

Abstract
A kinetic analysis has been made of the interaction of α-Hb chains with a mutant α-hemoglobin stabilizing protein, AHSPV56G, which is the first case of an AHSP mutation associated with clinical symptoms of mild thalassemia syndrome. The chaperone AHSP is thought to protect nascent α chains until final binding to the partner β-Hb. Rather than protecting α chains, the mutant chaperone is partially unfolded but recovers its secondary structure via interaction with α-Hb. For both AHSPWT and AHSPV56G, the binding to α-Hb is quite rapid relative to the α-β reaction, as expected because the chaperone binding must be quite competitive to complete the α chain folding process before α-Hb binds irreversibly to β-Hb. The main kinetic difference is a dissociation rate of AHSPV56G·α-Hb some four times faster relative to AHSP·α-Hb. Considering a role of protein folding, the AHSPV56G apparently does not bind long enough (0.5 s versus 2 s for the WT) to complete the structural modifications. The overall replacement reaction (AHSP·α-Hb + β-Hb → AHSP + αβ) can be quite long, especially if there is an excess of AHSP relative to β-Hb monomers.
Keywords: Chaperone Chaperonin, Fluorescence Resonance Energy Transfer (FRET), Hemoglobin, Kinetics, Protein Folding, Protein-Protein Interactions
Introduction
During the investigation of a child presenting with a mild thalassemia syndrome, normal globin gene pattern was observed while a structural abnormality of the α-hemoglobin stabilizing protein (AHSP)2 gene (AHSP Val56 → Gly or AHSPV56G) was found at the homozygous state (1). The child suffered from a microcytic, hypochromic anemia, which required a blood transfusion at the age of 1 month. This clinical observation was the first evidence that a homozygous defect of AHSP may be the etiology of thalassemia syndrome in human. AHSP is the α-hemoglobin (α-Hb) chaperone that binds the holo or apoα-Hb chain but not β-Hb or Hb tetramers (2). The steady state red blood cell showed values below the normal range for total Hb (11 g/decaliter), mean corpuscular volume (60 femtoliter), and mean corpuscular hemoglobin (20 pg).
AHSP is a small 102-residue protein expressed only in erythroid cells, which adopts a three helix bundle (3). Helices 1 and 2 and the intervening segment of AHSP specifically recognize the G and H helices of α-Hb, which also are involved in the α1β1 interface of Hb dimers (4). dos Santos et al. (5) have shown that, in human erythroid cells, the AHSP gene expression is related to that of the α-globin gene. AHSP concentration is estimated to be ∼0.1 mm at the end of erythropoiesis (2), whereas that of Hb is ∼5 mm. Nascent α chains are protected by the chaperone before binding to the partner β chains. Within the developing red blood cell precursors, the chaperone must first bind the α chain to complete its correct folding. Evidence that AHSP facilitates folding of the α-globin subunits for Hb synthesis has been brought by Yu et al. (6), which adds a key element to the reaction scheme of Hb synthesis.
If the chaperone simply acts as a buffer for an excess of α chains, an appropriate affinity is necessary, but there would be freedom in the protein-protein interaction rates. This role would allow storage under a harmless form of the excess α-Hb, known to be naturally synthesized at an excess of 10% relative to β-Hb. However, an active role of AHSP for the correct folding of α chains places strict conditions on the protein association and dissociation rates. The chaperone obviously must bind α chains before irreversible capture by the β chains, and the overall contact time becomes an important factor.
The aim of this work was to demonstrate how the altered biochemical and biophysical properties of the human mutant AHSPV56G may explain the observed thalassemic effect, by comparing the protein-protein interactions of α-Hb with the mutant AHSPV56G and wild type AHSP (AHSPWT). Using wild type and mutated AHSPs produced in Escherichia coli, we investigated their association and dissociation kinetics in forming the AHSP·α-Hb complex. Several biophysical approaches are available for studying complexes involving hemoproteins. The intense absorption bands, which depend on the ligation state, allow rapid kinetic studies, including the measurement of ligand binding. Heme is also an excellent energy acceptor, which strongly quenches the globin fluorescence and that of interacting partner proteins. Thus, quenching of the fluorescence of the single tryptophan (Trp) residue of AHSP (see Fig. 1A) may be used as a probe for its binding to α chains (7). In the AHSP·α-Hb complex (8), the distance between the Trp residue of AHSP and the heme molecule is ∼20 Å, which is well below the Förster distance (28 Å in this case). There is thus a drop in fluorescence of AHSP due to energy transfer to the heme of the α chain (see Fig. 1A). By the same method, after addition of the β-Hb, one can follow the dissociation of the AHSP·α-Hb complex.
FIGURE 1.

A, three-dimensional view of the complex of AHSP (green) and α-Hb (red). The molecular structure shows the position of the AHSP tryptophan residue Trp44 (orange) and the valine (violet) that is mutated (V56G) in the clinical case. The image was obtained from the crystallographic structure of the AHSP·α-Hb complex (Protein Data Bank code 3ia3) using PyMOL software. B, composite kinetics of the AHSP association with α chains and subsequent replacement of AHSP by β chains at 37 °C, pH 7. Mixing ratios were 1:1 in each case at 10 μm final protein concentration. The β chain and AHSP were premixed and loaded in one of the stopped-flow syringes, for mixing with the α chains: [β-Hb + AHSP] + α-Hb → β-Hb + AHSP·α-Hb → AHSP + αβ. The fluorescence of the single tryptophan (Trp44) residue of AHSP serves as a probe for the binding to α chains.
Using the fluorescence energy transfer technique, we investigated the dynamic association and dissociation of AHSPWT and AHSPV56G with α-Hb. The protein secondary structure and thermal stability of the WT and mutant AHSP were also studied by circular dichroism (CD) spectroscopy.
EXPERIMENTAL PROCEDURES
Preparation of α-Hb and β-Hb Chains
After reaction of human Hb with p-hydroxy-mercuribenzoic acid, the isolated Hb chains were purified by ion exchange chromatography, saturated with CO, and stored at −80 °C, as described previously (9–10).
Generation of Mutant AHSP
The AHSPV56G was generated by site-directed mutagenesis (QuikChange® II site-directed mutagenesis kit, Stratagene) using the pGEX6P-AHSP vector as described previously (7).
Recombinant Protein Expression and Purification
The normal and mutated AHSPs were expressed as fusion protein with S-transferase (GST) in E. coli BL21 (DE3) cells using the pGEX6P-AHSP expression plasmid (7). Briefly, the cell pellets were resuspended in phosphate-buffered saline (150 mm NaCl, 10 mm Na2HPO4, pH 7.4) and stored frozen at −80 °C. Then, the normal GST-AHSP (GST-AHSPWT) and mutated GST-AHSP (GST-AHSPV56G) were solubilized as described previously (7). After centrifugation, the soluble fraction containing the GST-AHSPV56G or GST-AHSPWT was purified in one step by affinity chromatography on glutathione-Sepharose 4B (GSTrap 4B column, GE Healthcare). The cleavage of the GST moiety was achieved in the tube by the addition of the PreScission Protease (2 units/100 μg of fusion protein) (GE Healthcare) in phosphate-buffered saline containing 1 mm dithiothreitol overnight at 4 °C under gentle agitation. Then, the mix was placed on glutathione-Sepharose 4B affinity support (GE Healthcare) to remove the GST moiety and PreScission protease. The AHSPV56G or AHSPWT was recovered in the flow-through and concentrated by ultracentrifugation (Amicon Ultra, Millipore, Billerica, MA).
Measurement of Auto-oxidation Rates
The α-Hb was prepared by a 15-min incubation on ice in 100 mm potassium phosphate buffer at pH 7.0 with 1 mm potassium ferricyanide, to accelerate removal of CO from the stock α-Hb. Then, to remove the excess of potassium ferricyanide, the solution was loaded on PD10 column (GE Healthcare) under air and eluted quickly with 100 mm potassium phosphate, pH 7.0. The AHSPWT or AHSPV56G were added to the α-Hb(Fe3+) in closed sample cuvettes. The solutions were immediately flushed under nitrogen and an excess of sodium dithionite was added to obtain the ferrous proteins. Just before starting the kinetics, the samples at 10 μm on a heme basis were saturated under oxygen. The auto-oxidation kinetics were followed by measuring the entire absorption spectra of the proteins versus time. We analyzed the auto-oxidation reaction at 576 nm for the α-Hb and the AHSP·α-Hb complexes. All spectra and kinetics were measured using a thermostated diode-array spectrophotometer (Hewlett Packard 8453); samples were in 4 × 10 mm quartz cuvettes at 37 °C.
Stopped-flow/Fluorescence
Rapid-mixing experiments were carried out in a SFM-3 stopped-flow instrument (Bio-Logic SAS, Claix, France), equipped with three syringes, and maintained at 37 °C in a circulating water bath. Stock protein concentrations used were 20 μm in phosphate-buffered saline. For experiments with β-Hb, 5 mm dithiothreitol was added. The final concentration of each protein was 10 μm (on a heme basis for α-Hb and β-Hb). Detection was made for both absorption changes through a 10-mm optical path length or fluorescence using a 1-mm path length FC-18 cuvette. Fluorescence excitation was at 280 nm with the monochromator bandwidth set to 5 nm, and the total emission signal of AHSP was collected. Results were the average of at least three measurements. The obtained parameters were analyzed and modeled with BioKine software (version 3.20).
Kinetics of CO Recombination
The CO recombination to the AHSPWT·α-Hb and AHSPV56G·α-Hb complexes were obtained after flash photolysis using 10 ns laser pulses at 532 nm (YAG laser, Quantel, Les Ulis, France). Absorption differences were measured at 436 nm, for samples at 10 μm on a heme basis in phosphate-buffered saline, 100 μm CO at 25 °C (11).
Circular Dichroism
CD spectra and thermal denaturation curves were measured with a Jasco J810 spectropolarimeter (Jasco, Tokyo, Japan), using a 0.5 mm optical path length cell. Full spectra from 190 to 260 nm were measured at several temperatures, at 5 and 10 μm, and were used to determine the % α helix.
For thermal denaturation curves, the cell temperature was programmed using a Jasco PTC-423S thermoelectric temperature controller. The ellipticity at 222.6 nm was monitored over a temperature range of 0–97 °C, using a bandwidth of 1 nm and a temperature gradient of 1 °C/min. The Hb samples were in the CO form, at a concentration of 18 μm (on a heme basis) in 2.5 mm Na2HPO4, 37 mm NaCl buffer at pH 7.4. The CD signal was normalized to obtain the unfolded fraction: fU = (yN − yobs)/(yN − yU), where yobs is the observed CD signal and yN and yU represent the CD signal of the native and unfolded protein, respectively. The temperature corresponding to 50% unfolded molecule is the melting temperature, Tm.
RESULTS
Both AHSPV56G and AHSPWT were expressed in E. coli. In whole cell lysate, we observed an identical expression of AHSPV56G and AHSPWT indicating a normal level of biosynthesis in bacteria. In contrast, the amount of GST-AHSPV56G obtained after purification was reduced by ∼25% compared with that obtained for the wild type protein, using the same experimental procedures (data not shown). This result suggests an instability of the mutant AHSP. After removing the GST moiety, we investigated the interaction of AHSPWT/V56G with α-Hb.
Protein-binding Kinetics
The fluorescence energy transfer technique was used to observe both the association and dissociation of AHSP with α-Hb. The stopped-flow mixing was measured by two methods. In the first method, the individual proteins were mixed: AHSP and α-Hb to study the association, followed by addition of β-Hb to the AHSP·α-Hb complex to follow the dissociation. We observed a rapid association of AHSP and α-Hb with a constant of 20 μm−1 s−1 at 37 °C for both normal and mutated AHSP (Table 1). This value is ∼40 times higher than that reported for the α and β association (12). Because β-Hb does not interact with AHSP, an alternate mixing sequence was used; α-Hb was mixed with a solution containing a premixed stock of AHSP and β-Hb (Fig. 1B). The same reaction kinetics were obtained, confirming that the AHSP reaction with α-Hb is much faster than the α-β association reaction. The experiment is simpler because it allows observation of both reactions after a single mixing, without change of the reactant concentrations or inner filter effects throughout two step kinetics.
TABLE 1.
Kinetic parameters for the AHSP/α-Hb interactions
The time coefficient τ = 1/rate is given for the dissociation, replacement (see Equation 1), and oxidation reactions, with A, B, and C referring to the reactants α-Hb, β-Hb, and the chaperone AHSP, respectively. Kinetic measurements were made in phosphate-buffered saline at 37 °C with a final concentration of each protein of 10 μm (on a heme basis for α-Hb and β-Hb). Oxidation kinetics of the heme iron were made in 100 mm phosphate buffer at pH 7 and 37 °C; in the same conditions, the oxidation of free α-Hb chains is ∼6000 s. An αβ association rate of 0.5 μm−1 s−1 (12) was used, which influences directly the AHSP·α-Hb dissociation rate extracted from the rate of the replacement reaction (Equation 2); however, it does not affect the ratio (mutant versus WT) of rates.
| Reaction | AHSPWT | AHSPV56G |
|---|---|---|
| k association, AHSP·α-Hb | 20 | 20 μm−1 s−1 |
| τd dissociation, AHSP·α-Hb | 2 | 0.5 s |
| τr replacement reaction | ||
| τr = 1/λr at 1:1:1 for A:B:C | 80 | 20 s |
| τr calculated at 1:1:20 for A:B:C | 1600 | 400 s |
| τoxid = 1/k of iron oxidation | 1610 | 2860 s |
CO Recombination Kinetics
After in vitro association of AHSPV56G with α-Hb, the CO rebinding kinetics of the AHSPV56G·α-Hb complex exhibit a single phase identical to that obtained with the AHSPWT·α-Hb complex (Fig. 2), described previously as an intermediate state “I” relative to the classical R and T allosteric conformations (7). When native β-Hb was added to the AHSPV56G·α-Hb complex, the kinetics became biphasic as expected for tetrameric Hb (α2β2) showing the two phases corresponding to the two allosteric states (Fig. 2). These results indicate that the properties of native α-Hb within the tetrameric Hb are not modified after interaction with AHSPV56G.
FIGURE 2.

Fraction of the ligand-rebinding phases R (black bars), I (gray bars), and T state (dotted white bars), after photodissociation of CO. Protein concentrations were 10 μm. The isolated α-Hb (left) shows a single phase typical of monomers, dimers and R state Hb tetramers with a rate of 6 μm−1 s−1; after addition of β-Hb chains, one observes two phases characteristic of the allosteric R and T states of tetrameric α2β2 Hb, with rates 6 μm−1 s−1 and 0.2 μm−1 s−1, respectively. The AHSPWT·α-Hb complex (center) shows a single phase for an intermediate form I with a rate of 2 μm−1 s−1, three times lower than for the isolated α-Hb, as reported previously (7). The kinetics for the AHSPV56G·α-Hb complex (right) were similar to those obtained with the WT complex; in both cases, addition of β-Hb leads to a loss of the I state and an increase in the usual R and T states of tetrameric Hb.
Replacement Reaction
Although a direct interaction of α-Hb and β-Hb would be rapid (<1 s), the nature of a replacement reaction leads to a greatly reduced rate for formation of αβ Hb dimers in the presence of AHSP. Effectively, each time α-Hb separates from AHSP, the same competition will favor rebinding to the chaperone; αβ formation thus requires nearly 1 min. With notation of A (α chain), B (β chain), C (chaperone protein), and considering B and C in excess relative to A to treat the association as a pseudo first order reaction, the overall rate (λ) for the replacement reaction depends on both the individual association and dissociation rates.
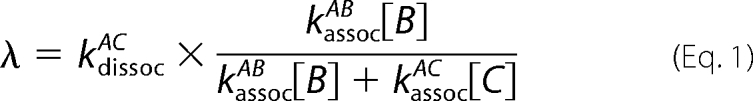 |
If the rate of association of AC is much greater than that for AB, the form simplifies to the AC dissociation rate modulated by the ratio of the competing association reactions.
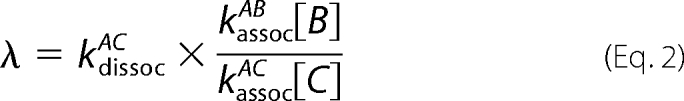 |
The observed replacement rate λ is thus the intrinsic AC dissociation modulated by the ratio of the concentration-dependent association rates. The AC association is favored by a factor of 40 relative to the AB reaction, and the local concentrations could produce an additional factor. Note that the important concentrations for the kinetics are the monomeric states, rather than the overall protein concentration, which accumulates mainly as the Hb tetramer.
For the mutant AHSPV56G, the main difference was in the dissociation from α-Hb being nearly 4 times faster than for AHSPWT, with a rate of 2/s for the wild type and 0.5/s for the mutant AHSP (Table 1 and Fig. 3A); note that the exact values depend on the rate of the AB reaction, but the ratio of dissociation rates (mutant versus WT) does not depend on this factor. Thus, the overall contact time will be reduced for this mutation. The replacement reaction for AHSPWT was also measured for the deoxy ferrous Hb, under anaerobic conditions, following the small absorption change for the R to T allosteric transition. Under these conditions, a very stable α2β2 tetramer is formed. A similar rate was observed for the replacement reaction at 25 °C, indicating that the state of the iron atom does not significantly change the rates of interactions between the proteins.
FIGURE 3.

A, kinetics for the replacement of AHSP by β-Hb, at 37 °C, pH 7. The rate for the mutant AHSPV56G is ∼4 times faster than for AHSPWT. B, the rate of replacement depends on the ratio of the concentrations of the competing proteins. At a ratio of AHSP to β-Hb of 20, the time required would be 20 times longer, approaching the time for oxidation of α-Hb within the complex with WT AHSP, but not the mutant AHSP. C, reaction scheme: nascent α chains (red) enter a local region where the number of AHSP (green) exceeds that of β-Hb (blue) monomers, due to formation of β4 at concentrations above a few μm. Eventually, the α and β chains bind, quasi irreversibly, to continue the formation of Hb tetramers.
Protein Secondary Structure Analyzed by Circular Dichroism
AHSPWT exhibits ∼74% of the α helix form at 25 °C, with a melting temperature (Tm) of 61 °C as reported by Gell et. al (13). For AHSPV56G, a poorly defined unfolding curve is observed (Fig. 4A) with an estimated Tm of 42 °C; the mutant AHSP thus appears to be partially unfolded at ambient temperatures, with a decreased percent of α helix (roughly 50% at 25 °C and 60% at 0 °C). However, in the presence of α-Hb chains, the complex displays ∼65% α helix at 25 °C with a transition centered at 53 °C (Fig. 4B), similar to that for the wild type complex (65% α helix and Tm of 56 °C) and free α chains (74% of α helix and Tm of 56 °C). These results suggest that α-Hb may aid in recovering the normal α helical content and protein conformation of the mutated AHSP. We observed a reversible thermal cycle for both wild type and mutated AHSP, with minor loss in helical content for AHSPV56G. The thermal unfolding cycle to 95 °C was irreversible for both isolated α-Hb and the complexes; the complex shows a partial reversibility with an important loss in helical content of about 40%, probably due to irreversible loss of the α-Hb.
FIGURE 4.

A, thermal unfolding curve. The mutant AHSPV56G displays a wider unfolding curve (red) versus temperature, as if the transition occurs progressively. The AHSPWT (dark red) shows a more distinct transition, but not as sharp as isolated α-Hb (black). AHSPV56G displays less α helix than AHSPWT; curves were normalized at lower temperatures to the fraction α helix determined from full CD spectra. B, thermal unfolding curves for the AHSPWT·α-Hb (dark red) and AHSPV56G·α-Hb (red) complexes. When complexed to α-Hb, AHSP and especially the mutant AHSP have a higher % α helix (based on the full CD spectra) and a sharper transition. C, the molecular graphic images show the solvent surface accessibility of the AHSPWT and AHSPV56G, based structure of Protein Data Bank code 1XZY (4) using PyMOL software. The mutation leads to open a cavity between helices 2 and 3, which allows a water molecule to access and possibly destabilize the two α helices. Such a perturbation of the core hydrogen bonds could explain the lower fraction of α helix found in the mutant AHSP and the extended thermal unfolding curve.
Auto-oxidation
The transition of oxy-α-Hb to the oxidized or metHb form also could be followed spectrophotometrically. Note that in the case of the complex AHSP·α-Hb, the final state of the α is a bis-histidyl hexacoordinated state with a distinct spectral form relative to the usual aquomet Hb ferric state (14–15). The observed time coefficient 2860 s of α-Hb complexed to mutant AHSPV56G is a bit less than 2-fold slower than the rate of 1610 s for the wild type complex (Table 1).
The switching from the ferric aquomet structure of α-Hb to the ferric bis-histidyl hexacoordinated form was followed by absorbance stopped flow by mixing met-α-Hb with AHSP. The observed kinetics are the same as for the association of ferric α-Hb to AHSP, as the absorbance change strictly followed the fluorescence decrease (data not shown); this indicates that the subsequent switch to the His-Fe-His form is fast relative to the protein association rate.
DISCUSSION
Molecular chaperones are crucial in the acquisition and maintenance of the native conformation of their target proteins and thereby help prevent their aggregation. The α-Hb, when associated with its chaperone AHSP, clearly displays a more robust form, relative to the isolated α-Hb. Certain properties of the complex are maintained, such as the decrease in the CO binding rate to ferrous α-Hb and the hexacoordination of the oxidized form of α-Hb. In this work, we demonstrate that the AHSP mutation V56G leads to a less stable protein, which may account for the observed pathology in the patient. In this case, binding of the α chain apparently helps protein folding of AHSPV56G. Surprisingly, the association rate is similar, despite the modified structure of the isolated AHSPV56G. In contrast, the dissociation rate of the complex is much higher for the mutant AHSP.
Protein Stability
Studies of thermal denaturation revealed a decreased stability of AHSPV56G; the thermal unfolding curve of AHSPV56G showed a wider transition (Fig. 4A), as if the unfolding involved several steps. Note that even AHSPWT has a wider transition relative to Hb and other proteins; this could indicate an exceptional example of a protein that has several stable states that are partially unfolded. As seen in the three-dimensional structure (Fig. 1A), the residue at position 56 is not at the AHSP·α-Hb interface but at the junction of two α helices. The decrease in Tm indicates an important alteration even at physiological temperature, which could explain the lower yield of mutated recombinant protein obtained after the purification step.
Unlike AHSPV56G, the two other known AHSP mutations, AHSPM45K and AHSPN75I, have normal stability, with a Tm of 58 °C and 54 °C, respectively (3). These two mutations do not modify the binding of AHSP for α-Hb, but AHSPN75I displays a reduced capacity to inhibit reactive oxygen species production by α-Hb (3, 16). Moreover, an unusually severe anemia has been reported for a patient heterozygous for both β-thalassemia and the AHSPN75I mutation (17). Different in vitro studies have shown that the binding of AHSP to α-Hb leads to the reorganization of the heme pocket with the final structure possessing a bis-histidyl hexacoordinated α-Hb form (14–15). This bis-His-α-Hb·AHSP complex inhibits α-Hb peroxidase activity, heme loss, and thus the production of reactive oxygen species (14).
Studies in silico of the modification of solvent accessibility resulting from the V56G mutation show that the removal of the valine side chain opens a direct solvent access to the core of two α helices of AHSP (Fig. 4C). It is known that water molecules near the core of an α helix significantly can disturb its secondary structure by destabilizing the core hydrogen bonds of the α helix. Another explanation could be that a large internal cavity is thermodynamically unfavorable for folding, thus disturbing the formation of these two helices. By either mechanism, the α helices near the mutation zone can be destabilized, leading to a temperature-dependent loss of the α helix, as measured by CD. Surprisingly, this mutant, when associated with α-Hb to form the AHSPV56G·α-Hb complex, returns to a normal conformation and stability (Fig. 4B). In silico studies show that the channel was closed when the complex is formed and so limits the solvent accessibility and the gap between the helices.
Protein-Protein Kinetics
The kinetic measurements show that the mutation does not perturb the association rate of AHSP with α-Hb, indicating that the altered structure does not disturb the contact and suggests that this step of the reaction does not require a highly specific conformation. The main difference for AHSPV56G is a much higher dissociation rate of the AHSPV56G·α-Hb complex, as evidenced from the replacement reaction.
The replacement reaction was measured by two methods. In the first method, three syringes were used; only α-Hb and AHSP were initially mixed, followed by addition of β-Hb after a fixed delay. The variable delay did not influence the rate of replacement, indicating there were no slow conformational changes of the complex that might modify the dissociation kinetics. In the second protocol, a premixing of AHSP and β-Hb chains was employed; this demonstrates directly a higher association rate of α-Hb to AHSP versus β-Hb, at equal concentrations. For both methods, the results at 37 °C indicate a rapid association of AHSP and α-Hb as shown in Table 1. This ensures that α-Hb initially will preferentially bind to AHSP to complete the protein folding process. At 25 °C, the association rate was slightly decreased, similar to the value reported by Mollan et al. (18).
This higher dissociation rate of the mutant complex, by typically a factor of 3–4, could impair the final protein folding of the α-Hb due to a reduced time of contact between the proteins. The dissociation rates were not found to depend strongly on the Hb oxidation state (oxy versus met versus deoxy). This leads to the question of the time necessary for the protein folding, and whether multiple binding contacts are involved. Based on the kinetic data, the complex dissociates every 2 s for AHSPWT and 0.5 s for the mutant. Note that the most likely result after AHSP·α-Hb dissociation is a reassociation of α-Hb with AHSP, due to the very high association rate compared with β-Hb. Thus, the overall contact time before finally binding to β-Hb is provided by the rate of replacement, which is >1 min for AHSPWT, at equal concentrations of all three proteins. For a detailed analysis, the local proteins concentrations are needed, taking into account the β tetramer (β4) formation. The local AHSP concentration has been reported as ∼100 μm; if 20% are unbound, one could approach relative concentrations of AHSP to β-Hb monomer of 20 to 1, and the replacement reaction would exceed 20 min.
Relation to Auto-oxidation
The time coefficient for auto-oxidation of the α-Hb within the AHSPWT·α-Hb complex, was ∼1610 s (Table 1 and Fig. 3B). In the case of the AHSPV56G·α-Hb complex, this rate was nearly 2-fold slower than for AHSPWT·α-Hb but remains faster than that observed for free α-Hb.
Different explanations could be proposed for the clinical implications of the mutant AHSP observed in the patient. The decreased stability of AHSPV56G, as evidenced by our results of thermal unfolding, could lead to a deficit in AHSP; a possible degradation of the free partially unfolded AHSPV56G by the cell could be considered. However, the mutant still binds efficiently to α chains and recovers its α helix conformation within the complex (Fig. 4A).
Alternately, the higher dissociation rate may not allow enough time to complete the necessary protein folding of the α chain. If protein folding requires iron oxidation, then the overall contact time, which could include multiple bindings, is of importance. This could explain the very high ratio of association rates of AHSP versus β-Hb. A lower ratio would be sufficient for initial binding, but the high ratio ensures multiple bindings and a slower replacement reaction. Note that when oxidized α-Hb was mixed with AHSP, the hexacoordinated form was produced at the same rate as complex formation, indicating a rapid transition to this state upon complex formation; this transition of α-Hb thus occurs on a ms time scale.
A reaction scheme is presented in Fig. 3C, displaying the competing proteins for binding to a single α-Hb chain. Because the β4 formation maintains a low concentration of β-Hb monomers, there may be a large excess of AHSP protein relative to β-Hb monomers; the replacement reaction (Equation 2) may thus be extremely slow by the combined factors favoring AHSP association with α chains: a higher intrinsic rate and a higher local concentration.
The kinetic model implies a minimum AHSP concentration necessary for the competitive reactions. One can pose the question as to whether there is enough AHSP to satisfy the high flux of α-Hb production. We consider a single red blood cell of volume 90 femtoliter to obtain the total amount of Hb per cell of 30 pg (270 million tetramer molecules). This quantity of Hb tetramer is produced in the developing cells in ∼50 h, corresponding to an average rate of α-Hb production of 3000/s. If the AHSP must remain bound to an α chain for a total of 15 min (accumulated binding time), then some 3 million AHSP molecules are required. The reported AHSP concentration of 100 μm (2) would correspond to 10 million molecules, satisfying this requirement. If only the shorter contact time of 1 s were required (sufficient for binding and conformational changes such as hexacoordination of the oxidized form) then the AHSP would seem to be present in great (over 100-fold) excess relative to the requirements.
A high AHSP concentration is not necessary for the initial competition with β-Hb, as the factor of 40 in intrinsic rates would already ensure that only a few percent of the α chains were not chaperoned. Thus, the high AHSP concentration would seem necessary to satisfy the additional requirement of a long overall contact time with the α-Hb chains.
The α-Hb chains bind to the AHSP chaperone protein much faster than to β-Hb chains, as expected if the chaperone protein must be competitive to intercept nascent α-Hb chains. At equal protein concentrations, the transfer to β-Hb is slow (80 s) but still faster than oxidation of α-Hb in the presence of AHSP; in this case, fabrication of Hb tetramers would not pass through the oxidized state. If the local concentration of free AHSP is high relative to β-Hb monomers, the slow replacement of AHSP by Hb could approach the AHSP·α-Hb auto-oxidation time, confirming such a mechanism as part of the correct folding of α-Hb. The dominant difference between AHSPWT and the mutant AHSPV56G is a faster dissociation for the mutant (Fig. 3A), suggesting that the overall aid in protein folding may be incomplete.
All of these results may reflect the pathology observed in the patient with this abnormal chaperone, but in vivo mechanisms are often more complex. Indeed, the level of AHSP gene expression varies from one individual to another (19, 20). In addition, the analysis of the AHSP gene of this patient homozygous for the AHSPV56G has shown the presence of haplotypes characterized by a decrease of the AHSP expression (1). Thus, the association of the altered expression of AHSP and the presence of structural abnormality, which could lead to a lower lifetime, appear to be relevant factors in the hematological phenotype of the patient.
This work was supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), the Délégation Générale pour l'Armement (DGA), and the University of Paris 7, 11, and 12 (France).
- AHSP
- α-hemoglobin stabilizing protein
- GST
- glutathione S-transferase
- Tm
- melting temperature
- WT
- wild type.
REFERENCES
- 1.Pissard S., Vasseur C., Toutain F., Silva M., Faubert-Laugé E., Domingues E., Marden M., Wajcman H., Baudin-Creuza V. (2009) 51st ASH Annual Meeting, New Orleans, December 5–8, 2009, Blood 114, 462 [Google Scholar]
- 2.Kihm A. J., Kong Y., Hong W., Russell J. E., Rouda S., Adachi K., Simon M. C., Blobel G. A., Weiss M. J. (2002) Nature 417, 758–763 [DOI] [PubMed] [Google Scholar]
- 3.Santiveri C. M., Pérez-Cañadillas J. M., Vadivelu M. K., Allen M. D., Rutherford T. J., Watkins N. A., Bycroft M. (2004) J. Biol. Chem. 279, 34963–34970 [DOI] [PubMed] [Google Scholar]
- 4.Feng L., Gell D. A., Zhou S., Gu L., Kong Y., Li J., Hu M., Yan N., Lee C., Rich A. M., Armstrong R. S., Lay P. A., Gow A. J., Weiss M. J., Mackay J. P., Shi Y. (2004) Cell 119, 629–640 [DOI] [PubMed] [Google Scholar]
- 5.dos Santos C. O., Duarte A. S., Saad S. T., Costa F. F. (2004) Exp. Hematol. 32, 157–162 [DOI] [PubMed] [Google Scholar]
- 6.Yu X., Kong Y., Dore L. C., Abdulmalik O., Katein A. M., Zhou S., Choi J. K., Gell D., Mackay J. P., Gow A. J., Weiss M. J. (2007) J. Clin. Invest. 117, 1856–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baudin-Creuza V., Vasseur-Godbillon C., Pato C., Préhu C., Wajcman H., Marden M. C. (2004) J. Biol. Chem. 279, 36530–36533 [DOI] [PubMed] [Google Scholar]
- 8.Gell D. A., Feng L., Zhou S., Jeffrey P. D., Bendak K., Gow A., Weiss M. J., Shi Y., Mackay J. P. (2009) J. Biol. Chem. 284, 29462–29469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geraci G., Parkhurst L. J., Gibson Q. H. (1969) J. Biol. Chem. 244, 4664–4667 [PubMed] [Google Scholar]
- 10.Parkhurst K. M., Parkhurst L. J. (1992) Int. J. Biochem. 24, 993–998 [DOI] [PubMed] [Google Scholar]
- 11.Marden M. C., Kister J., Bohn B., Poyart C. (1988) Biochemistry 27, 1659–1664 [DOI] [PubMed] [Google Scholar]
- 12.McGovern P., Reisberg P., Olson J. S. (1976) J. Biol. Chem. 251, 7871–7879 [PubMed] [Google Scholar]
- 13.Gell D., Kong Y., Eaton S. A., Weiss M. J., Mackay J. P. (2002) J. Biol. Chem. 277, 40602–40609 [DOI] [PubMed] [Google Scholar]
- 14.Feng L., Zhou S., Gu L., Gell D. A., Mackay J. P., Weiss M. J., Gow A. J., Shi Y. (2005) Nature 435, 697–701 [DOI] [PubMed] [Google Scholar]
- 15.Hamdane D., Vasseur-Godbillon C., Baudin-Creuza V., Hoa G. H., Marden M. C. (2007) J. Biol. Chem. 282, 6398–6404 [DOI] [PubMed] [Google Scholar]
- 16.dos Santos C. O., Zhou S., Secolin R., Wang X., Cunha A. F., Higgs D. R., Kwiatkowski J. L., Thein S. L., Gallagher P. G., Costa F. F., Weiss M. J. (2008) Am. J. Hematol. 83, 103–108 [DOI] [PubMed] [Google Scholar]
- 17.dos Santos C. O., Zhou S., Albuquerque D. M., Saad S., Weiss M. J., Costa F. F. (2005) 47th ASH Annual Meeting, Atlanta, December 10–13, 2005, Blood 106, 3629 [Google Scholar]
- 18.Mollan T. L., Yu X., Weiss M. J., Olson J. S. (2010) Antioxid. Redox. Signal 12, 219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galanello R., Perseu L., Giagu N., Sole G. (2003) 45th ASH Annual Meeting, San Diego, December 6–9, 2003, Blood 102, 1881 [Google Scholar]
- 20.Lai M. I., Jiang J., Silver N., Best S., Menzel S., Mijovic A., Colella S., Ragoussis J., Garner C., Weiss M. J., Thein S. L. (2006) Br. J. Haematol. 133, 675–682 [DOI] [PubMed] [Google Scholar]