

Abstract
Regulation of the number of Ca2+-activated K+ channels at the endothelial cell surface contributes to control of the endothelium-derived hyperpolarizing factor response, although this process is poorly understood. To address the fate of plasma membrane-localized KCa2.3, we utilized an extracellular epitope-tagged channel in combination with fluorescence and biotinylation techniques in both human embryonic kidney cells and the human microvascular endothelial cell line, HMEC-1. KCa2.3 was internalized from the plasma membrane and degraded with a time constant of 18 h. Cell surface biotinylation demonstrated that KCa2.3 was rapidly endocytosed and recycled back to the plasma membrane. Consistent with recycling, expression of a dominant negative (DN) RME-1 or Rab35 as well as wild type EPI64C, the Rab35 GTPase-activating protein, resulted in accumulation of KCa2.3 in an intracellular compartment. Expression of DN RME-1, DN Rab35, or wild type EPI64C resulted in a decrease in steady-state plasma membrane expression. Knockdown of EPI64C increased cell surface expression of KCa2.3. Furthermore, the effect of EPI64C was dependent upon its GTPase-activating proteins activity. Co-immunoprecipitation studies confirmed an association between KCa2.3 and both Rab35 and RME-1. In contrast to KCa2.3, KCa3.1 was rapidly endocytosed and degraded in an RME-1 and Rab35-independent manner. A series of N-terminal deletions identified a 12-amino acid region, Gly206–Pro217, as being required for the rapid recycling of KCa2.3. Deletion of Gly206–Pro217 had no effect on the association of KCa2.3 with Rab35 but significantly decreased the association with RME-1. These represent the first studies elucidating the mechanisms by which KCa2.3 is maintained at the plasma membrane.
Keywords: Endocytosis, Endosomes, Endothelium, Potassium Channels, Trafficking, KCa2.3, KCa3.1
Introduction
It has been nearly 30 years since it was first demonstrated that the endothelium plays an obligatory role in the acetylcholine-induced vascular smooth muscle relaxation (1). It is now clearly established that the intermediate (KCa3.1) and small (KCa2.3) conductance Ca2+-activated K+ channels play a critical role in this endothelium-dependent relaxation. Initially, the involvement of KCa2.3 and KCa3.1 was demonstrated using the combination of apamin, a selective KCa2.x blocker, and charybdotoxin (an inhibitor of KCa3.1 (2–4)), although recent studies have employed the more specific KCa3.1 inhibitor TRAM-34 (5, 6). Definitive evidence for the involvement of KCa2.3 in the regulation of vascular tone and hence blood pressure came from the work of Nelson and co-workers (7) in which it was demonstrated, using transgenic mice, that KCa2.3 was responsible for the sustained hyperpolarization that is communicated to the vascular smooth muscle, and suppression of KCa2.3 expression resulted in an elevation of blood pressure. Similarly, Kohler and co-workers (8) demonstrated that the targeted knock-out of KCa3.1 in mice resulted in an attenuated endothelium-derived hyperpolarizing factor (EDHF)2 response and mild arterial hypertension. Recent studies using mice deficient in both KCa2.3 and KCa3.1 have confirmed the critical role these channels play in the EDHF response and hence blood pressure regulation (9).
Although it is clear that KCa2.3 and KCa3.1 are required for the EDHF response, their exact roles are still being clarified. However, mounting evidence indicates that the hyperpolarization induced in the endothelial cells by Ca2+-mediated agonists, and hence activation of KCa channels, is directly communicated to the smooth muscle cells via myoendothelial gap junctions (10–12). Indeed, uncoupling of the gap junctions attenuated the acetylcholine-induced hyperpolarization of the underlying smooth muscle, consistent with a direct current flow between cells (10). Recent evidence, using double KCa2.3/KCa3.1 knock-out mice, demonstrates that KCa3.1 deficiency attenuates the acetylcholine-induced, EDHF-mediated vasodilation, whereas KCa2.3 knock-out impairs the NO-mediated dilation induced by acetylcholine (9). Additionally, KCa3.1 appears to play a critical role in the propagation of the acetylcholine-induced dilation along the arterioles (13). A second, perhaps parallel role for KCa channels is to conduct K+ ions into the intracellular space resulting in an activation of the Na+/K+-ATPase and ultimately vascular relaxation (10). Recent immunolocalization studies have suggested that KCa2.3 and KCa3.1 are perfectly positioned to play a role in the EDHF response as outlined above, i.e. KCa3.1 has been shown to be localized primarily to the myoendothelial gap junctions, whereas KCa2.3 is more uniformly distributed across the endothelial cell surface, including at the myoendothelial gap junctions (10, 14).
The magnitude of the EDHF response, induced by activation of KCa3.1 and KCa2.3, is a direct consequence of the total current flow generated by these channels, which in turn is directly proportional to the number of actively gating channels (N) in the plasma membrane, the probability that a channel is open (Po), and the current flow through an individual ion channel (i). Although a great deal has been learned about the second messenger-dependent regulation of these KCa channels in terms of altering Po (15–21), much less information exists regarding the mechanisms by which N is determined. We (20, 22–24) and others (25–27) have identified numerous motifs in the N and C termini of KCa2.3 and KCa3.1, which are required for the proper assembly and anterograde trafficking of these channels to the plasma membrane. However, to date, no studies have defined the retrograde transport of KCa2.3 or KCa3.1 from the plasma membrane. As these are typically dynamic processes, modulation of these endocytic and/or recycling events will alter N and therefore the physiological response of the cell. Here, we present the first studies demonstrating that KCa2.3 is rapidly endocytosed and recycled back to the plasma membrane, whereas KCa3.1 does not enter the recycling pathway. We further demonstrate that the recycling of KCa2.3 is dependent upon RME-1, Rab35, and the Rab GAP, EPI64C. Finally, we identify a 12-amino acid motif within the N terminus of KCa2.3 whose deletion results in the rapid endocytosis and targeting for degradation.
EXPERIMENTAL PROCEDURES
Molecular Biology
The GFP-tagged receptor-mediated endocytosis-1 (RME-1) construct was generously provided by Dr. Barth D. Grant (Rutgers University). The GFP-tagged and monomeric RFP-tagged (mRFP) variants of Rab35 and EPI64C as well as the S22N Rab35 and R141K EPI64C were generated as described previously (28). KCa2.3 (also referred to as SK3) and KCa3.1 (also referred to as IK1 or SK4) cDNAs were kindly provided by J. P. Adelman (Vollum Institute, Oregon Health Sciences University) and subcloned into pcDNA3.1(+) (Invitrogen). To insert the biotin ligase acceptor peptide (BLAP) sequence (GLNDIFEAQKIEWHE) into the extracellular loop between transmembrane domains S3 and S4 of KCa3.1 and KCa2.3, we first introduced a BglII (KCa2.3) or NotI (KCa3.1) site such that the BLAP sequence could be directly annealed. In the case of KCa2.3, the following primers were utilized: forward (gatcggtggcggtctgaacgacatcttcgaggctcagaaaatcgaatggcacgaagg) and reverse (gatcccttcgtgccattcgattttctgagcctcgaagatgtcgttcagaccgccacc), where the boldface indicates nucleotides corresponding to the BLAP sequence, the underlined nucleotides indicate those corresponding to the BglII overhang, the nucleotides in italics represent glycines added to provide flexibility at the front and back of the insert, and the nucleotides in normal type were added to maintain the correct reading frame (note that this will result in the addition of a glycine at the 3′ end of the BLAP sequence). Chimeras between KCa2.3 and KCa3.1 were generated by overlap extension PCR as described previously (19). The 26KCa3.1-KCa2.3 chimera was tagged with a C-terminal myc epitope (EQKLISEEDL) using a single step PCR. All deletions and mutations were generated using the Stratagene QuikChangeTM site-directed mutagenesis strategy (Stratagene, La Jolla, CA). The fidelity of all constructs utilized in this study was confirmed by sequencing (ABI PRISM 377 automated sequencer, University of Pittsburgh) and subsequent sequence alignment (NCBI BLAST) using GenBankTM accession numbers AF022150 (KCa3.1) and U69884 (KCa2.3).
Small hairpin RNA suppression vectors directed against EPI64C were described previously (28). Co-transfections of shRNA directed against EPI64C (sh-EPI) and KCa2.3 in HEK cells were performed 48 h prior to experimentation.
Cell Culture
Human embryonic kidney (HEK293) cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a humidified 5% CO2, 95% O2 incubator at 37 °C. Cells were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Stable cell lines were generated for all constructs by subjecting cells to antibiotic selection (1 mg/ml G418). Note that clonal cell lines were not subsequently selected from this stable population to avoid clonal variation. The human microvascular endothelial cell line, HMEC-1 (29), was generously provided by Dr. Edwin Ades (Centers for Disease Control and Prevention), Francisco J. Candal (Centers for Disease Control and Prevention), and Dr. Thomas Lawley (Emory University) and was cultured in MCDB-131 complete media (VEC Technologies, Rensselear, NY). The HMEC-1 cell line has been extensively studied and has been shown to have morphological, phenotypical, and functional characteristics of human microvascular endothelial cells (29, 30), making it an excellent in vitro model for our studies. HMEC-1 cells were transfected by electroporation using an AMAXA Nucleofector II (Lonza, Walkersville, MD). Electroporations were performed with 5 μg of total DNA per 2 × 106 cells.
Electrophysiology, Inside-out Patch Clamp Experiments
The effects of Ca2+, 5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one (DCEBIO), and clotrimazole on KCa2.3 and KCa3.1 were assessed with inside-out patch clamp experiments as a functional assay. During patch clamp experiments, the bath solution contained 145 mm potassium gluconate, 5 mm KCl, 2 mm MgCl2, 10 mm HEPES, and 1 mm EGTA (pH adjusted to 7.2 with KOH). Sufficient CaCl2 was added to obtain the desired free Ca2+ concentration (program kindly provided by Dr. Dave Dawson, Oregon Health Sciences University). To obtain a 0 Ca2+ bath solution, EGTA (1 mm) was added without CaCl2 (estimated free Ca2+ <10 nm). For Ca2+ concentration-response experiments, the Ca2+ was directly applied to the excised patch utilizing a BioLogic RSC-160 rapid solution changer (BioLogic Science Instruments, Claix, France). The subsequent data were fitted to the concentration-response Equation 1,
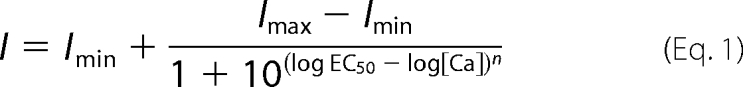 |
An apparent Ca2+ sensitivity (K0.5) and Hill coefficient (n) were determined. The pipette solution was 140 mm potassium gluconate, 5 mm KCl, 1 mm MgCl2, 10 mm HEPES, and 1 mm CaCl2 (pH adjusted to 7.2 with KOH). All experiments were performed at room temperature. All patches were held at a holding potential of −100 mV. Total channel current was determined using Clampfit 8.2 (Axon Instruments).
Antibodies
α-KCa2.3 Ab was obtained from Chemicon (Temecula, CA), α-GFP Ab was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), α-HA Ab was obtained from Covance (Princeton, NJ), and α-streptavidin Ab was obtained from GenScript Corp. (Piscataway, NJ). Antibodies against EPI64C and Rab35 were described previously (28).
Biotinylation of KCa2.3 and KCa3.1 Using BirA
Biotin ligase (BirA) was either purchased from Avidity (Aurora, CO) or expressed from pET21a-BirA (generously provided by Dr. Alice Y. Ting, Massachusetts Institute of Technology) in Escherichia coli according to methods published previously (31). HEK or HMEC-1 cells expressing BLAP-tagged KCa2.3 or KCa3.1 were incubated in PBS containing BirA (0.03 μg/μl), 10 mm ATP, 50 μm biotin, 1 mm Ca2+, and 2 mm Mg2+ for 30 min at room temperature. The cells were then washed three times with PBS to remove the BirA and incubated in PBS containing 1% BSA plus either streptavidin-Alexa488, streptavidin-Alexa555, or streptavidin-Cy5 (0.01 mg/ml) for 10 min at 4 °C. The cells were extensively washed with PBS/BSA to remove unbound streptavidin and then either incubated for various periods of time as indicated in the text at 37 °C or immediately fixed and permeabilized with 2% paraformaldehyde plus 0.1% Triton X-100. Nuclei were labeled with Hoechst 33258 (Sigma). Cells were imaged in one of two ways as indicated in the figure legends. In some cases, cells were subjected to laser confocal microscopy using an Olympus FluoView 1000. To ensure maximal X-Y spatial resolution, sections were scanned at 1024 × 1024 pixels, using sequential image collection to minimize cross-talk between the imaged channels. In other experiments, cells were imaged using an Olympus IX-81 wide field fluorescence microscope. Multiple planes were imaged, deconvolved using a point-spread function, and presented as a projection image.
Immunofluorescence (IF)
KCa2.3 was transiently transfected into HEK cells grown on poly-l-lysine (Sigma)-coated glass coverslips, and 16–24 h later, the cells were fixed and permeabilized as above. Blocking was performed with 1% BSA plus 10% goat serum. KCa2.3 was labeled sequentially with 1° Ab directed against the channel and 2° Cy3.18-conjugated goat anti-mouse IgG antibody. Imaging was carried out as above.
Recycling Assay
Cell surface proteins, including BLAP-tagged KCa2.3, were biotinylated using EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific, Rockford, IL) at 4 °C; the unreacted biotin was quenched (PBS plus 1% BSA), and the cells were warmed to 37 °C to allow channel endocytosis for various periods of time, as indicated. The remaining cell surface biotin was stripped using MESNA (100 mm sodium 2-mercaptoethanesulfonate, 50 mm Tris, 100 mm NaCl, 1 mm EDTA, 0.2% BSA) after which the cells were lysed (50 mm HEPES, pH 7.4, 150 mm NaCl, 1% v/v Triton X-100, 1 mm EDTA containing CompleteTM EDTA-free protease inhibitor mixture, Roche Applied Science), and the protected (endocytosed) biotin-tagged channels were subjected to pulldown using streptavidin-agarose (Sigma) following normalization of protein concentrations. Proteins were resolved by SDS-PAGE (8% gel) and transferred to nitrocellulose for immunoblot analysis of KCa2.3 using the SNAP i.d.TM system (Millipore, Billerica, MA), according to the manufacturer's instructions. In additional experiments, the NHS-SS-Biotin-tagged KCa2.3 channels were allowed to be endocytosed at 37 °C, after which the remainder of cell surface biotin was stripped, and the cells were then rewarmed to 37 °C to allow endocytosed KCa2.3 to recycle. The channels that returned to the cell surface were then stripped of biotin as above, and the remaining nonrecycling channels were quantified by pulldown and IB as above. In this case, a decrease in signal over time was indicative of recycling.
Immunoprecipitations (IP)
Cells were lysed as above, and protein concentrations were determined and normalized to achieve equivalent loading. Crude lysates were pre-cleared with protein A/G-Sepharose beads (Sigma) and incubated with the appropriate antibody. Immune complexes were precipitated with protein A/G-Sepharose beads, washed extensively, and resuspended in Laemmli sample buffer. Proteins were resolved by SDS-PAGE (8% gel) and transferred to nitrocellulose for immunoblot analysis.
Determination of Steady-state and Half-life Measurements for Plasma Membrane-localized KCa2.3 and KCa3.1
For KCa2.3, total plasma membrane protein was biotinylated as above, and the cells were returned to 37 °C for various periods of time, as indicated. Note that for steady-state measurements of plasma membrane-localized channel, the cells were not returned to 37 °C. The cells were then lysed, and the biotinylated proteins were pulled down using streptavidin-agarose, resolved by SDS-PAGE, transferred to nitrocellulose, and then subjected to IB for KCa2.3 as above. The IB was quantified by densitometry, and the data were fitted to either a first-order exponential decay or quadratic function to determine the time constant (τ) or half-life (t½) for protein degradation, where t½ = 0.693·τ. As an alternative method for determining the plasma membrane half-life of KCa2.3 as well as KCa3.1, the channel was specifically biotinylated using BirA as above after which the cells were incubated for various periods of time at 37 °C, as indicated. The cells were then lysed, and equivalent amounts of total protein were separated by SDS-PAGE, transferred to nitrocellulose, and probed for streptavidin. As streptavidin remains tightly coupled to the channel during SDS-PAGE, this provides a direct correlate to the IF studies detailed above.
Determination of Total Protein Half-life
To determine the time constant for channel degradation, HEK cells expressing the channel of interest were incubated in cycloheximide (400 μg/ml) for the indicated times at 37 °C, after which they were immediately cooled to 4 °C by washing in ice-cold PBS and prepared for IB as above.
Chemicals
All chemicals were obtained from Sigma, unless otherwise stated. DCEBIO was synthesized in the laboratory of R. J. Bridges (Rosalind Franklin University), as described previously (32). Both DCEBIO and clotrimazole were made as 10,000-fold stock solutions in DMSO.
Statistics
All data are presented as means ± S.E., where n indicates the number of experiments. Statistical analysis was performed using Student's t test. A value of p < 0.05 was considered statistically significant and is reported. Time constants for protein decay were determined by fitting the data to a single exponential decay function (SigmaPlot 2001).
RESULTS
Insertion of the BLAP Sequence into an Extracellular Loop Does Not Alter the Function of KCa2.3 or KCa3.1
To investigate the fate of plasma membrane-localized KCa2.3 and KCa3.1, we introduced the BLAP sequence into the second extracellular loop of each of these channels. Initially, we determined whether inserting the BLAP epitope altered the function of KCa2.3 or KCa3.1 expressed in HEK cells using the inside-out patch clamp technique. As shown in Fig. 1, both KCa2.3-BLAP (A–C) and KCa3.1-BLAP (D–F) demonstrated normal responses to both Ca2+ and the pharmacological opener, DCEBIO (32). Insertion of the BLAP sequence also had no effect on the clotrimazole-dependent inhibition of KCa3.1 (Fig. 1, D and E). Finally, the apparent Ca2+ sensitivity of these channels was not significantly affected by the introduction of the BLAP sequence as shown in Fig. 1, C and F. Taken together, these results suggest that these BLAP-tagged channels can be utilized to define the fate of plasma membrane-localized KCa2.3 and KCa3.1.
FIGURE 1.

Insertion of the BLAP sequence into the 2nd extracellular loop of KCa2.3 and KCa3.1 does not affect channel function. A, inside-out patch clamp recording at a holding potential of −100 mV in symmetric K+ of BLAP-tagged KCa2.3 from HEK cells. Changing free bath Ca2+ from 10 μm to Ca2+-free and back to 0.4 μm caused the channel to close and then activate as expected. Subsequent addition of DCEBIO (30 μm) caused a further increase in channel activity, which was eliminated in 0 Ca2+. B, average fold-increase in response to DCEBIO (30 μm) for KCa2.3 and BLAP-tagged KCa2.3. No significant difference in responses was observed. C, Ca2+ concentration-response curves for KCa2.3 (solid line) and BLAP-tagged KCa2.3 (dashed line). The data were fitted to the Hill equation to determine K0.5 and Hill coefficients (n). KCa2.3 had a K0.5 of 416 ± 42 nm with an n of 3.8 ± 0.3 (n = 8), whereas BLAP-tagged KCa2.3 had a K0.5 of 302 ± 18 nm with an n of 4.6 ± 0.4 (n = 9). D, inside-out patch clamp recording at a holding potential of −100 mV in symmetric K+ of BLAP-tagged KCa3.1 from HEK cells. Changing from 0 Ca2+ to 10 μm caused the channel to activate as expected. Subsequent addition of DCEBIO (30 μm) caused a further increase in channel activity, which was blocked by clotrimazole (CLT, 10 μm). E, average relative current response to DCEBIO and clotrimazole for KCa3.1 (open bars) and BLAP-tagged KCa3.1 (shaded bars). No significant difference in responses was observed. F, Ca2+ concentration-response curves for KCa3.1 (solid line) and BLAP-tagged KCa3.1 (dashed line). The data were fitted to Equation 1 as shown under the “Experimental Procedures” to determine K0.5 and Hill coefficients (n). KCa3.1 had a K0.5 of 679 ± 35 nm with an n of 2.3 ± 0.2 (n = 9), whereas BLAP-tagged KCa2.3 had a K0.5 of 653 ± 48 nm with an n of 2.0 ± 0.1 (n = 9).
KCa2.3 Enters a Rapidly Recycling Endosomal Compartment
To define the fate of plasma membrane-localized channels, we labeled KCa3.1 (Fig. 2A, top panels) and KCa2.3 (Fig. 2A, bottom panels) in HEK (left panels) and HMEC-1 (right panels) cells using BirA and streptavidin-Alexa555 and determined localization at various times after returning the cells to 37 °C. At time 0 h, the labeled channel is expressed at the cell surface as expected. Note that in the HMEC-1 cells, this labeling appears as a discontinuous pattern across the entire cell surface due to the fact these cells are only 2–3 μm thick. Importantly, no labeling was observed if we expressed wild type KCa2.3 or KCa3.1 (no BLAP tag) and carried out the labeling procedure as above. Similarly, if the BirA labeling step was omitted, streptavidin exposure alone failed to label BLAP-tagged KCa2.3 or KCa3.1 (data not shown). In both HEK and HMEC-1 cells, nearly all of the KCa3.1 channels had left the membrane after 1 h, after being localized to endosomes. After 5 h, some KCa3.1 was still visible in the endosomes, but significant channel had been degraded as assessed by the reduced fluorescence signal. In contrast, after 5 h in HEK cells and 1 h in HMEC-1 cells, virtually all of the KCa2.3 channel remained at the membrane. Indeed, even after 12 h in HEK cells and 8 h in HMEC-1 cells, the KCa2.3 channel was still present at the plasma membrane, although significant channel had been endocytosed. These results suggest that KCa2.3 has a much longer plasma membrane residency than KCa3.1 in both HEK and HMEC-1 cells. Although our results in HMEC-1 and HEK cells were qualitatively similar, it should be noted that KCa2.3 appeared to be more rapidly endocytosed in the HMEC-1 cell line. Unfortunately, given the poor transfection efficiency of KCa2.3 into HMEC-1 cells (<10%), this potential difference could not be biochemically quantified. However, these effects were quantified in HEK cells via biotinylation. As shown in Fig. 2C (upper blot), using a standard biotinylation protocol, plasma membrane-localized KCa2.3 had a time constant for degradation of 18.9 ± 0.2 h (t½ = 13.1 h, n = 3, solid line, right panel). We also determined the residency time for plasma membrane KCa2.3 by specifically biotinylating the channel using BirA (Fig. 2C, lower blot), labeling with streptavidin, and then allowing internalization (see “Experimental Procedures”). Most importantly, as shown in Fig. 2C, the association of streptavidin with the channel did not significantly influence the time constant for degradation of KCa2.3, being 17.5 ± 1.1 h (n = 3, dashed line, right panel), indicating that our IF data accurately reflects the fate of the channel. Similarly, we evaluated the degradation of KCa3.1 by specifically biotinylating with BirA. As shown in Fig. 2D, KCa3.1 was degraded much more rapidly than KCa2.3; however, this degradation could not be fit to an exponential decay function. Rather, these data indicate that channel protein remains stable during the initial endocytic process, after which the channel is rapidly degraded between 6 and 12 h. These initially stable protein levels are expected during the endocytic process and are also observed for KCa2.3 when this channel is evaluated over a short time frame (see Fig. 3A). These widely disparate half-lives suggest that KCa2.3 and KCa3.1 are uniquely anchored and/or endocytosed/recycled from the plasma membrane in both HEK and HMEC-1 cells.
FIGURE 2.

Time course for internalization and degradation of plasma membrane-localized KCa2.3 and KCa3.1 in HEK and HMEC-1 cells. HEK (A) or HMEC-1 (B) cells expressing either BLAP-tagged KCa3.1 (top panels) or KCa2.3 (bottom panels) were labeled with streptavidin-Alexa555, and localization was evaluated at the time points indicated. At time 0 h, both KCa3.1 and KCa2.3 were localized exclusively to the plasma membrane. After 1 h at 37 °C, nearly all of the KCa3.1 had been endocytosed, and after 5 h significant channel was degraded as evidenced by the reduced fluorescence signal. In contrast, KCa2.3 remains at the plasma membrane for an extended period of time, and even after 8 (B) or 12 (A) h, a significant channel is observed in both the plasma membrane as well as in an endosomal compartment. Nuclei are labeled with DAPI (blue). HEK cells are shown as single confocal sections, whereas HMEC-1 cells are shown as projection images from multiple z-sections. C, to quantify the time course for plasma membrane KCa2.3 degradation in HEK cells, we either randomly biotinylated plasma membrane proteins using EZ-Link Sulfo-NHS-SS-Biotin (upper blot) followed by streptavidin pulldown and blotted for KCa2.3 or specifically biotinylated BLAP-tagged KCa2.3 using BirA (lower blot), labeled with streptavidin, and then blotted for streptavidin at time 0, 12, 24, or 36 h at 37 °C (see “Experimental Procedures”). In the latter case, 20 μg of total protein was loaded per lane. Note that the channel runs at a different apparent molecular mass for these two conditions because of the addition of streptavidin in the lower blot. Blots were quantified by densitometry and fit to an exponential decay function with time constants of 18.9 ± 0.2 h (n = 3) for KCa2.3 (solid line, circles) and 17.5 ± 1.1 h (n = 3) for BLAP-tagged KCa2.3 (dashed line, triangles). D, degradation of plasma membrane-localized BLAP-tagged KCa3.1 was quantified in HEK cells by specifically biotinylating using BirA. Following labeling, the cells were returned to 37 °C for 0, 3, 6, 9, or 12 h, as indicated for the representative blot shown in the left panel. 20 μg of total protein was loaded per lane. The data were quantified by densitometry and plotted as shown. KCa3.1 protein levels initially decayed slowly and then exhibited rapid degradation between 6 and 12 h (right panel, n = 3).
FIGURE 3.
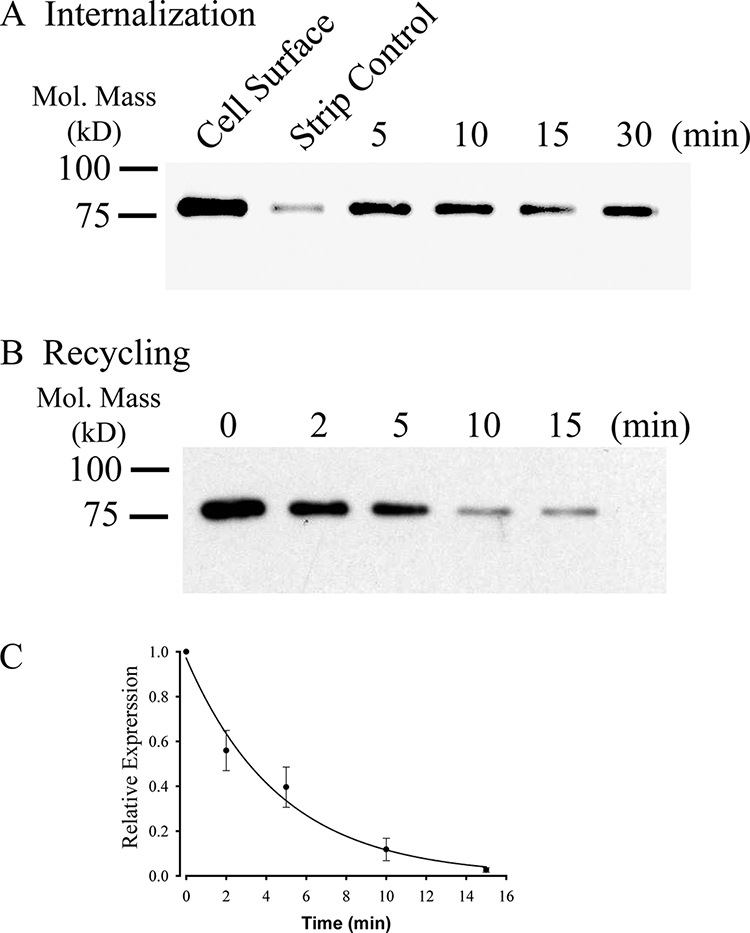
KCa2.3 recycles to the plasma membrane in HEK cells. A, representative IB demonstrating that plasma membrane KCa2.3 is rapidly endocytosed to a steady-state level in HEK cells. Plasma membrane proteins were biotinylated using EZ-Link Sulfo-NHS-SS-Biotin (see “Experimental Procedures”) after which the cells were incubated at 37 °C for the times indicated. Initial plasma membrane expression was determined by omitting the 37 °C incubation step (cell surface, 1st lane). The remaining cell surface biotin was stripped (MESNA), after which the endocytosed, biotinylated protein was pulled down using streptavidin-agarose, and the proteins were separated by SDS-PAGE and blotted for KCa2.3. The efficiency of stripping was determined by subjecting cells to MESNA in the absence of a 37 °C endocytosis step (strip control). The 3rd to 6th lanes demonstrate rapid endocytosis of KCa2.3 to a steady-state level. Similar results were observed in three separate experiments. B, representative IB demonstrating that KCa2.3 recycles back to the plasma membrane following endocytosis. Plasma membrane proteins were biotinylated and allowed to endocytose for 30 min at 37 °C as in A, after which the remaining cell surface biotin was stripped. As shown in the 1st lane, KCa2.3 was endocytosed as in A. The cells were returned to 37 °C for various periods of time as indicated, after which the cells were subject to a second round of biotin stripping from the cell surface. If KCa2.3 returns to the plasma membrane, the biotin associated with the channel will be stripped resulting in a decreasing signal upon IB, and this is what is observed. C, experiment in B was repeated three times, and the resulting IB was digitized, and band intensities for the various time points were determined as a percent change from time 0. The decrease in protein was fit to an exponential decay function with a time constant of 4.7 ± 0.3 min (n = 3).
Given the long residence time of KCa2.3 in the plasma membrane, we determined whether this was due to the rapid endocytosis and recycling of the channel rather than the channel remaining static in the membrane. Initially, we determined whether KCa2.3 was endocytosed to a steady-state level. For these studies, cell surface proteins were biotinylated and allowed to endocytose for 5, 10, 15, or 30 min at 37 °C, after which the remaining cell surface biotin was stripped. In this case, only proteins that were endocytosed remain biotinylated. These endocytosed and therefore biotinylated proteins are then pulled down followed by IB for KCa2.3. As shown in Fig. 3A, KCa2.3 rapidly achieved a steady-state level of accumulation inside the cell, demonstrating that the channel is rapidly endocytosed from the plasma membrane. To determine whether KCa2.3 recycles, the channel was biotinylated and allowed to endocytose for 30 min as above, after which the remaining cell surface biotin was stripped. The cells were then incubated for 2, 5, 10, or 15 min at 37 °C followed by a second round of biotin stripping from the cell surface. In this case, if the channel that was endocytosed, and is therefore still biotinylated, returns to the cell surface, its biotin will be stripped off such that following pulldown and IB the signal associated with KCa2.3 will decrease with time. As shown in Fig. 3B, this is the result obtained, with the signal decreasing as the channel recycles back to the plasma membrane. These data were fitted with a time constant of 4.7 ± 0.4 min (n = 3; Fig. 3C), conclusively demonstrating that KCa2.3 is rapidly endocytosed and recycled back to the plasma membrane.
KCa2.3 Enters an RME-1-positive Recycling Compartment
Lin et al. (33) demonstrated that RME-1 is required for the recycling of numerous proteins back to the cell surface, following endocytosis. Indeed, dominant negative RME-1 (DN RME-1) has been shown to inhibit the exit of membrane proteins from recycling endosomes resulting in a dramatic expansion of this compartment in both Caenorhabditis elegans and mammalian systems (33–35). Thus, we determined whether KCa2.3 would become localized to an RME-1 compartment following endocytosis from the plasma membrane. For these experiments, BLAP-tagged KCa2.3 and GFP-tagged DN RME-1 were co-transfected into either HEK or HMEC-1 cells. The plasma membrane-localized channel was labeled with streptavidin-Alexa555 and allowed to internalize for either 1, 5, or 12 h at 37 °C, after which co-localization was assessed by IF. As shown in Fig. 4A for HEK cells, KCa2.3 is initially located at the plasma membrane (time 0 h, upper panels) demonstrating that DN RME-1 does not preclude plasma membrane expression of the channel. After 1 h at 37 °C (Fig. 4A, middle panels), only a small amount of KCa2.3 had been endocytosed; however, after 12 h at 37 °C, KCa2.3 had accumulated into an RME-1-containing structure in the cell (lower panels), consistent with KCa2.3 recycling through an RME-1-containing compartment. Similarly, in HMEC-1 cells, after 5 h at 37 °C virtually all the channel that had been endocytosed from the plasma membrane was localized in the RME-1-containing vesicles (Fig. 4B). To confirm that co-localization of KCa2.3 and RME-1 is independent of the DN RME-1 phenotype, we carried out identical experiments in HEK cells expressing wild type GFP-tagged RME-1 and BLAP-tagged KCa2.3. As shown in Fig. 4C, after 3 h at 37 °C, the majority of KCa2.3 is localized to the plasma membrane as expected; however, KCa2.3 is also localized to small RME-1-positive vesicles, confirming KCa2.3 enters an RME-1-delimited recycling compartment. To confirm this localization is independent of the BLAP tag, we co-transfected either wild type (Fig. 4D) or DN (Fig. 4E) GFP-RME-1 and untagged KCa2.3 and evaluated steady-state localization by IF. As was apparent, a small fraction of KCa2.3 localized with wild type RME-1 in recycling endosomes, and the expression of DN RME-1 results in the accumulation of KCa2.3 in a dramatically expanded recycling compartment as described previously (35). To determine whether DN RME-1 expression alters the expression of KCa2.3, we carried out immunoblots following co-expression of KCa2.3 with either WT RME-1 or DN RME-1. As shown in Fig. 4F, DN RME-1 had a small but significant effect on total KCa2.3 expression, reducing it an average of 30 ± 3% (n = 3, p < 0.05) relative to WT RME-1. Our results suggest that RME-1 is required for recycling of KCa2.3 back to the plasma membrane, thus predicting that in the presence of a DN RME-1, the steady-state accumulation of KCa2.3 at the plasma membrane would be reduced. This was confirmed by cell surface biotinylations, as shown in Fig. 4G. By comparing the 0-h time points in the presence of WT (Fig. 4G, left) or DN (right) RME-1, it is clear that KCa2.3 expression at the plasma membrane is reduced when co-expressed with DN RME-1. In four experiments, DN RME-1 reduced plasma membrane expression of KCa2.3 an average 62 ± 3.5% (p < 0.05). This decrease is significantly more than the change in total KCa2.3 expression, consistent with RME-1 affecting plasma membrane expression of the channel via an effect on recycling. Furthermore, we determined the half-life of plasma membrane KCa2.3 in the presence of either WT or DN RME-1. As shown in Fig. 4G (bottom panel), in the presence of DN RME-1 (dashed line, triangles), KCa2.3 degradation followed an exponential decay having a t½ of 9.1 h (τ = 13.1 ± 1.5 h, n = 4), which is faster than the 13.1-h t½ for KCa2.3 in the absence of RME-1. In contrast, in the presence of WT RME-1, the degradation of plasma membrane KCa2.3 could not be fit to an exponential function (Fig. 4G, bottom panel, solid line, circles). Therefore, to estimate the t½ for KCa2.3 in the presence of WT RME-1, the data were fitted to a quadratic function, yielding a t½ of 22.4 h (n = 4), which is longer than in the absence of RME-1. These data are consistent with WT RME-1 fostering the recycling of KCa2.3 and hence increasing the t½ of plasma membrane KCa2.3, whereas DN RME-1 inhibits the recycling of KCa2.3, resulting in a reduced t½ for the plasma membrane-localized channel. Finally, we determined whether KCa2.3 is associated with RME-1 by co-immunoprecipitation. As shown in Fig. 4H, following IP of wild type GFP-tagged RME-1, we were able to detect KCa2.3 upon IB, confirming KCa2.3 enters the RME-1-positive recycling compartment. In contrast, KCa3.1, which is rapidly endocytosed and degraded, does not enter the recycling compartment defined by RME-1 (Fig. 4I).
FIGURE 4.

KCa2.3 enters RME-1-positive endosomes in HEK and HMEC-1 cells, whereas KCa3.1 does not. A, BLAP-tagged KCa2.3 was co-expressed with GFP-tagged DN RME-1 in HEK cells. KCa2.3 was labeled with streptavidin-Alexa555 and localization evaluated after 0, 1, and 12 h at 37 °C. KCa2.3 is localized to the plasma membrane at 0 h (upper panels), and after 1 h (middle panels) very little channel is localized to intracellular vesicles. However, after 12 h (lower panels), KCa2.3 has accumulated in the RME-1-expressing recycling compartment. B, BLAP-tagged KCa2.3 was co-expressed with GFP-tagged DN RME-1 in HMEC-1 cells, labeled as above, and localization evaluated after 0 (top panels) or 5 (bottom panels) h at 37 °C. At time 0, all of KCa2.3 is localized to the plasma membrane, whereas there is nearly complete co-localization of KCa2.3 with the RME-1-positive recycling compartment following endocytosis. C, BLAP-tagged KCa2.3 was co-expressed with GFP-tagged RME-1 in HEK cells. KCa2.3 was labeled as above, and localization was evaluated after 3 h at 37 °C. D and E, KCa2.3 (no BLAP tag) was co-expressed with either wild type (D) or DN (E) GFP-tagged RME-1 and steady-state localization assessed by IF. F, immunoblot of total lysate following co-expression of KCa2.3 with WT RME-1 (1st lane) or DN RME-1 (2nd lane). DN RME-1 reduced KCa2.3 expression an average of 30 ± 3% (n = 3). G, KCa2.3 was co-expressed with either WT (left panel) or DN (right panel) RME-1, and cell surface channel was biotinylated. At the indicated times, cells were lysed, subjected to streptavidin pull-down, and blotted for KCa2.3. At time 0, DN RME-1 reduced plasma membrane KCa2.3 expression an average of 62 ± 3.5% (p < 0.05, n = 4). The bands were quantified by densitometry, normalized, and plotted as a function of time (bottom panel) for DN (dashed line, triangles) and WT (solid line, circles) RME-1. H, co-immunoprecipitation of KCa2.3 with GFP-tagged RME-1 was carried out in HEK cells. RME-1 was immunoprecipitated via its GFP tag, and the subsequent blot was probed for KCa2.3. Cells expressed either RME-1 plus vector (1st lane), KCa2.3 plus vector (2nd lane), or KCa2.3 plus GFP-RME-1 (3rd and 4th lanes). The V5 Ab was used as an IgG control in the 3rd lane. 10 μg of total protein was loaded in the lysate lane to confirm the identity of KCa2.3. I, BLAP-tagged KCa3.1 was co-expressed with GFP-tagged DN RME-1 in HEK cells. KCa3.1 was labeled with streptavidin-Alexa555, and localization was evaluated after 0, 1, and 5 h at 37 °C. In contrast to KCa2.3, KCa3.1 does not enter the RME-1-positive compartment and is rapidly degraded, similar to what was observed in the absence of DN RME-1. In all experiments, co-localization is shown in the overlay image as yellow. Nuclei are labeled with DAPI (blue). Images are either single confocal sections (A and I) or projection images (B–E).
Recycling of KCa2.3 Is Dependent upon Rab35 and the Rab35 GAP, EPI64C
Recent studies have highlighted the role of Rab35 as a novel player in the recycling of membrane proteins (28, 36). All Rab proteins are paired with both guanine exchange factors and GAPs, although few Rab/GAP pairs have been clearly defined. Recently, we identified EPI64C as a Rab35-specific GAP (28). Based on these observations, we determined whether Rab35 and its GAP, EPI64C, play a role in the recycling of KCa2.3 to the plasma membrane in HEK and HMEC-1 cells. Initially, we confirmed by IB that both Rab35 and EPI64C are expressed in HEK and HMEC-1 cells (Fig. 5).
FIGURE 5.
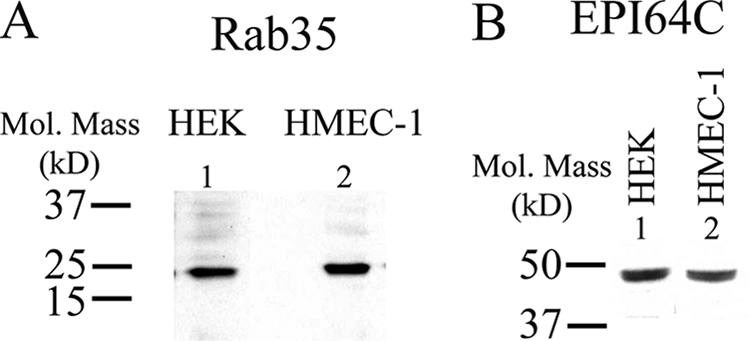
Rab35 and the Rab GAP EPI64C are expressed in HEK and HMEC-1 cells. A, IB demonstrating expression of Rab35 in HEK (1st lane) and HMEC-1 (2nd lane) cells. 20 μg of total protein was loaded per lane. B, IB demonstrating expression of EPI64C in HEK (1st lane) and HMEC-1 (2nd lane) cells. 25 μg of total protein was loaded per lane.
The role of Rab35 on the endocytosis and recycling of KCa2.3 was evaluated by co-transfecting either wild type GFP-Rab35 or the S22N dominant negative GFP-Rab35 with BLAP-tagged KCa2.3. We then determined the localization of KCa2.3 following labeling at the cell surface and then allowing endocytosis to proceed for 5 h at 37 °C. As shown in Fig. 6A, KCa2.3 remained primarily at the plasma membrane of HEK cells after 5 h in the presence of Rab35, demonstrating that overexpression of Rab35 alone does not influence the endocytosis of the channel. Overexpression of DN Rab35 resulted in the accumulation of large vacuoles (Fig. 6B), as reported previously (28, 36). These cells also display a multinucleate phenotype consistent with the observation that Rab35 is important in cytokinesis (36). At time 0 h, KCa2.3 is localized to the plasma membrane in these cells as shown in Fig. 6B (upper panels). However, in contrast to wild type Rab35, overexpression of the DN Rab35 caused KCa2.3 to be internalized onto the large DN Rab35-induced vacuoles (Fig. 6B, lower panels). Similar results were observed in HMEC-1 cells when KCa2.3 and DN Rab35 were coexpressed, as shown in Fig. 6C. These results suggest that Rab35 is required for the recycling of KCa2.3, and eliminating Rab35 GTPase activity prevents the recycling of KCa2.3 to the plasma membrane such that the channel is accumulated inside the cell. Given the large cellular perturbations induced by Rab35 expression, we determined whether this significantly altered the total expression of KCa2.3. Thus, we carried out immunoblots following co-expression of KCa2.3 with either WT Rab35 or DN Rab35. As shown in Fig. 6D, DN Rab35 had no effect on total KCa2.3 expression, averaging 98 ± 4% (n = 3), relative to WT Rab35. Our results predict that in the steady-state there would be less KCa2.3 at the plasma membrane in the presence of the DN Rab35 because the cumulative time of membrane expression would be reduced in the absence of recycling. This was confirmed by cell surface biotinylations, as shown in Fig. 6E, i.e. expression of DN Rab35 reduced plasma membrane KCa2.3 an average of 75 ± 9% (n = 3; p < 0.01) relative to wild type Rab35 expression. As with RME-1, it would be predicted that WT Rab35 would stimulate recycling of KCa2.3, whereas DN Rab35 would inhibit recycling such that the t½ for the plasma membrane-localized channel would be increased and decreased, respectively. As shown in Fig. 6F, in the presence of DN Rab35, the degradation of KCa2.3 could be well described by an exponential with a t½ of 9.8 h (τ = 14.2 ± 1.4 h, n = 3, solid line, triangles). In contrast, following expression of WT Rab35, the degradation of KCa2.3 was best fit to a quadratic function having a t½ of 24.1 h (Fig. 6F, dashed line, circles). These results are consistent with DN Rab35 retarding recycling of KCa2.3 to the plasma membrane resulting in an increased rate of degradation, whereas WT Rab35 enhanced recycling, such that the degradation of plasma membrane channel was slowed. Finally, using co-immunoprecipitation studies, we confirmed an association between KCa2.3 and Rab35 as shown in Fig. 6G.
FIGURE 6.

DN Rab35 co-localizes with KCa2.3 and prevents recycling to the plasma membrane. BLAP-tagged KCa2.3 was co-expressed with either GFP-tagged Rab35 (A) or DN Rab35 (B) in HEK cells; the plasma membrane channel was labeled with streptavidin-Alexa555, and localization was determined following incubation at 37 °C for the times indicated. A, in the presence of Rab35, KCa2.3 was localized primarily at the plasma membrane after 5 h at 37 °C, identical to what was observed when KCa2.3 is expressed alone. B, at time 0 h, KCa2.3 is localized to the plasma membrane (top panels). However, after 5 h at 37 °C, KCa2.3 localizes to the DN Rab35-induced intracellular vacuoles. C, BLAP-tagged KCa2.3 was co-expressed with GFP-tagged DN Rab35 in HMEC-1 cells, and the plasma membrane channel was labeled with streptavidin-Alexa555, and localization was determined following incubation at 37 °C for 5 h. KCa2.3 localizes with DN Rab35 after 5 h and is nearly absent from the membrane demonstrating that recycling is impaired. Nuclei are labeled with DAPI (blue). Projection images from multiple z-sections are shown. D, immunoblot of total lysate following co-expression of KCa2.3 with WT Rab35 (1st lane) or DN Rab35 (2nd lane). Rab35 expression had no effect on expression of KCa2.3. E, cell surface KCa2.3 expression was evaluated by biotinylation following co-expression with either Rab35 (1st lane) or DN Rab35 (2nd lane). DN Rab35 caused a significant decrease (75 ± 9%, n = 3; p < 0.01) in steady-state cell surface expression of KCa2.3. F, KCa2.3 was co-expressed with either WT (left panel) or DN (right panel) Rab35, and cell surface channel was biotinylated. At the indicated times, cells were lysed, subjected to streptavidin pulldown, and blotted for KCa2.3. The bands were quantified by densitometry, normalized, and plotted as a function of time (bottom panel) for DN (solid line, triangles) and WT (dashed line, circles) Rab35. G, co-immunoprecipitation of KCa2.3 with HA-tagged Rab35 was carried out in HEK cells. Rab35 was immunoprecipitated via an HA tag, and the subsequent blot was probed for KCa2.3. Cells expressed either Rab35 plus empty vector (1st lane), KCa2.3 plus vector (2nd lane), or KCa2.3 plus HA-Rab35 (3rd and 4th lanes). The V5 Ab was used as an IgG control in 3rd lane.
Our results demonstrate that KCa3.1 is rapidly endocytosed and targeted for degradation. Thus, Rab35 would not be expected to influence the endocytosis of KCa3.1. This was confirmed in a parallel series of studies to those above, as shown in Fig. 7. At time 0 h, KCa3.1 was localized to the plasma membrane when expressed with wild type GFP-Rab35 (Fig. 7A, upper panels), and after 1 h at 37 °C (Fig. 7A, lower panels), KCa3.1 was nearly completely endocytosed as above. As shown in Fig. 7B, DN Rab35 did not influence the endocytosis of KCa3.1, i.e. after 1 h (lower panels) KCa3.1 was endocytosed into vesicles that appeared identical to those observed when the channel was either expressed alone or with wild type Rab35, and these KCa3.1-containing endosomes failed to co-localize with the Rab35-induced vacuoles. These results confirm the specificity of Rab35 in the recycling of KCa2.3, while having no effect on a different member of the same gene family, KCa3.1.
FIGURE 7.

DN Rab35 does not alter the endocytic retrieval of KCa3.1 from the plasma membrane. Cells were co-transfected with BLAP-tagged KCa3.1 and either GFP-Rab35 (A) or DN GFP-Rab35 (B), and the channel was labeled with streptavidin-Alexa555, and the cells were imaged at time 0 or 1 h. Whether the cells expressed wild type (A) or DN (B) Rab35, KCa3.1 was initially expressed at the plasma membrane (time 0 h, upper panels). After 1 h at 37 °C (lower panels), KCa3.1 was endocytosed into small endosomes in the presence of both wild type (A) and DN (B) Rab35, identical to what was observed in the absence of Rab35. Nuclei are labeled with DAPI (blue). Projection images from multiple z-sections are shown.
As indicated above, the GAP associated with Rab35 function has been shown to be EPI64C in hematopoietic cells (28), and we demonstrate that both Rab35 and EPI64C are expressed in HEK and HMEC-1 cells (Fig. 5). Thus, overexpression of EPI64C would be expected to exhibit a phenotype similar to the overexpression of DN Rab35. Based on this, we determined the effect of EPI64C on KCa2.3 localization and steady-state plasma membrane expression. As shown in Fig. 8A, GFP-EPI64C expression induced vacuole formation as reported previously (28). Also, the cells became multinucleate, consistent with the inhibition of cytokinesis as expected if EPI64C is keeping Rab35 principally in the GDP-bound configuration (28). At time 0 h, KCa2.3 is expressed at the plasma membrane (Fig. 8A, upper panels). However, after 5 h at 37 °C, virtually all of the KCa2.3 channel has been endocytosed and is associated with the vacuolar membranes induced by EPI64C (Fig. 8A, lower panels), indicating the channel cannot recycle back to the plasma membrane. Elimination of the GAP activity in EPI64C by mutating a critical arginine (R141K) in the TBC domain (Tre/Bub2/Cdc16) required for GAP activity completely eliminated the effect of EPI64C overexpression on KCa2.3 endocytosis and recycling, as shown in Fig. 8B. After 5 h at 37 °C, KCa2.3 was predominantly localized to the plasma membrane in the presence of R141K EPI64C, similar to what was observed when the channel was expressed alone. Similar to Rab35, EPI64C induces large cellular perturbations, and thus we determined whether this significantly altered the total expression of KCa2.3. As above, we carried out immunoblots following co-expression of KCa2.3 with either WT EPI64C or R141K EPI64C. As shown in Fig. 8C, R141K EPI64C had no effect on total KCa2.3 expression, averaging 108 ± 3% (n = 3), relative to WT EPI64C. As with overexpression of DN Rab35, it would be predicted that overexpression of EPI64C would result in a decreased steady-state level of KCa2.3 in the plasma membrane because of a lack of recycling. As shown in Fig. 8D, EPI64C decreased plasma membrane expression of KCa2.3 compared with the expression observed in the presence of the R141K GAP mutant. In five experiments, this reduction averaged 41 ± 8% (p < 0.05). These results indicate that EPI64C is the GAP associated with the Rab35-dependent recycling of KCa2.3 to the plasma membrane.
FIGURE 8.

EPI64C co-localizes with KCa2.3 and prevents recycling to the plasma membrane. BLAP-tagged KCa2.3 was co-expressed with either GFP-tagged EPI64C (A) or R141K EPI64C (B) in HEK cells, and the plasma membrane channel was labeled with streptavidin-Alexa555, and localization was determined following incubation at 37 °C for the times indicated. A, at time 0 h, KCa2.3 is localized to the plasma membrane in the presence of EPI64C (top panels). Note that EPI64C induces large vacuoles as well as a multinucleate phenotype as described previously (see text). After 5 h at 37 °C, KCa2.3 was localized primarily on the EPI64C-induced vacuoles (bottom panels), indicating KCa2.3 cannot recycle back to the plasma membrane. B, after 5 h at 37 °C, KCa2.3 is localized primarily at the plasma membrane in the presence of the GAP-inactivated EPI64C (R141K) confirming GAP activity is required to inhibit the recycling of KCa2.3 back to the plasma membrane. Nuclei are labeled with DAPI (blue). Projection images from multiple z-sections are shown. C, immunoblot of total lysate following co-expression of KCa2.3 with WT EPI64C (1st lane) or R141K EPI64C (2nd lane). EPI64C expression had no effect on expression of KCa2.3. D, cell surface KCa2.3-BLAP expression was evaluated by biotinylation following coexpression with either EPI64C (1st lane) or R141K EPI64C (2nd lane). EPI64C caused a significant decrease (41 ± 8%, n = 5; p < 0.05) in steady-state cell surface expression of KCa2.3. E, shRNA construct directed against EPI64C (shEPI) decreased protein expression compared with a scrambled shRNA (Con). Tubulin was used as a loading control (bottom panel). F, cell surface KCa2.3 expression was evaluated by biotinylation following knockdown of EPI64C (shEPI) compared with scrambled shRNA (Cont). Knockdown of EPI64C expression caused a significant increase (76 ± 22%, n = 3; p < 0.05) in steady-state cell surface expression of KCa2.3.
To confirm a role for Rab35 and EPI64C in the trafficking of KCa2.3, we attempted to knock down expression of these proteins using shRNAs. As we reported previously in Jurkat cells (28), we were unable to suppress Rab35 expression by more than ∼50% in HEK cells (data not shown), such that this was not pursued. However, we were able to knock down expression of EPI64C by 72% after 48 h in two experiments when compared with a scrambled shRNA (Fig. 8E) in HEK cells, similar to what we reported in Jurkat cells (28). As knockdown of the Rab GAP would increase the amount of Rab35 in the GTP-bound state, this would be expected to increase the steady-state level of KCa2.3 plasma membrane expression as recycling would be accelerated. To confirm this, we biotinylated plasma membrane-localized KCa2.3 in the presence of a scrambled shRNA or an shRNA directed against EPI64C, pulled down KCa2.3 with streptavidin, and determined expression by IB. As shown in Fig. 8F, knockdown of EPI64C resulted in an increase in KCa2.3 cell surface expression. In three experiments, steady-state plasma membrane expression of KCa2.3 was increased an average of 76 ± 22% (p < 0.05). These results confirm a role for the Rab35 GAP, EPI64C, in the recycling of KCa2.3 to the plasma membrane.
Our results demonstrate that both RME-1 and Rab35/EPI64C are required for the recycling of KCa2.3 to the plasma membrane in both HEK and HMEC-1 cells, suggesting that the endosomal compartments in which these proteins reside are either in series or overlapping, rather than being distinct parallel pathways for recycling. This was evaluated by expressing BLAP-tagged KCa2.3 with GFP RME-1 and mRFP DN Rab35 (Fig. 9A) or mRFP EPI64C (Fig. 9B), labeling plasma membrane-localized channel with streptavidin-Cy5, and evaluating the localization of these three proteins following endocytosis of KCa2.3 for 5 h at 37 °C. As shown in Fig. 9, both KCa2.3 (shown in gray for clarity) and RME-1 localized to the vacuoles induced by overexpression of either DN Rab35 (Fig. 9A) or EPI64C (Fig. 9B) consistent with RME-1 and Rab35/EPI64C being part of the same endocytic pathway responsible for the recycling of KCa2.3 back to the plasma membrane.
FIGURE 9.

KCa2.3 and RME-1 co-localize to the vacuoles induced by either DN Rab35 (A) or EPI64C (B). BLAP-tagged KCa2.3 was co-transfected with GFP-tagged RME-1 and either mRFP-tagged DN Rab35 (A) or mRFP-tagged EPI64C (B) in HEK cells, and the channel was labeled with streptavidin-Cy5, and localization was assessed by IF after 5 h at 37 °C. As is apparent, KCa2.3 and RME-1 are both localized to the vacuoles induced by DN Rab35 (A) and EPI64C (B) indicating that the RME-1 and Rab35/EPI64C pathways intersect in the cell. KCa2.3 was pseudocolored gray for clarity. Nuclei are labeled with DAPI (blue). Projection images from multiple z-sections are shown.
Endocytosis and Recycling of KCa2.3 Is Dependent upon an N-terminal Domain
We previously demonstrated that KCa2.3 has a long total protein half-life, being in excess of 36 h, whereas KCa3.1 has a total protein half-life of ∼6 h (37). Here, we demonstrate that the plasma membrane half-lives of these two channels are similarly disparate. To determine which portion of KCa2.3 dictates its long half-life, and presumably its recycling, we generated two chimeras in which either the entire cytoplasmic C terminus of KCa3.1 (Arg287–Lys427) was appended onto S6 of KCa2.3 (Fig. 10A, KCa2.3–287KCa3.1) or the N terminus of KCa3.1 (Met1–Ala26) was appended to S1 of KCa2.3 (Fig. 10B, 26KCa3.1-KCa2.3). HEK cells stably expressing these chimeras were exposed to cycloheximide to inhibit protein synthesis, and channel protein expression was determined at various time points. As shown in Fig. 10A, channel expression stayed stable for 24 h when the C terminus of KCa3.1 was appended onto KCa2.3, similar to what we have previously reported for KCa2.3 (37). In contrast, when the N terminus of KCa3.1 was swapped for the N terminus of KCa2.3, the resultant chimera was degraded with a time constant of 3.7 ± 0.3 h (n = 3; Fig. 10C), similar to KCa3.1 (37). These results suggest that the N terminus of KCa2.3 is required for the long half-life of the channel in general and likely plays a role in the recycling of this channel.
FIGURE 10.

N terminus of KCa2.3 is deterministic for its long half-life. A, top panel illustrates a model in which the C terminus of KCa3.1 (amino acids Arg287–Lys427; shown in gray at arrow) is appended onto S6 of KCa2.3 (KCa2.3–287KCa3.1). Bottom panel shows a representative IB for the degradation of this chimera expressed in HEK cells in the presence of cycloheximide (400 μg/ml) for the times indicated. No apparent degradation was observed over a 24-h period. 20 μg of total protein was loaded per lane. Similar results were observed in three separate experiments. B, top panel illustrates a model in which the N terminus of KCa3.1 (amino acids Met1–Ala26; shown in gray at arrow) is appended on to S1 of KCa2.3 (26KCa3.1-KCa2.3). Bottom panel shows a representative IB for the degradation of this chimera expressed in HEK cells in the presence of cycloheximide (400 μg/ml) for the times indicated. 20 μg of total protein was loaded per lane. C, data from three separate experiments on 26KCa3.1-KCa2.3 (B) were digitized and plotted as a fraction of protein at time 0. The data were fitted to an exponential decay function with a time constant for degradation of 3.7 ± 0.3 h (n = 3).
Our data on the chimeras suggest that the molecular recognition signal(s) for recycling of KCa2.3 lie within the N terminus. Based on this, we made a series of N-terminal deletions of BLAP-tagged KCa2.3 and then evaluated plasma membrane expression in HEK cells transiently expressing these channels at time 0 and 3 h at 37 °C. As above, wild type BLAP-tagged KCa2.3 was localized almost exclusively to the plasma membrane after 3 h at 37 °C (Fig. 11A). Similar plasma membrane localization was observed for the Leu83–Asp135 deletion (ΔLeu83–Asp135) at time 0 and 3 h (Fig. 11B). The additional deletions, ΔPro22–Pro59, ΔGln62–Gln80, ΔHis136–Val185, and ΔHis236–Gln265 also had no effect on plasma membrane localization at time 0 or after 3 h at 37 °C (data not shown). In contrast, as shown in Fig. 11C, ΔMet186–Leu235 resulted in near complete endocytosis of KCa2.3 after 3 h at 37 °C, indicating that this portion of the channel is required for determining the endocytic fate of the channel. Additional deletions within this domain reveal that Met186–Glu205 (Fig. 11D) and Pro218–Leu235 (Fig. 11F) are not required for recycling, as these channels are normally localized at 3 h, whereas ΔGly206–Pro217 (Fig. 11E) is completely endocytosed, demonstrating that this 12-amino acid domain (206GQPLQLFSPSNP217) is required for the recycling of KCa2.3 back to the plasma membrane. Within this domain, the double mutations P214A/P217A and S213A/S215A had no effect on the localization of KCa2.3 (data not shown). However, the additional deletion, ΔGln207–Gln210 resulted in a channel that was nearly completely endocytosed within 3 h, as shown in Fig. 11G. Our IF data suggest that some ΔGln207–Gln210 (Fig. 11G) channel remains at the plasma membrane when compared with the ΔGly206–Pro217 channel (Fig. 11E), and cell surface biotinylations confirmed a small difference in the time constant for degradation of these channels (τ = 6.7 h for ΔGly206–Pro217 (Fig. 11, H and J) and τ = 8.1 h for ΔGln207–Gln210 (Fig. 11, I and J); n = 2 for each). To confirm that this molecular motif is similarly required for the endocytosis and recycling of KCa2.3 in endothelial cells, we transfected BLAP-tagged wild type and ΔGly206–Pro217 KCa2.3 into HMEC-1 cells and evaluated localization by IF at time 0 and 3 h. As shown in Fig. 11K, KCa2.3 remains at the membrane at 3 h, whereas ΔGly206–Pro217 (Fig. 11L) is clearly endocytosed. These results demonstrate that a small domain in the N terminus of KCa2.3, encompassing Gly206–Pro217, is required for the recycling of KCa2.3 back to the plasma membrane of both HEK and HMEC-1 cells.
FIGURE 11.

Small N-terminal motif is required for the proper endocytosis and recycling of KCa2.3 in HEK and HMEC-1 cells. BLAP-tagged full-length KCa2.3 (A) or the N-terminal deletions, ΔLeu83–Asp135 (B), ΔMet186–Leu235 (C), ΔMet186–Glu205 (D), ΔGly206–Pro217 (E), ΔPro218–Leu235 (F), and ΔGln207–Gln210 (G), were transiently transfected into HEK cells, and the plasma membrane channel was labeled with streptavidin-Alexa555 and localized by IF at either time 0 or 3 h. Each of these deletions was localized to the plasma membrane at time 0 h (left panels). After 3 h at 37 °C (right panels), the deletion ΔLeu83–Asp135 (B) was correctly localized at the plasma membrane, whereas the ΔMet186–Leu235 deletion (C) was completely endocytosed. Further deletions within this domain demonstrate that the amino acids Met186–Glu205 (D) and Pro218–Leu235 (F) localize normally at 0 and 3 h, whereas Gly206–Pro217 (E) as well as the smaller deletion ΔGln207–Gln210 (G) are mislocalized to intracellular endosomes. For these studies, single confocal sections are shown. H and I, to quantify the time course for plasma membrane, ΔGly206–Pro217 (H) and ΔGln207–Gln210 (I) degradation plasma membrane proteins were biotinylated followed by streptavidin pulldown and blotted for KCa2.3 at the times indicated. J, blots were quantified by densitometry and fit to an exponential decay function with time constants of 6.7 h (n = 2) for ΔGly206–Pro217 (dashed line, open triangles) and 8.1 h (n = 2) for ΔGln207–Gln210 (solid line, filled circles). To confirm a similar function for the N-terminal domain of KCa2.3 in endothelial cells, we transiently transfected HMEC-1 cells with either BLAP-tagged KCa2.3 (K) or ΔGly206–Pro217 (L) and evaluated localization as above. Each of these channels is correctly localized to the plasma membrane at time 0 h. However, after 3 h at 37 °C, KCa2.3 is almost exclusively localized to the plasma membrane, whereas ΔGly206–Pro217 (L) has been completely endocytosed. For these studies, projection images from multiple z-sections are shown. Nuclei are labeled with DAPI (blue).
Deletion of Gly206–Pro217 Alters the Association with RME-1 but Not Rab35
We demonstrate that both RME-1 and Rab35 are required for the proper recycling of KCa2.3 to the plasma membrane. Given the role of Gly206–Pro217 in this process, we determined whether deletion of these amino acids would alter these associations. As shown in Fig. 12A, ΔGly206–Pro217 (Δ12) KCa2.3 could still be immunoprecipitated with Rab35 indicating this domain is not required for the observed association. Note that the small decrease in KCa2.3 observed following Rab35 IP can be accounted for by the decreased expression of ΔGly206–Pro217 KCa2.3 compared with wild type KCa2.3 (Fig. 12A, lower blot). In contrast, the amount of KCa2.3 that could be immunoprecipitated with RME-1 was significantly decreased following deletion of ΔGly206–Pro217 (Fig. 12B). In three experiments, this decrease averaged 62 ± 11% (p < 0.05), whereas total ΔGly206–Pro217 protein was decreased only 26 ± 11% (not different) relative to wild type channel. More significantly, because channels that traffic through the recycling compartment originate at the plasma membrane, we evaluated the effect of the ΔGly206–Pro217 deletion on cell surface expression by biotinylation. As shown in Fig. 12C, the ΔGly206–Pro217 deletion (2nd lane) resulted in only a small decrease in cell surface expression (18 ± 9%; n = 3) relative to wild type KCa2.3 (1st lane). These results suggest that the ΔGly206–Pro217 deletion inhibits the recycling of KCa2.3 by altering the association with RME-1 in the recycling pathway.
FIGURE 12.

Gly206–Pro217 deletion does not affect the association of KCa2.3 with Rab35 but decreases the association with RME-1. A, co-immunoprecipitation of KCa2.3 or ΔGly206–Pro217 KCa2.3 (Δ12) with HA-tagged Rab35 was carried out in HEK cells as described under “Experimental Procedures.” Rab35 was immunoprecipitated via an HA tag, and the subsequent blot was probed for KCa2.3. Cells expressed either Rab35 plus empty vector (1st lane), KCa2.3 plus vector (2nd lane), KCa2.3 plus HA-Rab35 (3rd and 4th lanes) or ΔGly206–Pro217 KCa2.3 plus HA-Rab35 (5th lane). The V5 Ab was used as an IgG control in 3rd lane. Lysate for each of these was probed for KCa2.3 (bottom panel) to evaluate expression. Note that ΔGly206–Pro217 KCa2.3 is expressed at a lower level than full-length KCa2.3. Similar results were observed in three separate experiments. Thus, deletion of this 12-amino acid domain does not influence the association between KCa2.3 and Rab35. B, co-immunoprecipitation of KCa2.3 or ΔGly206–Pro217 KCa2.3 (Δ12) with GFP-tagged RME-1 was carried out in HEK cells as described under the “Experimental Procedures.” RME-1 was immunoprecipitated via a GFP tag, and the subsequent blot was probed for KCa2.3. Cells expressed either KCa2.3 plus vector (1st lane), RME-1 plus empty vector (2nd lane), KCa2.3 plus GFP-RME-1 (3rd and 4th lanes), or ΔGly206–Pro217 KCa2.3 plus GFP-RME-1 (5th lane). The V5 Ab was used as an IgG control in 3rd lane. Lysate for each of these was probed for KCa2.3 (bottom panel) to evaluate expression. Note that RME-1 pulled down significantly less ΔGly206–Pro217 KCa2.3 compared with wild type KCa2.3, which cannot be accounted for by the small decrease in total ΔGly206–Pro217 expression. C, cell surface expression of KCa2.3 and ΔGly206–Pro217 KCa2.3 was evaluated by biotinylation followed by streptavidin pulldown. ΔGly206–Pro217 KCa2.3 was expressed at a similar level to wild type KCa2.3, indicating this cannot account for the decreased association observed upon co-IP (A).
DISCUSSION
There is a clear consensus that both KCa3.1 and KCa2.3 play a pivotal role in the EDHF response in the microvasculature such that these channels are critical to the maintenance of vascular tone, and hence blood pressure regulation. The number of channels (N) in the membrane when an agonist is present is deterministic in any physiological response, and it is now clear that N can be directly modulated by physiological agonists (38, 39). However, to date there have been no studies detailing the mechanisms by which these channels are retrieved from the plasma membrane. To begin to address this question, we utilized an approach pioneered by Ting and co-workers (31) in which the BLAP sequence is inserted into an extracellular loop of the channel such that the channel can be rapidly and selectively biotinylated and then labeled using a fluorophore-conjugated streptavidin. Importantly, we demonstrate that the BLAP tag does not alter the response of KCa2.3 or KCa3.1 to Ca2+, the pharmacological agonist DCEBIO (32), or the KCa3.1 blocker clotrimazole (Fig. 1) (40, 41). Furthermore, using either a classic biotinylation approach, which does not require the BLAP tag, or tagging BLAP-tagged KCa2.3 with biotin directly using BirA followed by streptavidin results in an identical time constant for degradation of plasma membrane-localized KCa2.3 (Fig. 2C). Finally, we demonstrate that the addition of the BLAP tag does not alter the trafficking of KCa2.3 into an RME-1-positive compartment (Fig. 4). These results demonstrate that the addition of streptavidin to the channel does not influence its trafficking. Thus, the use of these BLAP-tagged channels represents a novel method for studying the endocytosis of KCa2.3 and KCa3.1.
Our data demonstrate that plasma membrane-localized KCa2.3 has a long half-life when compared with KCa3.1 (Fig. 2, C and D). Furthermore, our IF data demonstrate that KCa2.3 remains localized to the plasma membrane for several hours in both HEK and HMEC-1 cells, whereas KCa3.1 is rapidly endocytosed and targeted for degradation in each of these cell types (Fig. 2, A and B). Given the apparent differences in half-life between our HEK and HMEC-1 data, it will be important to define the residency time of this channel in native endothelia in the future. The long residency time of KCa2.3 could result from the channel being slowly removed from the plasma membrane or from the channel being rapidly endocytosed and recycled back to the plasma membrane. Using a biotinylation approach, we demonstrate that KCa2.3 is rapidly endocytosed (Fig. 3A) and that this endocytosed channel is recycled back to the plasma membrane with a time constant of 4.7 min (Fig. 3, B and C). Although we did not expressly measure the endocytic time constant, we do show that KCa2.3 has reached a steady-state within the endosomal compartment within 5 min (Fig. 3A), which is consistent with the rapid rate of recycling observed to maintain steady-state plasma membrane expression levels. As RME-1 (EHD1 in mammalian cells) has been shown to be an integral part of the recycling machinery (35), we determined whether KCa2.3 would co-localize with RME-1. Our results clearly demonstrate that KCa2.3 enters the RME-1-positive recycling compartment (Fig. 4C) and that this is independent of the BLAP tag (Fig. 4D). Furthermore, the DN form of RME-1 results in KCa2.3 being trapped inside the cell following endocytosis in both HEK (Fig. 4, A and E) and HMEC-1 (Fig. 4B) cells as has been shown for other recycling proteins (33, 34). We also demonstrate that both the steady-state expression and half-life of plasma membrane-localized KCa2.3 are affected by RME-1 expression., i.e. DN RME-1 both decreased cell surface expression of KCa2.3 and also accelerated the degradation of the channel, whereas WT RME-1 increased the half-life of cell surface KCa2.3 (Fig. 4G). Note that the decrease in plasma membrane KCa2.3 expression (62%) cannot be explained by the small decrease in total KCa2.3 expression observed (30%). These results are entirely consistent with RME-1 being an important factor in regulating the recycling of KCa2.3 back to the plasma membrane. Finally, we demonstrate that KCa2.3 is associated with the RME-1 complex by immunoprecipitation (Fig. 4H); confirming a role for RME-1 in the recycling of this channel. In contrast, KCa3.1 is excluded from this compartment (Fig. 4I). Indeed, expressing the DN RME-1 had no impact on KCa3.1 endocytosis, thus demonstrating the specificity of the effect for KCa2.3.
Echard and co-workers (36) first identified Rab35 as a novel Rab involved in the fast endocytic recycling pathway. These studies were extended by us identifying EPI64C as the GAP associated with Rab35 and further demonstrating that both Rab35 and EPI64C were involved in the recycling of the transferrin and T cell receptor in Jurkat T cells (28). Here, we demonstrate for the first time that both Rab35 and EPI64C are involved in the recycling of an ion channel to the plasma membrane, i.e. we demonstrate that KCa2.3 accumulates on the intracellular vacuoles induced by DN Rab35 (Fig. 6) overexpression in both HEK and HMEC-1 cells and that this effect requires the GAP activity of EPI64C (Fig. 8). Furthermore, we demonstrate that steady-state plasma membrane expression of KCa2.3 is decreased by expression of either DN Rab35 (Fig. 6E) or EPI64C (Fig. 8D) as predicted if recycling is interrupted. In addition, DN Rab35 accelerated the degradation of KCa2.3 from the plasma membrane, whereas WT Rab35 increased the half-life of plasma membrane channel (Fig. 6F); consistent with DN Rab35 excluding KCa2.3 from the recycling pathway and WT Rab35 fostering the recycling of the channel to the plasma membrane. Finally, we demonstrate that shRNA-induced knockdown of EPI64C increases steady-state expression of KCa2.3 at the plasma membrane (Fig. 8F) as predicted for the removal of GAP activity. The specificity of Rab35 and its GAP, EPI64C for KCa2.3, which recycles, is demonstrated by their lack of effect on KCa3.1 that does not recycle but rather is targeted for degradation. Finally, we demonstrate that RME-1 co-localizes with DN Rab35 and EPI64C (Fig. 9) consistent with the hypothesis that each of these proteins is involved in the recycling of KCa2.3. Based on this, we conclude that RME-1 and Rab35 minimally work in series to facilitate the recycling of KCa2.3 to the plasma membrane. We did not observe any co-localization between wild type Rab35 and RME-1 (data not shown) suggesting these two proteins may be remotely located within a vesiculotubular compartment or perhaps in separate endosomes in series, which transiently merge during the recycling process. In either of these scenarios, we would expect to observe co-localization when the DN Rab35 or EPI64C is expressed as they prevent exit from the recycling compartment and the formation of large vacuoles trapping the other components of the recycling pathway. Our observations are distinct from those in C. elegans, in which it was concluded that Rab35 and RME-1 are involved in parallel recycling pathways (42). However, it should be noted that C. elegans null for Rab35 are viable and fertile indicating Rab35 is not involved in cytokinesis in C. elegans, whereas Rab35 is involved in cytokinesis in mammalian systems as reported previously (36) and as apparent from the multinucleate cells observed in this study in the presence of DN Rab35 and EPI64C (Figs. 6–9). Also, homozygous Rab35 knock-out mice exhibit an embryonic lethal phenotype3 further suggesting that the role of Rab35 in C. elegans and mammalian cells is distinct.
Recently, Rabs have been shown to directly associate with the proteins being trafficked through recycling endosomes (43, 44). We demonstrate that Rab35 immunoprecipitates KCa2.3 confirming these proteins are part of the same complex. Our results also demonstrate that a small domain within the N terminus of KCa2.3, consisting of the amino acids Gly206–Pro217, is responsible for the rapid recycling of the channel (Fig. 11), i.e. deletion of this domain does not prevent the channel from being expressed at the cell surface nor does it prevent the rapid endocytosis of the channel. Rather, it prevents the return of the channel to the plasma membrane via a recycling compartment such that the channel is degraded with a time constant of ∼7 h, which is 2.5-fold faster than a full-length channel. Most importantly, the function of this domain is identical whether KCa2.3 is expressed in HEK or HMEC-1 cells suggesting the mechanisms involved in endocytosis of this channel are identical in endothelial cells.
Our observations led us to determine whether the N-terminal domain of KCa2.3 was important for its association with either Rab35 or RME-1. We demonstrate that the association between KCa2.3 and RME-1 is significantly abrogated following deletion of Gly206–Pro217 (Fig. 12B). The mammalian homolog of RME-1, EHD1, has been shown to interact with numerous proteins (35), including the cargo, insulin-like growth factor 1 receptor (45). Thus, we speculate that RME-1 associates with the N terminus of KCa2.3 and that this is critical to the proper sorting of the channel into the recycling pathway. Our data further suggest that when this association is altered the channel is removed to the degradative pathway. Additional studies are required to determine whether this is a direct association between RME-1 and KCa2.3, however. In contrast, immunoprecipitation confirms this deletion does not preclude the association between KCa2.3 and Rab35 (Fig. 12A). In total, our results demonstrate that KCa2.3, RME-1, and Rab35 converge at the same compartment during recycling of the channel (Fig. 9) and that KCa2.3 associates, minimally indirectly, with both of these proteins critical to the recycling process. Although our results provide molecular insight into the association between RME-1 and KCa2.3, further studies are required to delineate the associations required for the Rab35-dependent recycling observed.
It is also important to recognize that a chimera in which the N terminus of KCa3.1 is replaced by the N terminus of KCa2.3 does not imbue KCa3.1 with a prolonged protein half-life4 indicating this domain in itself cannot recapitulate the endocytic paradigm associated with KCa2.3. Thus, it is likely that the endocytosis and recycling of KCa2.3 requires a larger three-dimensional domain of which the Gly206–Pro217 motif identified is one critical component. Whether this involves an interaction with a separate domain within KCa2.3 or the interaction with another protein (e.g. RME-1) that is dependent upon the cytosolic structure of KCa2.3 in total is not clear. In this regard, it is interesting to note that the N and C termini of KCa2.2 have been shown to be complexed with casein kinase-2 (16), whereas yeast two-hybrid analysis has shown a direct association between the N and C termini of KCa2.3 (17).
Recent studies have immunolocalized KCa3.1 to the myoendothelial gap junctions within the endothelial cell projections, although KCa2.3 is more evenly distributed across the endothelial cell membrane (10, 14). Whether these distinct localizations can be attributed to the reported association of KCa2.3 with caveolin-rich domains (46) is not clear. However, these differences in plasma membrane localization likely help to explain the variable inputs these channels have to the EDHF response during agonist stimulation (2, 10, 13). A critical component to this response is the mechanism by which the number of KCa channels is regulated at the cell surface and therefore contributes to the agonist-induced response. For the first time, we demonstrate that KCa2.3 is rapidly endocytosed and recycled back to the plasma membrane in a Rab35/EPI64C- and RME-1-dependent manner, whereas KCa3.1 is rapidly endocytosed and targeted for degradation. In preliminary studies, we demonstrate that the short half-life of KCa3.1 is the result of this channel being targeted to the lysosome via the ESCRT-dependent pathway (47). We speculate that the differences in the functional half-life of these channels in the plasma membrane are directly related to their common yet unique roles in the EDHF response. Furthermore, given the dynamic nature of these channels at the plasma membrane, any change in the rate constants for endocytosis or recycling would be expected to have dramatic effects on the number of channels in the membrane and thus affect the EDHF response and hence blood pressure regulation.
Acknowledgments
We gratefully acknowledge the assistance of Caitlin E. Devor, who generated numerous deletion constructs and point mutations critical to the delineation of the N terminus as being required for the recycling of KCa2.3. We also acknowledge Dr. Kirk Hamilton for critically reading the manuscript and for many helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants HL083060 and HL092157 (to D. C. D.). This work was also supported by American Heart Fellowship 0825542D (to C. M. B.).
G. Patino-Lopez and S. Shaw, unpublished observations.
Y. Gao and D. C. Devor, unpublished observations.
- EDHF
- endothelium-derived hyperpolarizing factor
- BSA
- bovine serum albumin
- PBS
- phosphate-buffered saline
- HA
- hemagglutinin
- Ab
- antibody
- GFP
- green fluorescent protein
- mRFP
- monomeric red fluorescent protein
- BLAP
- biotin ligase acceptor peptide
- DAPI
- 4′,6-diamidino-2-phenylindole
- WT
- wild type
- DN
- dominant negative
- HEK
- human embryonic kidney
- shRNA
- small hairpin RNA
- IF
- immunofluorescence
- IP
- immunoprecipitation
- IB
- immunoblot
- GAP
- GTPase-activating protein
- DCEBIO
- 5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one.
REFERENCES
- 1.Furchgott R. F., Zawadzki J. V. (1980) Nature 288, 373–376 [DOI] [PubMed] [Google Scholar]
- 2.Crane G. J., Gallagher N., Dora K. A., Garland C. J. (2003) J. Physiol. 553, 183–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crane G. J., Garland C. J. (2004) Br. J. Pharmacol. 142, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkington H. C., Chow J. A., Evans R. G., Coleman H. A., Tare M. (2002) J. Physiol. 542, 929–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eichler I., Wibawa J., Grgic I., Knorr A., Brakemeier S., Pries A. R., Hoyer J., Köhler R. (2003) Br. J. Pharmacol. 138, 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinton J. M., Langton P. D. (2003) Br. J. Pharmacol. 138, 1031–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor M. S., Bonev A. D., Gross T. P., Eckman D. M., Brayden J. E., Bond C. T., Adelman J. P., Nelson M. T. (2003) Circ. Res. 93, 124–131 [DOI] [PubMed] [Google Scholar]
- 8.Si H., Heyken W. T., Wölfle S. E., Tysiac M., Schubert R., Grgic I., Vilianovich L., Giebing G., Maier T., Gross V., Bader M., de Wit C., Hoyer J., Köhler R. (2006) Circ. Res. 99, 537–544 [DOI] [PubMed] [Google Scholar]
- 9.Brähler S., Kaistha A., Schmidt V. J., Wölfle S. E., Busch C., Kaistha B. P., Kacik M., Hasenau A. L., Grgic I., Si H., Bond C. T., Adelman J. P., Wulff H., de Wit C., Hoyer J., Köhler R. (2009) Circulation 119, 2323–2332 [DOI] [PubMed] [Google Scholar]
- 10.Dora K. A., Gallagher N. T., McNeish A., Garland C. J. (2008) Circ. Res. 102, 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffith T. M. (2004) Br. J. Pharmacol. 141, 881–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mather S., Dora K. A., Sandow S. L., Winter P., Garland C. J. (2005) Circ. Res. 97, 399–407 [DOI] [PubMed] [Google Scholar]
- 13.Wolfle S. E., Schmidt V. J., Hoyer J., Köhler R., de Wit C. (2009) Cardiovasc. Res. 82, 476–483 [DOI] [PubMed] [Google Scholar]
- 14.Sandow S. L., Neylon C. B., Chen M. X., Garland C. J. (2006) J. Anat. 209, 689–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen D., Fakler B., Maylie J., Adelman J. P. (2007) J. Neurosci. 27, 2369–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bildl W., Strassmaier T., Thurm H., Andersen J., Eble S., Oliver D., Knipper M., Mann M., Schulte U., Adelman J. P., Fakler B. (2004) Neuron 43, 847–858 [DOI] [PubMed] [Google Scholar]
- 17.Frei E., Spindler I., Grissmer S., Jäger H. (2006) Cell. Physiol. Biochem. 18, 165–176 [DOI] [PubMed] [Google Scholar]
- 18.Gerlach A. C., Gangopadhyay N. N., Devor D. C. (2000) J. Biol. Chem. 275, 585–598 [DOI] [PubMed] [Google Scholar]
- 19.Gerlach A. C., Syme C. A., Giltinan L., Adelman J. P., Devor D. C. (2001) J. Biol. Chem. 276, 10963–10970 [DOI] [PubMed] [Google Scholar]
- 20.Jones H. M., Hamilton K. L., Papworth G. D., Syme C. A., Watkins S. C., Bradbury N. A., Devor D. C. (2004) J. Biol. Chem. 279, 15531–15540 [DOI] [PubMed] [Google Scholar]
- 21.Srivastava S., Li Z., Ko K., Choudhury P., Albaqumi M., Johnson A. K., Yan Y., Backer J. M., Unutmaz D., Coetzee W. A., Skolnik E. Y. (2006) Mol. Cell 24, 665–675 [DOI] [PubMed] [Google Scholar]
- 22.Jones H. M., Bailey M. A., Baty C. J., MacGregor G. G., Syme C. A., Hamilton K. L., Devor D. C. (2007) Channels 1, 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones H. M., Hamilton K. L., Devor D. C. (2005) J. Biol. Chem. 280, 37257–37265 [DOI] [PubMed] [Google Scholar]
- 24.Syme C. A., Hamilton K. L., Jones H. M., Gerlach A. C., Giltinan L., Papworth G. D., Watkins S. C., Bradbury N. A., Devor D. C. (2003) J. Biol. Chem. 278, 8476–8486 [DOI] [PubMed] [Google Scholar]
- 25.Joiner W. J., Khanna R., Schlichter L. C., Kaczmarek L. K. (2001) J. Biol. Chem. 276, 37980–37985 [DOI] [PubMed] [Google Scholar]
- 26.Lee W. S., Ngo-Anh T. J., Bruening-Wright A., Maylie J., Adelman J. P. (2003) J. Biol. Chem. 278, 25940–25946 [DOI] [PubMed] [Google Scholar]
- 27.Roncarati R., Decimo I., Fumagalli G. (2005) Mol. Cell. Neurosci. 28, 314–325 [DOI] [PubMed] [Google Scholar]
- 28.Patino-Lopez G., Dong X., Ben-Aissa K., Bernot K. M., Itoh T., Fukuda M., Kruhlak M. J., Samelson L. E., Shaw S. (2008) J. Biol. Chem. 283, 18323–18330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ades E. W., Candal F. J., Swerlick R. A., George V. G., Summers S., Bosse D. C., Lawley T. J. (1992) J. Invest. Dermatol. 99, 683–690 [DOI] [PubMed] [Google Scholar]
- 30.Bouïs D., Hospers G. A., Meijer C., Molema G., Mulder N. H. (2001) Angiogenesis 4, 91–102 [DOI] [PubMed] [Google Scholar]
- 31.Chen I., Howarth M., Lin W., Ting A. Y. (2005) Nat. Methods 2, 99–104 [DOI] [PubMed] [Google Scholar]
- 32.Singh S., Syme C. A., Singh A. K., Devor D. C., Bridges R. J. (2001) J. Pharmacol. Exp. Ther. 296, 600–611 [PubMed] [Google Scholar]
- 33.Lin S. X., Grant B., Hirsh D., Maxfield F. R. (2001) Nat. Cell Biol. 3, 567–572 [DOI] [PubMed] [Google Scholar]
- 34.Picciano J. A., Ameen N., Grant B. D., Bradbury N. A. (2003) Am. J. Physiol. Cell Physiol. 285, C1009–C1018 [DOI] [PubMed] [Google Scholar]
- 35.Grant B. D., Caplan S. (2008) Traffic 9, 2043–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kouranti I., Sachse M., Arouche N., Goud B., Echard A. (2006) Curr. Biol. 16, 1719–1725 [DOI] [PubMed] [Google Scholar]
- 37.Gao Y., Chotoo C. K., Balut C. M., Sun F., Bailey M. A., Devor D. C. (2008) J. Biol. Chem. 283, 9049–9059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewarchik C. M., Peters K. W., Qi J., Frizzell R. A. (2008) J. Biol. Chem. 283, 28401–28412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butterworth M. B., Edinger R. S., Johnson J. P., Frizzell R. A. (2005) J. Gen. Physiol. 125, 81–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devor D. C., Singh A. K., Gerlach A. C., Frizzell R. A., Bridges R. J. (1997) Am. J. Physiol. 273, C531–C540 [DOI] [PubMed] [Google Scholar]
- 41.Rufo P. A., Jiang L., Moe S. J., Brugnara C., Alper S. L., Lencer W. I. (1996) J. Clin. Invest. 98, 2066–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato M., Sato K., Liou W., Pant S., Harada A., Grant B. D. (2008) EMBO J. 27, 1183–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parent A., Hamelin E., Germain P., Parent J. L. (2009) Biochem. J. 418, 163–172 [DOI] [PubMed] [Google Scholar]
- 44.Wikström K., Reid H. M., Hill M., English K. A., O'Keeffe M. B., Kimbembe C. C., Kinsella B. T. (2008) Cell. Signal. 20, 2332–2346 [DOI] [PubMed] [Google Scholar]
- 45.Rotem-Yehudar R., Galperin E., Horowitz M. (2001) J. Biol. Chem. 276, 33054–33060 [DOI] [PubMed] [Google Scholar]
- 46.Absi M., Burnham M. P., Weston A. H., Harno E., Rogers M., Edwards G. (2007) Br. J. Pharmacol. 151, 332–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balut C. M., Gao Y., Devor D. C. (2008) Biophys. J. 96, 472a [Google Scholar]