3 and H4 status using our samples. Results demonstrated that the acetylation levels of histone H3 and H4 on the LDLR promoter decreased significantly upon hnRNP K depletion as compared with control samples (Fig. 7A). This provided additional evidence to support the activator role of hnRNP K in LDLR gene transcription.
DISCUSSION
In the present study, a previously undescribed transactivation mechanism for LDLR gene is identified, through which hnRNP K positively regulates LDLR transcription by direct binding to a single-stranded CT-rich element residing in the repeat 3 of the LDLR promoter. Through comparing the LDLR promoter sequence with defined promoter elements of hnRNP K target genes (34), we initially found a putative CT-rich like element within the repeat 3 of LDLR promoter. Our results presented here clearly demonstrated that hnRNP K can indeed regulate LDLR transcription through this putative CT-rich element independent of the classical SRE-1/SREBP regulatory pathway.
First, we used specific siRNA to deplete hnRNP K expression in HepG2 cells to examine the effect of hnRNP K on LDLR expression. We found that hnRNP K was required for LDLR expression as evidenced by real-time RT-PCR and Western blotting (Fig. 1, C and D). By means of the Dil-LDL uptake assay, we observed that LDLR-mediated DiI-LDL uptake was attenuated after hnRNP K depletion (Fig. 1, E and F). In addition, Northern blotting revealed that endogenous LDLR mRNA turnover rate was not changed regardless of hnRNP K levels (Fig. 2, A and B). Moreover, LDLR promoter luciferase assays showed that hnRNP K was indispensable for luciferase activity of the reporter gene (Fig. 2C). Thus, our results firmly established that hnRNP K levels are positively correlated with LDLR expression at the transcriptional level. By performing luciferase assays using a series of reporters containing wild type, mutational, and deletional constructs of LDLR promoters, we found that only mutations at R3 that disturb the CT sequence (wild type R3: AAACTCCTCCCCCTGC → R3D AAACTCCTCttCCTGC) are not affected by hnRNP K depletion. These results are consistent with previous findings that hnRNP K binds CT-rich elements of gene promoters (34, 37, 38).
It is well established that LDLR transcription is mainly regulated by sterol through SREBPs and its binding sequence SRE-1 within the LDLR promoter (4). When the sterol is present, transcription is repressed; while when sterol is depleted, the SREBPs are activated and bind to the SRE site of the LDLR promoter, thus activating the LDLR transcription (4). hnRNP K is required for the LDLR transcription, as the knockdown of the K protein attenuates the LDLR transcription and protein levels in HepG2 cells (Fig. 1), but it is independent of SREBPs and SRE site as evidenced by the observations that the depletion of hnRNP K further repressed transcription in the presence of sterol, and the promoter construct harboring an R2 mutation to disrupt SREBP binding responded to the siRNA-mediated depletion of hnRNP K in a manner similar to the wild-type promoter. A previous study of structural characterization has found that the third KH domain of hnRNP K interacts with single-stranded DNA through its recognition of a tetrad of sequence 5′-d-TCCC (39). It is interesting to note that R3 contains this TCCC within the CT-stretch whereas this sequence motif is absent in R1, which might explain the fact that an R1 mutation disrupted Sp1 binding but had no effect in hnRNP K-mediated LDLR transcription. Our data of EMSAs suggest that hnRNP K regulates the basal level transcription of LDLR through its interaction with the single-stranded CT-rich stretch within R3 (Fig. 5, A and C). The fact that hnRNP K has been demonstrated to bind the CT-element of the c-myc promoter as a single-stranded DNA-binding protein in vivo and may have the ability to modify the transcriptional activity of an adjacent gene through the interconversion of double and single strands (26) and that unlabeled c-Myc probe completely inhibited the binding of the K protein to biotinylated LDLR probe suggest that hnRNP K protein may likely bind to the single-stranded CT-rich element of LDLR promoter in vivo to regulate its transcriptional activity (Fig. 5B).
It has been well established that Sp1 binds to R3, and this binding is important to the basal transcription of LDLR in the absence of SREBPs. The binding of Sp1 to R3 is also required for a cooperative interaction of SREBP2 with Sp1 to activate LDLR transcription under sterol-depleted conditions. Interestingly, unlike hnRNP K, our results showed that Sp1 only binds the double-stranded DNA sequence (Fig. 6). It has been reported that hnRNP K interacts with Sp1 and Sp3 differentially to affect the transactivation of beta4 promoter activity (40). Currently, it is unknown whether Sp1 and hnRNP K interacts with R3 cooperatively or competitively. Further studies are required to delineate the relationship between these two transcription factors with regard to their functional role in LDLR transcription.
hnRNP K can either activate gene transcription (26) or repress transcription by interacting with differential factors (37, 41, 42). We also found that hnRNP K coimmunoprecipitates with LDLR promoter DNA and the hnRNP K-bound LDLR promoter region has a higher level of acetylated histone H3 and H4 as compared with the DNA without the bound K protein, providing additional evidence for an activator role of hnRNP K in LDLR transcription.
In summary, our present study has uncovered a new CT-rich element residing in the R3 sequence of the LDLR promoter and its interacting transactivator hnRNP K that recognizes the single-stranded CT-rich element of the LDLR promoter.
Acknowledgments
We thank Dr. Michael R. Briggs for the critical review of the manuscript and Wei Chen for technical assistance in reporter transfection assays.
This work was supported, in whole or in part, by National Institutes of Health (Grants 1R01 AT002543-01A1 and 1R21AT003195-01A2, to J. L.) from NCCAM. This study was also supported by the Dept. of Veterans Affairs (Office of Research and Development, Medical Research Service, to J. L.).
- LDL
- low density lipoprotein
- hnRNP
- heterogeneous ribonucleoprotein
- EMSA
- electrophoretic mobility shift assay
- LDLR
- LDL receptor
- ChIP
- chromatin immunoprecipitation
- LPDS
- lipoprotein-depleted serum
- R1
- repeat 1
- R2
- repeat 2
- R3
- repeat 3
- SRE
- sterol response element
- ds
- double-stranded
- ss
- single-stranded
- FACS
- fluorescent-activated cell sorting
- siRNA
- small interfering RNA
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
REFERENCES
- 1.Rader D. J., Cohen J., Hobbs H. H. (2003) J. Clin. Invest. 111, 1795–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldstein J. L., Brown M. S. (2001) Science 292, 1310–1312 [DOI] [PubMed] [Google Scholar]
- 3.Goldstein J. L., Brown M. S. (2009) Arterioscler. Thromb. Vasc. Biol. 29, 431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith J. R., Osborne T. F., Goldstein J. L., Brown M. S. (1990) J. Biol. Chem. 265, 2306–2310 [PubMed] [Google Scholar]
- 5.Liu J., Ahlborn T. E., Briggs M. R., Kraemer F. B. (2000) J. Biol. Chem. 275, 5214–5221 [DOI] [PubMed] [Google Scholar]
- 6.Zhang F., Ahlborn T. E., Li C., Kraemer F. B., Liu J. (2002) J. Lipid Res. 43, 1477–1485 [DOI] [PubMed] [Google Scholar]
- 7.Horton J. D., Cohen J. C., Hobbs H. H. (2009) J. Lipid Res. 50, (suppl.), S172–S177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seidah N. G. (2009) Exp. Opin. Ther. Targets 13, 19–28 [DOI] [PubMed] [Google Scholar]
- 9.Davidson M. H. (2009) Curr. Atheroscler Rep. 11, 67–70 [DOI] [PubMed] [Google Scholar]
- 10.Li H., Li H., Ziegler N., Cui R., Liu J. (2009) Recent Pat. DNA Gene Seq. 3, 201–212 [DOI] [PubMed] [Google Scholar]
- 11.Li H., Chen W., Zhou Y., Abidi P., Sharpe O., Robinson W. H., Kraemer F. B., Liu J. (2009) J. Lipid Res. 50, 820–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matunis M. J., Michael W. M., Dreyfuss G. (1992) Mol. Cell Biol. 12, 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makeyev A. V., Liebhaber S. A. (2002) Rna. 8, 265–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi H. S., Hwang C. K., Song K. Y., Law P. Y., Wei L. N., Loh H. H. (2009) Biochem. Biophys. Res. Commun. 380, 431–436 [DOI] [PubMed] [Google Scholar]
- 15.Paziewska A., Wyrwicz L. S., Bujnicki J. M., Bomsztyk K., Ostrowski J. (2004) FEBS Lett. 577, 134–140 [DOI] [PubMed] [Google Scholar]
- 16.Klimek-Tomczak K., Wyrwicz L. S., Jain S., Bomsztyk K., Ostrowski J. (2004) J. Mol. Biol. 342, 1131–1141 [DOI] [PubMed] [Google Scholar]
- 17.Expert-Bezançon A., Le Caer J. P., Marie J. (2002) J. Biol. Chem. 277, 16614–16623 [DOI] [PubMed] [Google Scholar]
- 18.Michael W. M., Eder P. S., Dreyfuss G. (1997) EMBO J. 16, 3587–3598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ostareck D. H., Ostareck-Lederer A., Wilm M., Thiele B. J., Mann M., Hentze M. W. (1997) Cell 89, 597–606 [DOI] [PubMed] [Google Scholar]
- 20.Ostrowski J., Wyrwicz L., Rychlewski L., Bomsztyk K. (2002) J. Biol. Chem. 277, 6303–6310 [DOI] [PubMed] [Google Scholar]
- 21.Ritchie S. A., Pasha M. K., Batten D. J., Sharma R. K., Olson D. J., Ross A. R., Bonham K. (2003) Nucleic Acids Res. 31, 1502–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takimoto M., Tomonaga T., Matunis M., Avigan M., Krutzsch H., Dreyfuss G., Levens D. (1993) J. Biol. Chem. 268, 18249–18258 [PubMed] [Google Scholar]
- 23.Michelotti E. F., Michelotti G. A., Aronsohn A. I., Levens D. (1996) Mol. Cell Biol. 16, 2350–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau J. S., Baumeister P., Kim E., Roy B., Hsieh T. Y., Lai M., Lee A. S. (2000) J. Cell Biochem. 79, 395–406 [DOI] [PubMed] [Google Scholar]
- 25.Hsieh T. Y., Matsumoto M., Chou H. C., Schneider R., Hwang S. B., Lee A. S., Lai M. M. (1998) J. Biol. Chem. 273, 17651–17659 [DOI] [PubMed] [Google Scholar]
- 26.Tomonaga T., Levens D. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 5830–5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Seuningen I., Ostrowski J., Bustelo X. R., Sleath P. R., Bomsztyk K. (1995) J. Biol. Chem. 270, 26976–26985 [DOI] [PubMed] [Google Scholar]
- 28.Bomsztyk K., Van Seuningen I., Suzuki H., Denisenko O., Ostrowski J. (1997) FEBS Lett. 403, 113–115 [DOI] [PubMed] [Google Scholar]
- 29.Moumen A., Masterson P., O'Connor M. J., Jackson S. P. (2005) Cell 123, 1065–1078 [DOI] [PubMed] [Google Scholar]
- 30.Bomsztyk K., Denisenko O., Ostrowski J. (2004) Bioessays. 26, 629–638 [DOI] [PubMed] [Google Scholar]
- 31.Mikula M., Dzwonek A., Karczmarski J., Rubel T., Dadlez M., Wyrwicz L. S., Bomsztyk K., Ostrowski J. (2006) Proteomics 6, 2395–2406 [DOI] [PubMed] [Google Scholar]
- 32.Kong W., Wei J., Abidi P., Lin M., Inaba S., Li C., Wang Y., Wang Z., Si S., Pan H., Wang S., Wu J., Wang Y., Li Z., Liu J., Jiang J. D. (2004) Nat. Med. 10, 1344–1351 [DOI] [PubMed] [Google Scholar]
- 33.Nelson J. D., Denisenko O., Bomsztyk K. (2006) Nat. Protoc. 1, 179–185 [DOI] [PubMed] [Google Scholar]
- 34.Lynch M., Chen L., Ravitz M. J., Mehtani S., Korenblat K., Pazin M. J., Schmidt E. V. (2005) Mol. Cell Biol. 25, 6436–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skalweit A., Doller A., Huth A., Kähne T., Persson P. B., Thiele B. J. (2003) Circ. Res. 92, 419–427 [DOI] [PubMed] [Google Scholar]
- 36.Li H., Dong B., Park S. W., Lee H. S., Chen W., Liu J. (2009) J. Biol. Chem. 284, 28885–28895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michelotti E. F., Tomonaga T., Krutzsch H., Levens D. (1995) J. Biol. Chem. 270, 9494–9499 [DOI] [PubMed] [Google Scholar]
- 38.Stains J. P., Lecanda F., Towler D. A., Civitelli R. (2005) Biochem. J. 385, 613–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braddock D. T., Baber J. L., Levens D., Clore G. M. (2002) EMBO J. 21, 3476–3485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du Q., Melnikova I. N., Gardner P. D. (1998) J. Biol. Chem. 273, 19877–19883 [DOI] [PubMed] [Google Scholar]
- 41.Miau L. H., Chang C. J., Shen B. J., Tsai W. H., Lee S. C. (1998) J. Biol. Chem. 273, 10784–10791 [DOI] [PubMed] [Google Scholar]
- 42.Da Silva N., Bharti A., Shelley C. S. (2002) Blood 100, 3536–3544 [DOI] [PubMed] [Google Scholar]