

Abstract
AIMS
Catumaxomab is the first EMEA approved trifunctional anti-EpCAM×anti-CD3 antibody for the treatment of cancer patients with malignant ascites. A phase II pharmacokinetic study was conducted to determine local and systemic antibody concentrations and anti-drug antibody (ADA) development.
METHODS
Thirteen cancer patients with symptomatic malignant ascites were treated with four ascending doses of 10, 20, 50, and 150 µg catumaxomab intraperitoneally (i.p.) infused on days 0, 3, 6 or 7 and 10. The pharmacokinetics of catumaxomab were studied by implementation of supportive data from a non clinical mouse tumour model. Additionally, ADA development was monitored.
RESULTS
Ten out of 13 patients were evaluable for pharmacokinetic analysis. Catumaxomab became increasingly concentrated in ascites during the course of treatment, attaining effective concentrations in the ng ml−1 range. Catumaxomab remained immunologically active even after several days in the circulation. The observed systemic catumaxomab exposure was low (<1%), with a maximal median plasma concentration (Cmax) of 403 pg ml−1. The mean elimination half-life in the plasma was 2.13 days. All patients developed ADA, but not before the last infusion. High observed inter-individual variability and low systemic exposure may be explained by the inverse correlation between tumour burden, effector cell numbers and systemic antibody bioavailability as demonstrated in a defined mouse tumour model.
CONCLUSIONS
Based on the high and effective local concentrations, low systemic exposure and acceptable safety profile, we confirmed that the i.p. application scheme of catumaxomab for the treatment of malignant ascites is appropriate.
Keywords: catumaxomab, immunogenicity, intraperitoneal infusion, malignant ascites, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The trifunctional antibody catumaxomab is a highly effective anti-cancer therapeutic that is administered to patients suffering from malignant ascites intraperitoneally (i.p.) in microgram (µg) doses. So far, no clinical pharmacokinetic data are available.
WHAT THIS STUDY ADDS
Catumaxomab attains effective local concentrations in the ascites fluid and shows low systemic exposure with an acceptable safety profile confirming the appropriateness of the i.p. application scheme.
Introduction
Catumaxomab is a novel trifunctional rat/murine hybrid antibody that has bi-specificity for the epithelial cell adhesion molecule, EpCAM and the T-cell antigen, CD3. In addition, it has a functional constant Fc region that preferentially binds to Fc-gamma receptors (FcγR) I/IIa and III [1–4]. This trifunctionality leads to effective destruction of EpCAM-positive tumour cells even at antibody concentrations in the pg ml−1 range [5]. During clinical development, the antibody demonstrated convincing efficacy as an intraperitoneal (i.p.) treatment for advanced carcinomas associated with malignant ascites (MA) [6]. In MA, the abnormal accumulation of fluid within the peritoneal cavity is primarily caused by the spread of epithelial tumour cells. The majority of these tumour cells (70–100%) express EpCAM [7–11]. Therefore, the rationale for i.p. application of catumaxomab was to obtain high local concentrations at the site of action. Efficient tumour cell elimination in the ascites fluid followed by absence of ascites re-accumulation confirmed the treatment concept [6, 12]. In a dose escalating study, the maximal tolerated dose (MTD) was defined at five i.p. infusions of 10, 20, 50, 200, and 200 µg, administered within 9–13 days [12]. The main side-effects, including fever, nausea, and vomiting, were fully reversible and consistent with the mode of action of catumaxomab. The dose escalation scheme employed a low starting dose based on the facts that i) i.p. administration of catumaxomab was associated with symptoms attributed to cytokine release and ii) the patients did not tolerate high starting doses. In subsequent infusions, high doses of catumaxomab were generally well tolerated. Most of the patients developed anti-drug antibodies (ADA) against the mouse and rat sequences of catumaxomab, but not before the last infusion [6, 12, 13]. Finally, these clinical parameters were implemented in a pivotal phase II/III study that showed that catumaxomab significantly prolonged puncture-free survival in patients with MA [14] leading to market approval of catumaxomab in Europe. Here, we report the results from the corresponding pharmacokinetic trial. The main study objectives were to determine local and systemic concentrations of catumaxomab and establish its safety profile. In addition, we present supporting data on the bioavailability of catumaxomab gained from a mouse tumour model.
Methods
Patients
Thirteen patients with cancer, symptomatic MA, and immunocytochemically confirmed EpCAM-positive tumour cells (≥400 in 106 analysed ascites cells) were treated. The most frequent type of primary tumour was ovarian (69%), followed by pancreatic (23%), and gastric (8%) carcinoma. Two patients who received only three infusions, and one patient who was ADA positive before treatment, were excluded from the pharmacokinetic evaluation. The study was conducted in accordance with the Declaration of Helsinki (Addendum from Washington, 2002 to the Edinburgh Version of 2000), and it was approved by the local authorities and independent ethics committees relevant to the centres involved. Written informed consent was obtained from each patient.
Pharmacokinetic study design
An open-label, phase II pharmacokinetic study was carried out at two centres in Romania and three centres in Germany. Patients with cancer and MA were treated with four ascending doses of the therapeutic antibody catumaxomab (10, 20, 50, and 150 µg) on days 0, 3, 6 or 7, and 10. The antibody was diluted in 0.9% NaCl solution and administered i.p. in a 6 h constant rate infusion via an indwelling catheter. To allow optimal distribution, 500 ml of 0.9% NaCl solution was given to patients before the application of the therapeutic antibody. To reduce adverse events related to cytokine release, 1000 mg of paracetamol was administered 30 min before each infusion as a premedication. Efficacy measurements and immunocytochemical analysis of ascites tumour cells were performed as described previously [12]. The primary study objective was the determination of local and systemic catumaxomab concentrations. Therefore, ascites samples were taken at screening and prior to each catumaxomab infusion. Blood samples (7.5 ml) were collected pre-infusion, 24 h after the first infusion, and at the indicated time points after the third and fourth infusions. Additional samples were collected from two patients after the second infusion, but no catumaxomab antibody was detected. The heparin plasma preparations were performed at the study site, and aliquoted samples were frozen within 1 h of collection. All samples were stored frozen (−80°C) until measurement.
Determination of catumaxomab concentrations
Plasma catumaxomab concentrations were measured by a validated two-site ELISA. Briefly, catumaxomab was captured by an anti-rat IgG λ light chain-specific antibody (LA1B12, TRION Research, Munich, Germany). Bound catumaxomab was then detected via an anti-mouse IgG2a-specific biotin-labeled detection antibody (BD Pharmingen, San Diego, CA). Then, streptavidin-β-galactosidase and its corresponding substrate, chlorphenolred-β-D-galactopyranosid (Roche Diagnostics, Mannheim, Germany), were added, and the colorimetric reaction was measured at 570 nm. Catumaxomab concentrations were calculated by interpolation on a standard curve. The lower limit of quantification (LLOQ) of the assay was determined to be 125 pg ml−1; the upper limit of quantification was 4000 pg ml−1. All samples were diluted 1:2 and subjected to a delipidation step before measuring in duplicate. Spike recoveries were performed with pre-therapy samples of all patients and the reciprocal values were used as a correction factor. The mean spike recovery was 107% (±17.21 SD). Post-study quality control (QC) sample analysis demonstrated an overall inter-assay precision of 5.5% CV and an overall assay accuracy of 99.2%. Assay-interfering ADA were effectively blocked with the use of an in-house developed blocking agent that consisted of isotype-matched rat and mouse monoclonal antibodies. All ADA-positive samples were re-measured in the presence of the blocking agent. In about 9% of the plasma samples with very high ADA concentrations, assay interference could not be completely eliminated. The results from these samples were considered invalid and were not used for PK calculations.
For quantification of catumaxomab in ascites fluid, the ELISA was adapted to the ascites matrix. The analytical range of the assay was 250–8000 pg ml−1.
Evaluation of bioactivity in a potency assay
The immunological activity, also referred to as cytotoxcity in ascites and plasma samples, was indirectly evaluated with an XTT (tetrazolium hydroxide) proliferation assay. Briefly, 1 × 105 peripheral blood mononuclear cells (PBMC) were mixed with 1 × 104 EpCAM-positive HCT-8 tumour cells (ATCC CCL-244) in 96-well flat bottomed plates. Samples were added at a 1:10 (ascites) or 1:5 (plasma) dilution, and plates were cultivated at 37 °C and 5% CO2. After 3 days, the culture supernatants were collected for cytokine measurement by Th1/Th2 cytometric bead array (CBA) assay, according to the manufacturer's instructions (BD Pharmingen, San Diego, CA). The cytokine concentrations of IL-2, IL-4, IL-6, Il-10, IFN-γ and TNF-α were determined. The remaining adherent tumour cells were stained with the XTT cell proliferation kit II (Roche Diagnostics GmbH, Mannheim, Germany) after soluble PBMC had been removed by washing twice with PBS. The tumour cell killing efficiency was assessed in two independent experiments with triplicate determinations of each sample, as described previously [5]. Final catumaxomab assay concentrations ranged between 300 and 4000 pg ml−1, and resulted in significant tumour cell killing (32–90%). Spiked controls were made by adding fresh catumaxomab to pre-therapy ascites or plasma samples from the corresponding patients. Additionally, pre-therapy non-spiked samples served as matrix controls. The % bioactivity was calculated according to the formula: (% killing sample/% killing spiking control) × 100%.
Analysis of anti-drug antibodies
Anti-drug antibodies in plasma samples were analysed with a validated bridging ELISA at Experimentelle und Diagnostische Immunologie (EDI) GmbH (Reutlingen, Germany). Briefly, catumaxomab was immobilized to the surface of a multititre plate as a capture antibody. Samples were added, and antibodies that bound catumaxomab were detected by adding biotinylated catumaxomab. Next, a streptavidin-β-galactosidase conjugate was added, and the ADA were quantified photometrically at 570 nm. The monoclonal mouse antibody LA1B12 (TRION Research, Germany) was used to calibrate the ADA concentrations. This antibody recognizes the rat λ light chain of catumaxomab, and it was shown to generate dose–response curves similar to those generated by polyclonal anti-catumaxomab sera. The LLOQ of the assay was 13 ng ml−1.
Pharmacokinetic evaluation
All kinetic parameters were determined independently of a model with a non-compartmental method in the WinNonlin, version 4.1 (Pharsight corporation, Mountain View, CA) software program. Pharmacokinetic characteristics were determined directly from the measured concentrations. Actual sampling times were used for pharmacokinetic evaluations. The pharmacokinetic parameters used for systemic catumaxomab were AUC(0,tlast), Cmax, tmax, Cmax3, and Cmax4 (the latter two parameters are the maximal plasma concentrations after the third and fourth infusions, respectively). The AUC was calculated by a log linear·trapezoidal method. The apparent terminal elimination rate constant λz was determined by log-linear regression analysis using three time points. The terminal elimination half-life t1/2 was calculated according to the equation t1/2= ln2/λz.
Non-clinical mouse study
A non-clinical pharmacokinetic study with catumaxomab antibody was performed in female C.B-17 SCID mice at Aurigon Life Science GmbH (Tutzing, Germany) in accordance with the local government requirements. Catumaxomab was administered either i.v. (group I) or i.p. (groups II-IV) at 100 µg kg−1 body weight. Additionally, groups III and IV received SKOV-3 tumour (ATCC HTB-77) and PBMC effector cells, both at 2 × 106 (group III) or 1 × 107 (group IV) per mouse. Cells were applied i.p. shortly before the catumaxomab injection; the total injection volumes were 500 µl i.p. and 100 µl i.v. For pharmacokinetic evaluation, blood samples were taken from the retro-orbital plexus under slight ether anaesthesia at 10 min, 2, 4, 8, 24, and 72 h after catumaxomab administration. Plasma samples were prepared, frozen (−80 °C), and stored until catumaxomab quantification was performed at TRION Research by applying the above-mentioned ELISA, adapted to mouse plasma matrix. The number of EpCAM surface molecules on SKOV-3 ovarian cancer cells was quantified on the day of injection by means of immunofluorescence staining using the Qifikit (DakoCytomation, Glostrup, Denmark) and the EpCAM-specific antibody, HO-3 [5]. Pharmacokinetic parameters were based on the mean plasma concentrations of catumaxomab (n= 5 per timepoint) and were calculated with the WinNonlin® software (version 5.0.1; Pharsight Corporation, Mountain View, CA) in a non-compartmental manner. The observed i.p. bioavailability was calculated relative to the i.v. route with the equation: Fobs= AUC(0,last)i.p. route/AUC(0,last)i.v. route
Results
Ascites catumaxomab concentrations increased during the course of treatment
Ten out of 13 patients with MA who received all four catumaxomab infusions of 10, 20, 50, and 150 µg delivered into the peritoneal cavity were evaluable for pharmacokinetic analysis. Before each infusion, ascites fluid was drained. Thus, samples were available pre-treatment and each 3 to 4 days after the 10, 20, and 50 µg dosages. Analysis of the samples revealed quantifiable catumaxomab concentrations in all patients except patient 108/102 (Figure 1). All pre-treatment samples were below LLOQ (125 pg ml−1). After the first infusion of 10 µg, catumaxomab was readily detectable in most patients. There was an overall trend of increasing concentrations with the increasing doses. Median antibody concentrations in the ascites rose from 552 to over 1721 and then to 6121 pg ml−1, in proportion to the dosing scheme of 10, 20, and 50 µg. The inter-individual variability was high; the ascites catumaxomab concentrations ranged from 0 to 39 912 pg ml−1 at day 10 (SD = 12 414).
Figure 1.
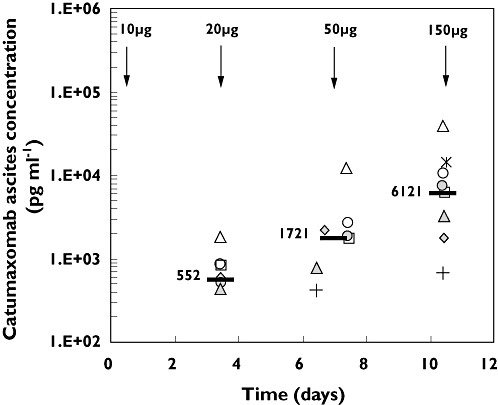
Individual (symbols) and median (–) catumaxomab ascites concentrations observed during the course of treatment. Intraperitoneal infusions are indicated by arrows. The third infusion (50 µg) was administered either on day 6 or 7. Ascites samples were taken immediately before the second, third, and fourth infusions. 108/102 ( ); 118/101 (
); 118/101 ( ); 805/101 (
); 805/101 ( ); 805/102 (
); 805/102 ( ); 805/103 (
); 805/103 ( ); 805/107 (
); 805/107 ( ); 805/108 (
); 805/108 ( ); 805/112 (
); 805/112 ( ); 805/113 (
); 805/113 ( ); 808/101 (
); 808/101 ( ); Median (
); Median ( )
)
Systemic catumaxomab exposure was low
Because unbound catumaxomab was observed in the ascites fluid of most patients, we investigated whether the antibody might have escaped into the systemic circulation. Indeed, quantifiable plasma concentrations were observed in nine patients after the third and fourth infusions (Table 1). However, in general, plasma catumaxomab concentrations were much lower than those measured in the ascites fluid. The median maximum concentration (Cmax) was 403 pg ml−1. Again, inter-individual variability was high; the systemic catumaxomab Cmax ranged from 0 to 2290 pg ml−1, and the individual area under the curve over the treatment period (AUC(0,tlast)) varied from 0 to 10 020 pg ml−1 day. The presence of high concentrations in the ascites did not always correlate with increased plasma concentrations. For example, patient 805/101 showed the highest measurable catumaxomab concentrations in ascites fluid, but had no detectable antibody in the plasma. Individual concentration profiles over time exhibited increasing concentrations in plasma after the third antibody infusion; however, the concentrations declined before the fourth antibody application (Figure 2). In most patients, plasma catumaxomab concentrations peaked after the last (fourth) infusion, reflecting the increase in dose from 50 to 150 µg. The median plasma Cmaxafter the fourth infusion was about five times higher than the median after the third infusion (398 vs. 82 pg ml−1, respectively). No catumaxomab was detected in the plasma after the first (10 µg) or second (20 µg) infusion. The maximum systemic exposure was reached at approximately 19 h (tmax) from the end of the last infusion. The mean terminal elimination half-life (t1/2) was 2.13 days, based on data from seven patients.
Table 1.
Pharmacokinetic parameters of systemic catumaxomab exposure after i.p. administrations of 10, 20, 50, and 150 µg
| Patient ID | AUC(0,tlast) (pg ml−1day) | Cmax (pg ml−1) | % estimated systemic exposure* | tmax (h) | Cmax3 (pg ml−1)† | Cmax4 (pg ml−1)‡ | λz (1 day−1) | t1/2 (days) |
|---|---|---|---|---|---|---|---|---|
| 108/102 | 177 | 171 | 0.28 | 2.64 | 0 | 171 | 0.9441 | 0.73 |
| 118/101 | 942 | 198 | 0.33 | 16.32 | 0 | 198 | 0.3340 | 2.08 |
| 805/101 | 0 | 0 | 0 | – | 0 | 0 | – | – |
| 805/102 | 10 020 | 2290 | 3.82 | 17.04 | 1108 | 2290 | 0.4288 | 1.62 |
| 805/103 | 1 819 | 431 | 0.72 | 26.40 | 0 | 431 | 0.2170 | 3.19 |
| 805/107 | 2 270 | 375 | 0.62 | 17.76 | 163 | 375 | 0.3003 | 2.31 |
| 805/108 | 3 386 | 968 | 1.61 | 27.60 | 248 | 968 | 0.1995 | 3.47 |
| 805/112 | 63 | 101 | 0.17 | 17.28 | 0 | 101 | – | – |
| 805/113 | 3 890 | 531 | 0.89 | 3.36 | 531 | 420 | – | – |
| 808/101 | 2 616 | 620 | 1.03 | 41.52 | 231 | 620 | 0.4634 | 1.5 |
| Statistics | ||||||||
| n | 10 | 10 | 10 | 9 | 10 | 10 | 7 | 7 |
| Mean | 2 518 | 569 | 0.95 | 19 | 228 | 557 | 0.4124 | 2.13 |
| SD | 2 978 | 668 | 1.11 | 12 | 354 | 670 | 0.2542 | 0.96 |
| Median | 2 045 | 403 | 0.67 | 17 | 82 | 398 | 0.3340 | 2.08 |
| Minimum | 0 | 0 | 0 | 2.64 | 0 | 0 | 0.1995 | 0.73 |
| Maximum | 10 020 | 2290 | 3.82 | 41.52 | 1108 | 2290 | 0.9441 | 3.47 |
Based on the last and highest infusion dose (150 µg) and assuming a total plasma volume of 2500 ml, the systemic exposure was calculated as follows: Cmax× 2500/(150 × 106) × 100%.
maximal measured concentration after the third infusion (50 µg).
maximal measured concentration after the fourth infusion (150 µg).
Figure 2.
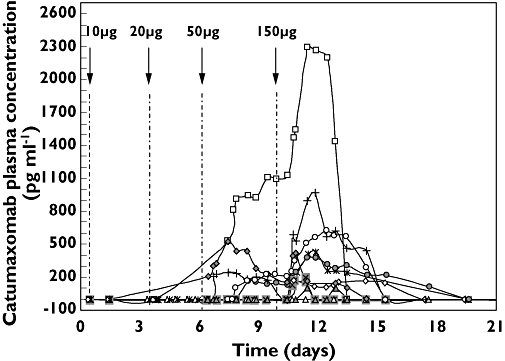
Individual catumaxomab plasma concentration vs. time profiles in 10 patients. Intraperitoneal infusions of catumaxomab are indicated by arrows. The third (50 µg) infusion was administered either on day 6 or 7. Catumaxomab was detectable in the plasma only after the third and fourth infusions. 108/102 ( ); 118/101 (
); 118/101 ( ); 805/101 (
); 805/101 ( ); 805/102 (
); 805/102 ( ); 805/103 (
); 805/103 ( ); 805/107 (
); 805/107 ( ); 805/108 (
); 805/108 ( ); 805/112 (
); 805/112 ( ); 805/113 (
); 805/113 ( ); 808/101 (
); 808/101 ( )
)
Tumour burden and effector cells influenced the bioavailability of catumaxomab
Catumaxomab has binding sites for EpCAM on tumour cells, for CD3 on T-cells, and for FcγR on accessory immune cells; thus, this trifunctional antibody is expected to exhibit complex pharmacokinetics. As anticipated, we observed large inter-individual differences in local and systemic antibody concentrations. Tumour load and immune effector cell numbers at the site of application were expected to influence the pharmacokinetics. Indeed, there were large differences among patients in the amounts of EpCAM-positive tumour cells in the ascites fluid (range 0.8–39 × 106) at the time of screening. However, it is difficult to estimate the total tumour burden in patients with progressive cancer. Therefore, we investigated this question in a defined non-clinical mouse tumour model. Severe combined immunodeficient (SCID) mice were pretreated with an i.p. dose of human peripheral blood mononuclear cells (PBMC) and EpCAM-positive human ovarian tumour cells (SKOV-3). Then mice were treated with catumaxomab, given intravenously (i.v.) or i.p. at a dose of 100 µg kg−1 body weight. In the absence of binding partners, the observed bioavailability (Fobs) of catumaxomab was 82%. The bioavailability significantly declined to 68% and 27% in the presence of low or high levels, respectively, of tumour and effector cells (Table 2). Similar decreases were observed in Cmax. By quantifying the number of EpCAM molecules per SKOV-3 tumour cell (3 × 105), we calculated the total number of EpCAM binding sites and thus, the theoretical maximal reduction in bioavailable catumaxomab (Fexp= 75 and 51%). However, the observed reduction in bioavailability was about twice as high as expected. This can be readily explained by the additional binding of catumaxomab to the co-injected PBMC effector cells. In summary, both the tumour cell load and the effector cell count strongly influenced the systemic availability of catumaxomab. In addition, catumaxomab demonstrated quantitative binding. Consequently, these data may at least partly explain the heterogenic pharmacokinetics from patients who received i.p. catumaxomab therapy.
Table 2.
Influence of tumour (SKOV-3) and immune effector cells (PBMC) on the pharmacokinetics of catumaxomab in SCID mice
| Group (n) | Application route | SKOV-3 cell number | EpCAM binding sites* | PBMC number | Cmax (ng ml−1) | tmax(h) | Clast (ng ml−1) | tlast(h) | AUC(0,last) (ng ml−1 h) | Fobs(%) | Fexp(%)† |
|---|---|---|---|---|---|---|---|---|---|---|---|
| I (5) | i.v. | – | – | 1879 | 0.17 | 488 | 72 | 45 669 | 100 | – | |
| SE | 132 | 2 253 | |||||||||
| II (5) | i.p. | – | – | 733 | 4 | 565 | 72 | 37 465 | 82 | – | |
| SE | 56 | 3 489 | |||||||||
| III (5) | i.p. | 2 × 106 | 6 × 1011 | 2 × 106 | 665 | 8 | 278 | 72 | 30 879 | 68 | 75 |
| SE | 60 | 2 437 | |||||||||
| IV (5) | i.p. | 1 × 107 | 3 × 1012 | 1 × 107 | 287 | 2 | 81 | 72 | 12 483 | 27 | 51 |
| SE | 123 | 3 461 |
Catumaxomab was applied at 100 µg kg−1 body weight, which corresponds to 8 × 1012 antibody molecules per 20 g mouse. Groups III and IV additionally received EpCAM-positive tumour and PBMC effector cells pre-injected i.p. The influence on the bioavailability (F) of i.p. administered catumaxomab was evaluated. SE, Standard error; SKOV-3, human ovarian cancer cell line; EpCAM, epithelial cell adhesion molecule; PBMC, peripheral blood mononuclear cells; SCID, severe combined immunodeficiency.
Total EpCAM binding sites: SKOV-3 cell number × 3 × 105 (quantified EpCAM molecules per cell);
expected bioavailability assuming quantitative binding to EpCAM: 82% – (EpCAM binding sites/8 × 1012 (applied catumaxomab molecules) × 100%.
High in vivo stability of catumaxomab
The ELISA-based quantification of catumaxomab does not allow conclusions about the antibody's maintenance of in vivo immunological activity; thus, we addressed this question by analysing suitable samples in a potency assay. According to its mode of action, the clinical efficacy of catumaxomab is exerted by the destruction of tumour cells in the ascites fluid via activation and redirection of different types of immune effector cells [3]. Therefore, ascites and plasma samples were tested for their killing activity against EpCAM-positive tumour cells. The biological activity in the samples was compared with control samples that were freshly spiked with matched concentrations of catumaxomab. As depicted in Figure 3a, analysed ascites samples revealed complete or nearly complete biological activity relative to the spiked controls, ranging between 111 and 84%. In contrast, catumaxomab negative pre-therapy samples displayed only background levels or low nonspecific matrix effects. Remarkably, the plasma samples exhibited 50–60% biological activity at the end of the last infusion and 2 days later. Moreover, the cytokine release induced by ascites and plasma samples was comparable with that observed with spiked controls, as demonstrated by measurement of TNF-α concentrations (Figure 3b). Similar results were obtained for IL-2, IL-6, Il-10, and IFN-γ (data not shown). Taking into account the long interval between drug application and sampling, these data confirmed the high in vivo stability of i.p. administered catumaxomab and its immunological activity even after several days in the circulation. Moreover, despite low systemic plasma concentrations in the pg ml−1 range, these concentrations were sufficiently potent to induce tumour cell killing.
Figure 3.
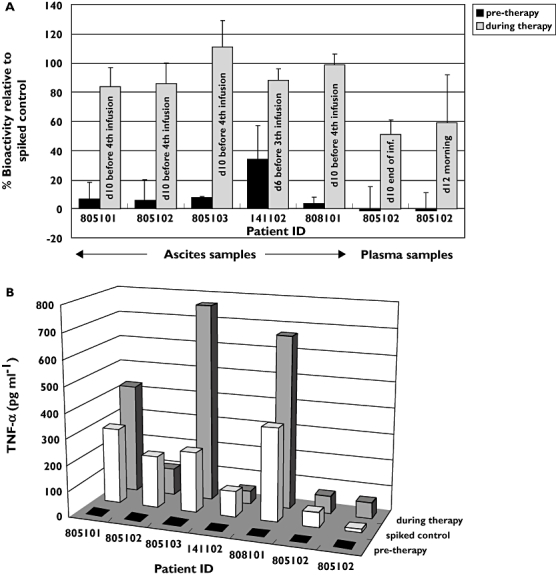
Bioactivity of catumaxomab in ascites and plasma samples. Bioactivity was determined in a potency assay that evaluated ascites and plasma samples for the abilities to A) kill EpCAM-positive HCT-8 tumour cells and B) secrete TNF-α cytokine, relative to controls with freshly spiked catumaxomab. Ascites samples were obtained before the third or fourth infusion from five patients, and two plasma samples were obtained from one patient
Evaluation of anti-drug antibody (ADA) development and safety
The rat/murine chimeric antibody catumaxomab is immunogenic in man [6, 12, 13]. This finding was confirmed in this study, because all patients developed ADA (Figure 4). ADA development was highly dynamic, with measured concentrations that differed by several orders of magnitude, probably reflecting the diverse immune status of patients with late stage cancer. The highest observed value was 60 000 ng ml−1. Of note, none of the patients developed significant ADA responses (>100 ng ml−1) before the time of the last infusion. In most patients, ADA onset occurred between days 11 and 16. These findings support the appropriateness of the clinical application schedule that terminates catumaxomab treatment before ADA development. Thus, ADA-based safety or efficacy issues are circumvented.
Figure 4.
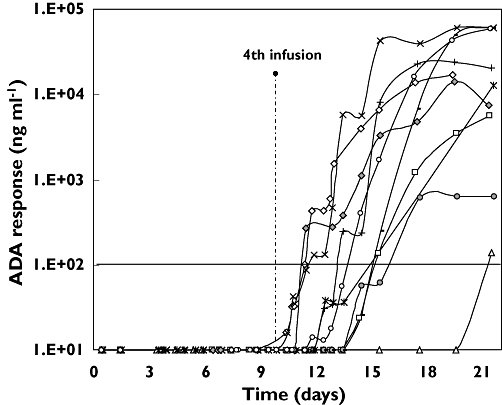
Anti-drug antibody response in plasma vs. time profiles in 10 patients within the first 3 weeks after the beginning of the treatment. 108/102 ( ); 118/101 (
); 118/101 ( ); 805/101 (
); 805/101 ( ); 805/102 (
); 805/102 ( ); 805/103 (
); 805/103 ( ); 805/107 (
); 805/107 ( ); 805/108 (
); 805/108 ( ); 805/112 (
); 805/112 ( ); 805/113 (
); 805/113 ( ); 808/101 (
); 808/101 ( )
)
Moreover, catumaxomab infusions were well tolerated with acceptable toxicities. Commonly observed adverse events, including pyrexia/chills, nausea/vomiting, dyspnoea, and hypotension/hypertension occurred at mild to moderate levels and were reversible. Cytokine release and transient increases in liver enzymes were similar to the safety findings of previous studies [6, 12] and consistent with the mode of action of catumaxomab. Importantly, there was no apparent relationship between systemic exposure and the adverse clinical findings.
Discussion
MA is an end stage manifestation, mainly observed in patients with progressive ovarian and gastrointestinal cancer, and it is associated with poor prognosis [7]. Affected patients experience a dramatic impairment in quality of life due to abdominal pain, nausea, anorexia, dyspnoea and other symptoms [15]. The spread of tumour cells infiltrating the peritoneum represents the main cause of abnormal accumulation of body fluid in the peritoneal cavity. Consequently, a direct attack on these infiltrating tumour cells should lead to effective relief of MA symptoms. This approach has been successfully pursued with the trifunctional antibody catumaxomab (anti-EpCAM × anti-CD3) that, when given i.p., specifically targets EpCAM-positive tumour cells [6, 13]. As EpCAM is also expressed in normal epithelium [16], the rationale for i.p. application was to achieve high local concentrations and thus diminish potential negative side-effects.
Indeed, effective catumaxomab concentrations were determined in nine of 10 evaluable patients in the ascites fluid. A median concentration of 552 pg ml−1 was measured 3 days after the starting dose of 10 µg. This represents an effective dose level, as catumaxomab has demonstrated significant capacity for killing tumour cells even at concentrations of 1000–100 pg ml−1[5]. Efficient tumour cell reduction in the ascites was consistently observed after the first 10 µg dose; the median number of EpCAM-positive tumour cells was reduced from 9362 (screening value) to 49 (before the second infusion) per 106 total ascites cells in this study. In the course of treatment, catumaxomab concentrations in the ascites further increased in proportion to the escalating infusion doses. Concomitantly, the median number of tumour cells in the ascites dropped to zero. Furthermore, catumaxomab exhibited a remarkable in vivo stability. Even after 3 days in ascites, the antibody remained biologically active without significant loss in its ability to induce tumour cell killing and cytokine release. These data support the clinical efficacy of catumaxomab previously observed in the treatment of MA [6,12,14]) and peritoneal carcinomatosis [17]. In accordance with the antibody stability in ascites, we observed a mean half-life (t1/2) of 2.13 days in plasma. This is relatively long, compared with most other murine IgG antibodies [18–20], and may be ascribed to the rat/murine hybrid nature of catumaxomab.
Compared with the amount of antibody detected in the ascites, much lower concentrations were detected in the plasma, and only after the third and particularly the fourth infusion. For the last and highest infusion dose (150 µg), a mean of only 0.95% of the i.p. administered catumaxomab was detected in the periphery as unbound antibody (Table 1). In contrast to the obvious assumption that most of the antibody in the peripheral blood may be bound to T cells via its CD3 binding arm spiking experiments revealed that 80–90% of catumaxomab were not cell bound (data not shown). This can be readily explained by the intermediate affinity of catumaxomab to CD3 (KD= 4.4 nm) which does not allow quantitative T-cell binding at very low antibody concentrations. Interestingly, there was no correlation between the systemic plasma concentrations and adverse clinical findings. This corroborates the notion that side-effects were mainly derived from locally acting catumaxomab and the observed low systemic concentrations of catumaxomab did not raise any safety concerns. On the other hand, despite the low levels of antibody concentrations in the plasma, these levels could effectively cause the destruction of tumour cells in vitro. It is tempting to speculate that patients might benefit from additional systemic effects under these conditions. This suggests that the third and fourth infusion doses may play an important role in the clinical effects. As expected from previous findings, all patients developed ADA, but not before termination of treatment; this obviated problems with safety or efficacy. ADA appearance was highly dynamic and potent which reflects the intrinsic immunogenicity of the T-cell engaging catumaxomab, that was probably enhanced by its repeated i.p. application [21]. Interestingly, when a single i.v. infusion dose of 2–7.5 µg of catumaxomab was given to patients with non-small cell lung cancer, it did not provoke ADA development [22].
As expected, considering the different degrees of tumour burden, immune cell status and permeability of the diseased peritoneum, there was high inter-patient variability in the local and systemic catumaxomab concentrations. Although individual ascites volumes had no major impact on the variability (data not shown), the number of available binding sites was important. This was demonstrated in the pharmacokinetic study performed in mice. High local numbers of EpCAM-positive tumour and PBMC effector cells significantly reduced the bioavailability of catumaxomab in the systemic circulation. An inverse correlation between systemic antibody concentrations and tumour bulk has also been reported and discussed in studies of other tumour-specific monoclonal antibodies, including rituximab and alemtuzumab [23–25]. The trifunctional antibody catumaxomab additionally binds to T-cells and FcγR positive immune cells; thus, the presence of these binding partners is of equal importance for its distribution. Of note, an increased number of CD45-positive leukocytes appeared in the peritoneal cavity after catumaxomab application [12]. These results suggested that high local tumour burden and immune effector cell accumulation might be the major causes for inter-individual variability and the observed low systemic exposure.
In summary, catumaxomab administered i.p. for the treatment of MA demonstrated high in vivo stability, effective local concentrations in ascites fluid, minor systemic exposure, and an acceptable safety profile.
Acknowledgments
We thank the contract research organizations Socratec R&D GmbH and Omega Mediation GmbH for their professional support. We gratefully appreciate the assistance provided by medical writers Drs Hans-Joachim Kremer and Barry Drees. Additional participating investigators included: Prof Dr Hermann Einsele, Uniklinik Würzburg, Würzburg, Germany; Dr Elena Doina Ganea-Motan, Spitalul Judetean de Urgenta ‘Sf. Ioan cel Nou’, Suceava, Romania; and Dr Doris Bucur Pelau, Oncologic Institut ‘Ion Chiricuta’, Cluj-Napoca, Romania.
Competing interests
M.K. and B.B. are employees of Fresenius Biotech GmbH. H.L. has shares in TRION Pharma GmbH and owns patents for the manufacture and application of trifunctional antibodies. P.W. and M.M.H. have been re-imbursed by Fresenius Biotech for speaking, attending a symposium, research and consulting. The other authors declared no competing interests. The study was funded by Fresenius Biotech GmbH.
REFERENCES
- 1.Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubal S, Schmitt B, Lindhofer H. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol. 1999;163:1246–52. [PubMed] [Google Scholar]
- 2.Zeidler R, Mysliwietz J, Csanady M, Walz A, Ziegler I, Schmitt B, Wollenberg B, Lindhofer H. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer. 2000;83:261–6. doi: 10.1054/bjoc.2000.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen J, Zhu Z. Catumaxomab, a rat/murine hybrid trifunctional bispecific monoclonal antibody for the treatment of cancer. Curr Opin Mol Ther. 2008;10:273–84. [PubMed] [Google Scholar]
- 4.Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98:2526–34. doi: 10.1182/blood.v98.8.2526. [DOI] [PubMed] [Google Scholar]
- 5.Ruf P, Gires O, Jager M, Fellinger K, Atz J, Lindhofer H. Characterisation of the new EpCAM-specific antibody HO-3: implications for trifunctional antibody immunotherapy of cancer. Br J Cancer. 2007;97:315–21. doi: 10.1038/sj.bjc.6603881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heiss MM, Strohlein MA, Jager M, Kimmig R, Burges A, Schoberth A, Jauch KW, Schildberg FW, Lindhofer H. Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer. 2005;117:435–43. doi: 10.1002/ijc.21165. [DOI] [PubMed] [Google Scholar]
- 7.Parsons SL, Watson SA, Steele RJ. Malignant ascites. Br J Surg. 1996;83:6–14. doi: 10.1002/bjs.1800830104. [DOI] [PubMed] [Google Scholar]
- 8.Ayantunde AA, Parsons SL. Pattern and prognostic factors in patients with malignant ascites: a retrospective study. Ann Oncol. 2007;18:945–9. doi: 10.1093/annonc/mdl499. [DOI] [PubMed] [Google Scholar]
- 9.az-Arias AA, Loy TS, Bickel JT, Chapman RK. Utility of BER-EP4 in the diagnosis of adenocarcinoma in effusions: an immunocytochemical study of 232 cases. Diagn Cytopathol. 1993;9:516–21. doi: 10.1002/dc.2840090509. [DOI] [PubMed] [Google Scholar]
- 10.De AM, Buley ID, Heryet A, Gray W. Immunocytochemical staining of serous effusions with the monoclonal antibody Ber-EP4. Cytopathology. 1992;3:111–7. doi: 10.1111/j.1365-2303.1992.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 11.Passebosc-Faure K, Li G, Lambert C, Cottier M, Gentil-Perret A, Fournel P, Perol M, Genin C. Evaluation of a panel of molecular markers for the diagnosis of malignant serous effusions. Clin Cancer Res. 2005;11:6862–7. doi: 10.1158/1078-0432.CCR-05-0043. [DOI] [PubMed] [Google Scholar]
- 12.Burges A, Wimberger P, Kumper C, Gorbounova V, Sommer H, Schmalfeldt B, Pfisterer J, Lichinitser M, Makhson A, Moiseyenko V, Lahr A, Schulze E, Jager M, Strohlein MA, Heiss MM, Gottwald T, Lindhofer H, Kimmig R. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: a phase I/II study. Clin Cancer Res. 2007;13:3899–905. doi: 10.1158/1078-0432.CCR-06-2769. [DOI] [PubMed] [Google Scholar]
- 13.Sebastian M, Kiewe P, Schuette W, Brust D, Peschel C, Schneller F, Rühle KH, Nilius G, Ewert R, Lodziewski S, Passlick B, Sienel W, Wiewrodt R, Jäger M, Lindhofer H, Friccius-Quecke H, Schmittel A. Treatment of malignant pleural effusions with the trifunctional antibody catumaxomab (removab) (anti-EpCAM × anti-CD3): results of a phase 1/2 study. J Immunother. 2009;32:195–202. doi: 10.1097/CJI.0b013e318195b5bb. [DOI] [PubMed] [Google Scholar]
- 14.Parsons S, Murawa PX, Koralewski P, Kutarska E, Kolesnik OO, Stroehlein MA, Lahr A, Jaeger M, Heiss M. Intraperitoneal treatment of malignant ascites due to epithelial tumors with catumaxomab: a phase II/III study. J Clin Oncol. 2008;26(Suppl.) abstr 3000. [Google Scholar]
- 15.Becker G, Galandi D, Blum HE. Malignant ascites: systematic review and guideline for treatment. Eur J Cancer. 2006;42:589–97. doi: 10.1016/j.ejca.2005.11.018. [DOI] [PubMed] [Google Scholar]
- 16.Went PT, Lugli A, Meier S, Bundi M, Mirlacher M, Sauter G, Dirnhofer S. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004;35:122–8. doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 17.Stroehlein MA, Gruetzner KU, Tarabichi A, Jauch KW, Bartelheim K, Lindhofer H, Von Roemeling R, Heiss MM. Efficacy of intraperitoneal treatment with the trifunctional antibody catumaxomab in patients with GI-tract cancer and peritoneal carcinomatosis: a matched-pair analysis. J Clin Oncol. 2006;24:2544. 2006 ASCO Annual Meeting Proceedings Part I. Vol 24, No. 18S (June 20 Supplement) [Google Scholar]
- 18.Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93:2645–68. doi: 10.1002/jps.20178. [DOI] [PubMed] [Google Scholar]
- 19.Frodin JE, Lefvert AK, Mellstedt H. Pharmacokinetics of the mouse monoclonal antibody 17-1A in cancer patients receiving various treatment schedules. Cancer Res. 1990;50:4866–71. [PubMed] [Google Scholar]
- 20.Ternant D, Paintaud G. Pharmacokinetics and concentration-effect relationships of therapeutic monoclonal antibodies and fusion proteins. Expert Opin Biol Ther. 2005;5(Suppl 1):S37–47. doi: 10.1517/14712598.5.1.s37. [DOI] [PubMed] [Google Scholar]
- 21.Mobus VJ, Baum RP, Bolle M, Kreienberg R, Noujaim AA, Schultes BC, Nicodemus CF. Immune responses to murine monoclonal antibody-B43.13 correlate with prolonged survival of women with recurrent ovarian cancer. Am J Obstet Gynecol. 2003;189:28–36. doi: 10.1067/mob.2003.347. [DOI] [PubMed] [Google Scholar]
- 22.Sebastian M, Passlick B, Friccius-Quecke H, Jager M, Lindhofer H, Kanniess F, Wiewrodt R, Thiel E, Buhl R, Schmittel A. Treatment of non-small cell lung cancer patients with the trifunctional monoclonal antibody catumaxomab (anti-EpCAM x anti-CD3): a phase I study. Cancer Immunol Immunother. 2007;56:1637–44. doi: 10.1007/s00262-007-0310-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis TA, White CA, Grillo-Lopez AJ, Velasquez WS, Link B, Maloney DG, Dillman RO, Williams ME, Mohrbacher A, Weaver R, Dowden S, Levy R. Single-agent monoclonal antibody efficacy in bulky non-Hodgkin's lymphoma: results of a phase II trial of rituximab. J Clin Oncol. 1999;17:1851–7. doi: 10.1200/JCO.1999.17.6.1851. [DOI] [PubMed] [Google Scholar]
- 24.Hale G, Rebello P, Brettman LR, Fegan C, Kennedy B, Kimby E, Leach M, Lundin J, Mellstedt H, Moreton P, Rawstron AC, Waldmann H, Osterborg A, Hillmen P. Blood concentrations of alemtuzumab and antiglobulin responses in patients with chronic lymphocytic leukemia following intravenous or subcutaneous routes of administration. Blood. 2004;104:948–55. doi: 10.1182/blood-2004-02-0593. [DOI] [PubMed] [Google Scholar]
- 25.Dayde D, Ternant D, Ohresser M, Lerondel S, Pesnel S, Watier H, Le PA, Bardos P, Paintaud G, Cartron G. Tumor burden influences exposure and response to rituximab: pharmacokinetic–pharmacodynamic modelling using a syngeneic bioluminescent murine model expressing human CD20. Blood. 2008;113:3765–72. doi: 10.1182/blood-2008-08-175125. [DOI] [PubMed] [Google Scholar]