

Abstract
The mammalian hippocampus, together with subcortical and cortical areas, is responsible for some forms of learning and memory. Proper hippocampal function depends on the highly dynamic nature of its circuitry, including the ability of synapses to change their strength for brief to long periods of time. In this study, we focused on a transient depression of glutamatergic synaptic transmission at Schaffer collateral synapses in acute hippocampal slices. The depression of evoked excitatory postsynaptic current (EPSC) amplitudes, herein called transient depression, follows brief trains of synaptic stimulation in stratum radiatum of CA1 and lasts for 2–3 min. Depression results from a decrease in presynaptic glutamate release, as NMDA-receptor–mediated EPSCs and composite EPSCs are depressed similarly and depression is accompanied by an increase in the paired-pulse ratio. Transient depression is prevented by blockade of metabotropic glutamate and acetylcholine receptors, presumably located presynaptically. These two receptor types—acting together— cause depression. Blockade of a single receptor type necessitates significantly stronger conditioning trains for triggering depression. Addition of an acetylcholinesterase inhibitor enables depression from previously insufficient conditioning trains. Furthermore, a strong coincident, but not causal, relationship existed between presynaptic depression and postsynaptic internal Ca2+ release, emphasizing the potential importance of functional interactions between presynaptic and postsynaptic effects of convergent cholinergic and glutamatergic inputs to CA1. These convergent afferents, one intrinsic to the hippocampus and the other likely originating in the medial septum, may regulate CA1 network activity, the induction of long-term synaptic plasticity, and ultimately hippocampal function.
INTRODUCTION
The hippocampal formation, through its communication with a host of neural regions, contributes to long-term storage and retrieval of some forms of memory. The ability to carry out these functions depends on network activity both intrinsic and extrinsic to the hippocampus, which in turn depends on spatial and temporal patterns of synaptic activity, and on the capacity for dynamic changes in synaptic strength (Martin et al. 2000; Yeckel and Berger 1998). There are a number of loci and timescales over which synaptic function can be altered, and myriad mechanisms are known to contribute to these processes. Although the properties of synaptic plasticity in the hippocampus have traditionally focused on long-term potentiation and long-term depression (LTP and LTD, respectively), many other forms of synaptic plasticity were previously identified that have shorter durations (Zucker and Regehr 2002). Of particular interest are mechanisms of short-term synaptic plasticity at hippocampal synapses that may hold functional implications for the hippocampus, both directly by determining patterns of activity relevant to ongoing behavior and indirectly by modulating the ability to induce LTP and LTD (Abraham and Bear 1996; Zucker and Regehr 2002).
Short-term changes in synaptic transmission lasting from 2 to 10 min at hippocampal pyramidal neuron synapses have been reported to depend either on alteration of presynaptic glutamate release or on changes to postsynaptic glutamate receptor function (Fernandez de Sevilla and Buno 2003; Fernandez de Sevilla et al. 2002; Grishin et al. 2004; Grover and Teyler 1993b; Manzoni et al. 1994; Sekino and Koyama 1992). For example, trains of synaptic stimulation or application of Gq-coupled receptor agonists elicits a brief depression of synaptic transmission by directly or indirectly decreasing glutamate release from presynaptic terminals (Fernandez de Sevilla and Buno 2003; Manzoni et al. 1994). In contrast to this presynaptic form of depression, others reported a postsynaptic form of depression arising from Ca2+- and/or G-protein–dependent changes in postsynaptic N-methyl-D-aspartate receptor (NMDAR) function triggered by increases in intracellular Ca2+ concentration ([Ca2+]i) (Grishin et al. 2004; Markram and Segal 1990). Common to both of these pre- and postsynaptic forms of short-term synaptic depression is their dependence on the activation of G-protein–coupled receptors, which can be activated by many neurotransmitters and neuromodulators (Gilman 1987; Hille 1992).
Based on the role of G-proteins in short-term synaptic depression and their responsiveness to multiple ligands, we examined whether coincident activation of different Gq-coupled receptors by convergent afferent inputs might act cooperatively to elicit synaptic depression at hippocampal CA1 synapses. Using evidence gathered from whole cell patch-clamp recordings and Ca2+ fluorescence imaging in acute hippocampal slices, we describe a short-lived, 2- to 3-min depression (transient depression) of evoked glutamatergic synaptic responses. This transient depression was elicited by brief trains (30 stimuli at 100 Hz) of stimulation in stratum radiatum and was expressed presynaptically. Despite its high correlation with postsynaptic internal Ca2+ release, transient depression was independent of internal Ca2+ release. Depression of test response amplitudes depended on coincident activation of presumed presynaptic metabotropic glutamate receptors (mGluRs) and muscarinic acetylcholine receptors (mAChRs). Our data raise the possibility that convergent afferent inputs arising in different brain regions, through the release of neurotransmitters that activate a common G-protein subtype, can act cooperatively to depress neurotransmission at glutamatergic synapses.
METHODS
Acute hippocampal slice preparation
Hippocampal slices (350 μm) were prepared from 3- to 5-wk-old male rats (Sprague–Dawley, Charles River) using experimental procedures consistent with those outlined in National Institutes of Health publication 91–3207, Preparation and Maintenance of Higher Animals During Neuroscience Experiments, and approved by the Institutional Animal Care and Use Committee at the Yale School of Medicine. An anesthetic consisting of ketamine (125 mg/kg), xylazine (6.25 mg/kg), and acepromazine (1.25 mg/kg) was injected intraperitoneally and animals were decapitated after they showed no response to a foot pinch. Brains were removed in ice-cold dissecting solution containing (in mM): 87 NaCl, 58.5 sucrose, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 7.0 MgCl2, 0.5 CaCl2, and 10 dextrose (296–301 mOsm), then blocked rapidly and glued to a cold slicing chamber insert. Horizontal slices were cut on a Vibratome 1500 (Vibratome, St. Louis, MO) using a freshly broken glass blade (Ralph Knife Breaker; Ted Pella) in a Peltier-cooled slicing chamber (model 900R, Vibratome). The slicing chamber was filled with dissecting solution bubbled with 95% O2-5% CO2 and maintained at −1 to −2°C by a VS-35M power supply (Astron) and digital thermometer (Fisher Scientific). Trimmed slices including the hippocampus were incubated for 10–20 min at 35°C in dissecting solution before being transferred to 35°C recording solution containing (in mM): 126 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 10 dextrose, and 0.01 glycine, bubbled with 95% O2-5% CO2, and placed at room temperature. The slices were allowed to stabilize for 1 h before recording. Mg2+ was omitted from, and glycine was added to, the recording solution to facilitate the activation of NMDARs, except where otherwise indicated (Johnson and Ascher 1987; Nowak et al. 1984). Test excitatory postsynaptic currents (EPSCs) were recorded in the presence of γ-aminobutyric acid type A or type B receptor (GABAAR and GABABR, respectively) antagonists, except where indicated, and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX, 20 μM) as needed to isolate NMDAR-mediated EPSCs.
Electrophysiological recordings
For recording, a slice was submerged in a recording chamber (bath volume ≃ 1 ml) that was perfused continuously (about 1 ml/min) with recording solution saturated with 95% O2-5% CO2. The slice was elevated off the chamber bottom using a circular platinum wire that supported a taut nylon mesh. Another, slightly smaller circular platinum harp crossed by four to five recessed pairs of nylon strands stabilized the slice from above. An in-line flow heater (Warner Instruments) and a custom heated stage insert maintained chamber temperature at 32–35°C. In an effort to limit outgassing, the recording solution reservoir was warmed to about 30°C with a band heater (Meriden Cooper) controlled by a VS-35M DC power supply (Astron). Slices were viewed through a Zeiss Axioskop 2 fitted with a ×40 water-immersion objective and differential interference contrast (DIC) optics. Whole cell patch-clamp recording pipettes (3- to 5-MΩ resistance) were pulled (model P-97 pipette puller, Sutter Instruments, Novato, CA) from thick-walled borosilicate glass (1.0 ID, 2.0 OD) and fire-polished. Recordings were made with an SEC 05L amplifier (npi, Tamm, Germany) in bridge mode or discontinuous voltage-clamp mode. Cells recorded in CA1 stratum pyramidale were identified morphologically as having a single large apical dendrite in stratum radiatum and smaller basal dendrites in stratum oriens and electrophysiologically as displaying spike-frequency adaptation and a hyperpolarization-activated current (Ih) (Magee 1998). Synaptic currents were recorded in voltage clamp at −63 mV (−73 mV if we account for a measured −10-mV junction potential). Series resistance was typically 10–20 MΩ. Whole cell recording electrodes were filled with (in mM) 134 KMeSO4, 3 KCl, 10 HEPES, 1.0 MgCl2, 4 Mg-ATP, 0.5 Na-GTP, 5 K2-creatine phosphate, 5 Na2-creatine phosphate, 0.1 bis-fura-2, 0.015 Alexa 488, and 50 units/ml creatine phosphokinase (pH 7.53 at 24°C, about 288 mOsm). KMeSO4, Mg-ATP, Na-GTP, Na2-creatine phosphate, and creatine phosphokinase were from Sigma–Aldrich (St. Louis, MO). K2-creatine phosphate was from Calbiochem (San Diego, CA). Bis-fura-2 and Alexa 488 were from Molecular Probes/Invitrogen (Carlsbad, CA).
Afferent stimulation
Schaffer collateral axons were stimulated by applying brief unipolar currents (100–200 μs, 5–60 μA) through a glass microelectrode (3- to 10-μm tip diameter) with a fine tungsten rod glued to its side to provide for more local current flow. Stimulating electrodes were filled with extracellular solution and 2.5 μM Alexa 488 for visualization. Two stimulating electrodes were placed in stratum radiatum, each about 50 μm from the apical dendrite of the recorded pyramidal cell, one 50 and the other 100 μm from the cell body layer, to stimulate independent synaptic inputs onto the recorded cell. The independence of the two pathways was verified using a paired-pulse test (50-ms interval; Barrionuevo and Brown 1983) in each experiment examining depression on an independent test pathway. Test synaptic currents (amplitude ≃ 500 pA) were evoked separately on each stimulus pathway at 20-s intervals using single-, paired- (20 Hz), or triple-pulse (100 Hz) stimuli. Short conditioning trains (30 stimuli, 100 Hz) of synaptic stimulation on either or both pathways were used to elicit transient depression of test currents. This stimulus pattern was used because it elicited robust transient depression while being weaker than stimuli typically used to induce long-term changes in synaptic strength. No difference in test stimulus artifact amplitude was observed during depression, confirming proper functioning of the stimulator after a conditioning train (model 2200 analog stimulus isolator, A–M Systems; artifact amplitude was 99.6 ± 0.4% of control at the peak of depression, n = 78).
Ca2+ fluorescence imaging
To measure relative changes in [Ca2+]i, the Ca2+ indicator dye bis-fura-2 (100 μM) was included in the pipette solution. Using a cooled CCD camera (Quantix 57; Photometrics, Tuscon, AZ) in a sequential frame transfer mode (50-Hz frame rate; binned in a 5 × 5 array), fluorescence images were recorded with single wavelength measurements (excitation ≃ 380 nm) from the somata and dendrites of filled cells. Synchronous acquisition of electrical and optical data was performed with custom software written in IGOR Pro. Relative changes in [Ca2+]i were quantified as changes in ΔF/F, where F is fluorescence intensity before stimulation and ΔF is the relative reduction from this value during neuronal activity. Data representing tissue autofluorescence and dye bleaching were collected for each experiment (although not always applied to the analysis of imaging data). Tissue autofluorescence was determined with an equivalent fluorescence measurement at a parallel location in the slice that was away from the dye-filled cell. Dye bleaching was assessed as the change in fluorescence of the recorded cell observed during an imaging run of standard duration (4 s) with no stimulation of the cell.
Pharmacology
The following drugs were included in the recording pipette when needed to affect biochemical processes in the recorded neuron: 10 or 50 mM K4-BAPTA [1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid], 5 mM QX-314 [N-(2,6-dimethylphenyl carbamoyl-methyl)triethylammonium bromide], and 1 mM GDP-β-S [guanosine 5′-O-(2-thiodiphosphate)]. BAPTA and QX-314 were substituted for equiosmolar amounts of KMeSO4 and GDP-β-S replaced Na-GTP. BAPTA and GDP-β-S were confirmed to block internal Ca2+ release in each recorded cell using Ca2+ imaging and QX-314 blocked Ih. K4-BAPTA and GDP-β-S trilithium salt were from Sigma. QX-314 bromide was from Tocris Cookson (Ellisville, MO) and was used in only three recorded cells.
Bath-applied drugs were typically present in the recording solution as soon as the slice was transferred to the recording chamber, usually 0.5–2 h before establishing a recording. Sometimes drugs were applied as a wash-in during the experiment, as indicated. The following bath-applied drugs were from Tocris Cookson (stock solution in parentheses): 100 μM D,L-APV, 25 mM in H2O [D,L-2-amino-5-phosphonovaleric acid], 1–2 μM CGP55845, 20 mM in DMSO [(2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenyl-methyl)phosphinic acid], 50 μM CPA, 100 mM in DMSO [cyclopiazonic acid], 20 μM DNQX, 40 mM in DMSO [6,7-dinitroquinoxaline-2,3-dione], 0.5–5 μM DPCPX, 5 mM in DMSO, made fresh [8-cyclopentyl-1,3-dipropylxanthine], 100 μM LY367385, 100 mM in 1.1 N NaOH [(+)-2-methyl-4-carboxyphenylglycine], 20 μM MK-801, 10 mM in H2O [(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate], 10 μM MPEP, 100 mM in DMSO [2-methyl-6-(phenylethyl)-pyridine], 3 μM thapsigargin, 30 mM in DMSO, 50 μM SR95531, gabazine, 10 mM in H2O [2-(3-carboxypropyl)-3-amino-6-methoxyphenyl-pyridazinium bromide], 1 μM WIN 55,212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone], and 5 μM AM251 [N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] (both stored at −20°C as 100 mM DMSO stocks, then prediluted in DMSO to 0.5–2.5 mM in the presence of other drugs before addition to aqueous solution to prevent precipitate formation, final DMSO concentration 0.2%). In a control experiment, bath application of 5 μM AM251 was observed to antagonize the depression of evoked inhibitory postsynaptic currents (IPSCs) produced by bath application of WIN 55,212-2 (data not shown). The following bath-applied drugs were from Sigma–Aldrich (St. Louis, MO): 10 μM atropine, 5 mM in H2O, 10 μM bicuculline methiodide, 2 mM in recording solution, 50 μM calmidazolium, 50 mM in DMSO, 5 μM eserine, 5 mM in H2O, made fresh, and 10 μM picrotoxin, 50 mM in ethanol, made fresh. When atropine was present in the recording solution, GABAAR antagonists were omitted to prevent slices from becoming hyperexcitable.
In control experiments to assess the efficacy of DPCPX, a pressure pipette was used to depress synaptic responses by applying 200 μM adenosine (from Sigma, freshly dissolved in puffer solution). The puffer solution contained (in mM): 124 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, and 10 HEPES (pH 7.35 at 24°C). The pressure pipette and stimulating electrode were placed 100 and 50 μm from the cell body layer in CA1, respectively, and 50 μm from the apical dendrite of the recorded cell. The effect of adenosine on NMDAR-mediated synaptic test current amplitudes was assessed in the absence and presence of 5 μM DPCPX.
Data analysis
Electrical data were analyzed both on- and off-line using routines written to accompany our existing data collection and optical data analysis software. Current amplitudes were measured from a smoothed (10-point Gaussian) current trace to limit the contribution of noise to the current peak and each measurement was confirmed visually. To assess the possibility that changes in resting dendritic conductances after conditioning trains may produce an apparent or real depression of synaptic current amplitudes measured at the soma, we followed the postsynaptic input resistance (Rin) over time using a 5-mV hyperpolarizing voltage step every 20 s. In agreement with a previous report (Grover and Teyler 1993a), Rin was unchanged from baseline during transient depression in our whole cell recordings (to 100.5 ± 1.2% of baseline 20 s after conditioning train, n = 54).
In experiments involving two stimulus pathways, data from trials in which depression occurred on a given test pathway were grouped into a data set. Data from trials within a data set were averaged, after which data were combined across data sets to generate summary data for a group of experiments. However, for simplicity we report n values as the number of cells recorded rather than the number of data sets. Current amplitude, Rin, stimulus artifact, and paired-pulse ratio (PPR) data were normalized for presentation so that the summary data represent variability of measurements within trials, rather than variability of baseline values between data sets or between cells. PPR data were averaged across trials in a data set as previously described (Kim and Alger 2001). Weighted exponential fits of summary data were performed in IGOR Pro.
Data from paired-pulse independence tests were analyzed by averaging raw traces from ten trials per condition. The first and second responses to a pair of stimuli on the test pathway were taken to be the baseline and potentiated responses, respectively. The second response to a pair of stimuli, in which the first stimulus was delivered on the conditioning pathway and the second stimulus was delivered on the test pathway, was taken as the test response. When a significant difference between baseline and potentiated responses was present and when the test response was not discernibly different from the baseline response, depression data from the independent pathway were included in the average (Fig. 3C). Data describing depression on a separate test pathway not meeting these criteria were discarded.
FIG. 3.
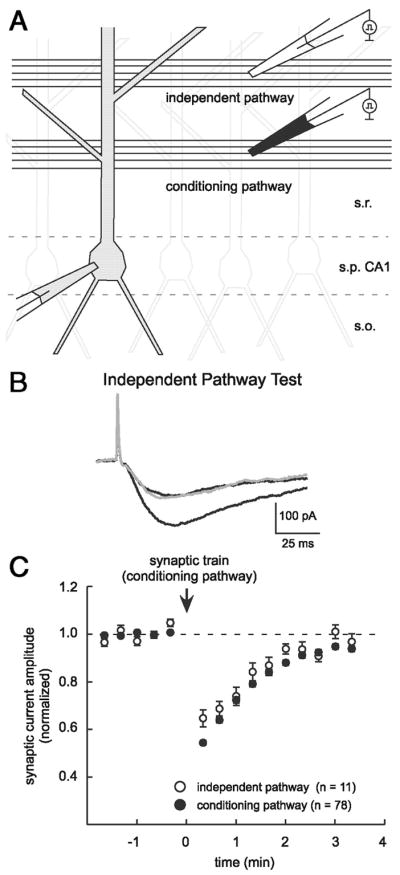
Neurotransmission on an independent test pathway can also be depressed. A: 2 stimulating electrodes were placed in stratum radiatum (s.r.) at 50 and 100 μm from stratum pyramidale (s.p.) to stimulate independent glutamatergic inputs to the recorded cell. A conditioning train (30 stimuli at 100 Hz) was delivered on the conditioning pathway, whereas test responses were followed over time on each pathway. s.o., stratum oriens. B: test for independence of the stimulus pathways. Traces are averages of 10 responses where a paired stimulus (50-ms interval) on the test pathway was used to establish naïve and potentiated responses (black traces). A test response (gray) was preceded by a stimulus on the conditioning pathway. Test response was not distinguishable from the naïve response, consistent with nonoverlapping axonal pathways. C: summary plot of depression observed on an independent test pathway (open symbols, n = 11) and on the conditioning pathway (filled symbols, n = 78).
Optical data from Ca2+ imaging were analyzed to determine the correlation between internal Ca2+ release events and transient depression and to verify the effect of internal BAPTA and GDP-β-S, as well as bath-applied CPA, thapsigargin, LY367385, MPEP, and atropine. Ca2+ transients elicited by synaptic trains included three components: 1) increases in Ca2+ during the stimulus train associated with depolarization and the opening of voltage-gated Ca2+ channels with fast exponential decay beginning at the end of the train (Christie et al. 1995), 2) increases in Ca2+ as the result of influx through NMDARs with a slower exponential decay that begins at the end of the train and depends on NMDAR closure (Regehr and Tank 1992), and 3) increases in Ca2+ as the result of IP3-mediated Ca2+ release from internal stores, involving a continued increase in Ca2+ after the end of the train, especially when involving an inflection point in the optical trace and an accelerating Ca2+ increase or propagation of a Ca2+ wave in the dendrites of the recorded cell (Kapur et al. 2001; Nakamura et al. 1999). Trials where Ca2+ transients had characteristics of this third component were considered to include an internal Ca2+ release event.
Conditioning train stimulus intensity data (Fig. 7A) were gathered post hoc by examining the conditioning trains used in trials that are included in the summary data for each recording condition. We calculated the charge commanded [nanocoulomb (nC)] through the stimulator during each stimulus of the 30-stimulus train as the product of the stimulus amplitude (microamperes) and duration (milliseconds). Only experiments using NMDAR-mediated EPSCs, single- or paired-test stimuli, and depression on the conditioning pathway were used for comparison. We pooled stimulus intensity data (Fig. 7A) from mGluR or mAChR blockade experiments (Fig. 6A) with corresponding experiments that included adenosine receptor antagonism (Fig. 6B) because the data were not significantly different.
FIG. 7.

Coincident glutamatergic and cholinergic inputs trigger transient depression. A: blockade of either mGluRs or mAChRs (from experiments in Fig. 6) increased the intensity of conditioning trains needed to elicit transient depression, suggesting cooperativity between these receptor types. Ordinate indicates the current passed per stimulus in the conditioning train. B: acetylcholinesterase inhibitor eserine faciltates transient depression, consistent with a cooperative contribution of the cholinergic input. Conditioning trains that were too weak to trigger depression in control conditions began triggering depression in the presence of eserine and the effect was reversed by atropine (n = 3).
FIG. 6.

Both metabotropic glutamate receptors (mGluRs) and muscarinic acetylcholine receptors (mAChRs) participate in eliciting transient depression. A: depression was not blocked after preincubation with the adenosine receptor antagonist DPCPX (0.5–5 μM, black symbols, n = 7), mGluR antagonists [LY367385 (100 μM) and MPEP (10 μM) open symbols, n = 5], or the mAChR antagonist atropine (10 μM, gray symbols, n = 3). B: preincubation with combinations of the antagonists in A had different effects on transient depression. Depression was blocked when mGluR and mAChR activation were blocked simultaneously (black symbols, n = 6), but depression was observed during blockade of adenosine receptors (DPCPX, 5 μM) plus either mGluRs (open symbols, n = 3) or mAChRs (gray symbols, n = 3). C: an increase in the PPR of AMPAR EPSCs accompanied bath application of the Group I mGluR agonist DHPG (50 μM), consistent with a presynaptic location of this receptor type (n = 3). Measurements were combined in 1-min bins for plotting and data were aligned according to the onset of DHPG action. D: 5 μM DPCPX blocks the synaptic depression induced by a pressure application (puff) of 200 μM adenosine, verifying that DPCPX blocks adenosine receptors in our experiments (n = 3).
All data are presented as mean ± SE. Statistical significance (P < 0.05) was tested using paired or unpaired two-tailed Student’s t-tests as appropriate, except for the series of experiments examining stimulation intensity requirements (Fig. 7A), which used a one-way ANOVA with Dunnett’s multiple comparison post hoc analysis.
RESULTS
Brief trains of synaptic stimulation elicit a transient depression of CA1 glutamatergic synapses
We describe a transient depression of glutamatergic neurotransmission at Schaffer collateral synapses using whole cell recordings in acute hippocampal slices from 3- to 5-wk-old male rats. NMDAR-mediated or composite (AMPAR + NMDAR) EPSCs were evoked in the recorded CA1 pyramidal cell by brief currents through a small (3- to 10-μm tip diameter) stimulation pipette placed in stratum radiatum. Short conditioning trains (30 stimuli at 100 Hz) of synaptic stimulation were followed by a transient reduction in EPSC amplitude (Fig. 1A) in 86% of the cells tested (96 of 111 cells). This stimulus pattern was used because it reliably elicited transient depression but was not expected to induce long-term changes in synaptic strength. Depression was maximal within 40 s after the conditioning train (to 57.5 ± 1.0% of baseline 20 s after train, n = 83) and recovered monoexponentially (to 94.9 ± 1.0% of baseline after 180–200 s, τ = 76.5 s; Fig. 1B). Depression could be elicited repeatedly through recordings of ≥1 h (Fig. 1C). Additional conditioning trains delivered while responses were depressed prolonged the duration of depression without increasing its amplitude (n = 3; data not shown). We frequently examined the transient depression of isolated NMDAR test currents because they are known targets for diffusible modulators of channel function that may be liberated during conditioning trains (e.g., nitric oxide; see following text) (Yamakura and Shimoji 1999) and are also targets for intracellular Ca2+ and other postsynaptic effectors (Kotecha and MacDonald 2003). As such, most experiments were conducted without extracellular Mg2+ and with GABAAR antagonists. However, transient depression was also observed in more physiologic conditions (Fig. 1D).
FIG. 1.

Transient depression of EPSCs follows conditioning trains in stratum radiatum. A: EPSC amplitude is reduced after conditioning trains. Example traces are averages of 3 consecutive NMDAR–mediated test responses 20–60 s before (baseline), 20–60 s after, and 160 –200 s after (recovery) a synaptic conditioning train. B: data averaged across 83 experiments illustrate the magnitude and time course of transient depression. Recovery is fit by a monoexponential curve (gray line, τ = 76.5 s). SE error bars are smaller than the plot symbols. C: depression can be elicited repeatedly over a long experiment without extinction. This plot of raw EPSC amplitudes over time illustrates 3 consecutive trials of depression after 45 min of recording and 5 previous episodes of depression. D: although most experiments in this study were performed with GABAAR antagonism and without extracellular Mg2+, normal transient depression was also observed without GABAAR antagonism (open symbols, n = 17) and with 2 mM Mg2+ and no GABAAR or GABABR antagonism (filled symbols, n = 5). All experiments in which GABABRs were left unblocked (n = 14) are included in the 2 groups of data plotted here.
Transient depression results from a decrease in presynaptic glutamate release
To begin to understand the mechanism of transient depression, we next examined whether it was expressed presynaptically or postsynaptically. One prediction about a presynaptic depression is that the associated reduction of glutamate release will reduce the activation of all types of glutamate receptors. Therefore we tested for transient depression of currents through two types of postsynaptic glutamate receptors and used paired test stimuli to assess relative changes in the probability of neurotransmitter release (Pr). Consistent with a previous report (Grover and Teyler 1993a), both NMDAR-mediated and composite EPSC amplitudes were similarly depressed after conditioning trains (composite depression to 55.8 ± 2.7% of baseline, τ = 79.4 s, n = 19; NMDAR depression to 57.1 ± 1.1% of baseline, τ = 74.9 s, n = 64; Fig. 2A). Composite EPSC amplitudes mainly reflect the level of synaptic AMPAR activation (Collingridge et al. 1988; Hestrin et al. 1990). Transient depression of current amplitudes was accompanied by a transient increase in the paired-pulse ratio (PPR) and the two effects recovered with similar time courses (Fig. 2, B and C). The PPR was increased during depression for both composite (to 139.1 ± 7.9% of baseline 20 s after train, n = 19, P < 0.001) and isolated NMDAR EPSCs (to 110.2 ± 2.1% of baseline, n = 52, P < 0.001). To illustrate the time course similarity between amplitude depression and PPR increase, we fit each group of PPR data with an exponential constrained to the time course of depression (τ = 76.5 s, Fig. 2B, gray lines). The similar depression of NMDAR- and AMPAR-mediated currents and the coincidence of depression with a PPR increase led us to conclude that transient depression is mediated presynaptically through a reduction in Pr. The PPR increase for isolated NMDAR currents, although significant, was smaller than that observed with composite EPSCs (P < 0.001). This difference may represent receptor-specific differences in PPR, like a reduction of the contribution of AMPAR desensitization during depression (Colquhoun et al. 1992) or different presynaptic properties of silent versus functional synapses (Cabezas and Buno 2006; Fernandez de Sevilla et al. 2002), but does not affect our conclusion that transient depression is expressed presynaptically.
FIG. 2.
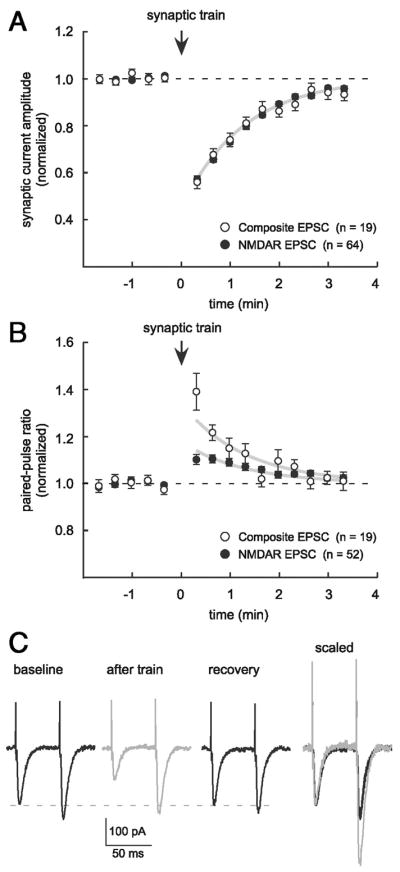
Transient depression results from a decrease in presynaptic glutamate release. A: transient depression similarly affects AMPAR– and NMDAR-mediated postsynaptic currents. Plot shows depression of composite (open symbols, n = 19) and NMDAR-mediated EPSCs (filled symbols, n = 64). Together, these data are the same as Fig. 1A and the gray line is an exponential fit to the combined data (τ = 76.5 s). B: paired-pulse ratio (PPR) increases during depression for both composite (n = 19) and NMDAR-mediated EPSCs (n = 52) and the time courses of the 2 phenomena are similar. Gray lines illustrate the similarity of time courses between amplitude depression and PPR increase and are exponential fits to PPR data that are constrained to the time course of recovery from depression (τ = 76.5 s). C: example traces are averages of 3 consecutive responses to paired-test stimulation (50-ms interval) before and after a synaptic conditioning train, illustrating depression of composite EPSCs and the change in PPR that accompanies depression. Right: “after train” trace (gray) is scaled and overlayed on the baseline trace to show the relative difference in the amplitude of the second response.
Depression is observed on an independent test pathway
One possible mechanism for transient depression is a use-dependent depression of the presynaptic terminal, in which the action potentials of the conditioning train may exhaust neurotransmitter release by, for example, depleting the readily releasable pool of synaptic vesicles (Wang and Kaczmarek 1998; Zucker and Regehr 2002). To explore this possibility, we tested whether depression could be observed at test synapses that were not stimulated during the conditioning train. More specifically, two small stimulating electrodes were placed in stratum radiatum at different distances (50 and 100 μm) from the pyramidal cell layer and response amplitudes were measured on a test pathway before and after a conditioning train of stimuli on an independent conditioning pathway (Fig. 3A). The independence of the two pathways was verified using a paired-pulse test (50-ms interstimulus interval; Fig. 3B) (Barrionuevo and Brown 1983) in each experiment. Depression was observed on an independent test pathway in 20 – 40% of such experiments (to 64.7 ± 3.6% of baseline, τ = 69.8 s, n = 11; Fig. 3C) and was similar to depression observed on the conditioning pathway (described earlier). Either of the two stimulators could trigger transient depression on the other pathway and conditioning stimuli were delivered on the proximal pathway (n = 6) or the distal pathway (n = 7) in these experiments. Our ability to observe synaptic depression on an independent test pathway was inconsistent with a use-dependent mechanism intrinsic to the axon terminal, thus leading us to consider alternative traveling or diffusible signals capable of contributing to a presynaptic depression.
Depression does not require postsynaptic Ca2+- or G-protein signaling
Given the suggestion of a traveling signal extrinsic to the axon terminal, we examined the possible dependence of transient depression on some kinds of postsynaptic and network signaling. One candidate mechanism was internal Ca2+ release or a propagating Ca2+ wave in the recorded cell or in neighboring neurons and glia (Araque et al. 2002; Kapur et al. 2001; Nakamura et al. 1999). Using Ca2+ fluorescence imaging of the recorded cell, we found a strong correlation between synaptic depression and postsynaptic internal Ca2+ release after conditioning trains (Fig. 4, A and B). An internal Ca2+ release event or propagating Ca2+ wave was detected following 93% of conditioning trains that elicited transient depression (53 of 57 trials, n = 25 cells; Fig. 4B), whereas only 25% of conditioning trains that failed to trigger depression were accompanied by internal Ca2+ release (14 of 55 trials, n = 25 cells). In spite of this correlation, transient depression occurred normally when internal Ca2+ release was blocked pharmacologically, either via the patch pipette or by bath application (Fig. 4C). Internal Ca2+ release was effectively blocked, as verified using Ca2+ imaging, by including a Ca2+ chelator (10 or 50 mM BAPTA, n = 24) and/or a G-protein antagonist (1 mM GDP-β-S, n = 14) in the recording pipette or by bath application of an antagonist of the sarcoplasmic/endoplasmic Ca2+ ATPase [SERCA, 50 μM cyclopiazonic acid (CPA), or 3 μM thapsigargin, n = 5; Fig. 4D]. The BAPTA data agree with a previous report (Grover and Teyler 1993a). Additionally, transient depression never followed trains of postsynaptic action potentials elicited by brief depolarizing pulses (2-nA steps lasting 2 ms; 30–100 action potentials at 100 Hz; n = 19; data not shown). The experiments performed in the presence of BAPTA, GDP-β-S, or SERCA antagonists demonstrate that the depression we observed does not require postsynaptic Ca2+ waves or internal Ca2+ release. Furthermore, these experiments suggest that no postsynaptic signal involving Ca2+ or G-protein activation is required for transient depression.
FIG. 4.

Transient depression does not require postsynaptic internal Ca2+ release. A: conditioning trains frequently elicit postsynaptic Ca2+ waves. Left: image of a CA1 pyramidal neuron filled with bis-fura-2 (100 μM) and Alexa 488 (15 μM), plus colored regions of interest (ROIs). A stimulating electrode filled with Alexa 488 (2.5 μM) is visible in the top left. Right: Ca2+ imaging: colored traces correspond to changes in relative intracellular concentration of Ca2+ ([Ca2+]i) in ROIs. Current trace (black): a train (30 stimuli at 100 Hz) of synaptic stimulation elicits an intracellular Ca2+ wave. B: occurrence of transient depression correlates with postsynaptic internal Ca2+ release. Bar widths represent the number of trials in 25 recorded neurons with depression (57 trials, top bar) or no depression (55 trials, bottom bar). Placement of the bar along the abscissa indicates the fraction of trials in which internal Ca2+ release was observed (left of center) or not observed (right of center). C: depression was observed in experiments where postsynaptic Ca2+ waves were verifiably blocked. Ca2+ chelator BAPTA (black symbols, 10 or 50 mM, n = 24) and/or G-protein antagonist GDP-β-S (1 mM, n = 14; red symbols), was included in the patch pipette for 30–75 min of recording, or SERCA antagonists were bath applied (blue symbols, wash-in of 50 μM CPA or preincubation with 3 μM thapsigargin, n = 5). D: SERCA antagonist CPA (50 μM) blocks internal Ca2+ release. A conditioning train evokes a propagating Ca2+ wave in control conditions (top optical and current traces) but not when CPA is present. During this experiment AMPAR-mediated responses were isolated with antagonists of GABARs [gabazine, 50 μM; CGP55845 (1 μM)]; and NMDARs (100 μM (D,L-APV)).
Evidence against the involvement of glia or neighboring dendrites in depression
In the experiments described earlier, SERCA antagonists were expected to block internal Ca2+ release not only in the recorded cell, but also in neighboring neurons and glia (Araque et al. 2002; Kapur et al. 2001; Nakamura et al. 1999; Yeckel et al. 1999). These data begin to address the possibility that Ca2+-dependent release of modulatory factors from neighboring cells during conditioning trains may mediate the presynaptic depression (Araque et al. 1998; Kreitzer and Regehr 2002; Ledo et al. 2004; Lowenstein et al. 1994). To further investigate this possibility, we examined whether the retrograde messengers nitric oxide (NO) (Boulton et al. 1994) and endocannabinoids contributed to transient depression (Bernard et al. 2005; Domenici et al. 2006; Wilson and Nicoll 2001).
Because NO production is frequently triggered through NMDAR activation and the action of Ca2+-calmodulin (Bredt and Snyder 1990; Garthwaite et al. 1988), we first examined whether antagonists expected to diminish the production of NO influenced transient depression. Bath application of NMDAR antagonists (100 μM D,L-APV and 20 μM MK-801, n = 3; Fig. 5A) had no effect on transient depression. NMDAR antagonists were effective in these experiments because they blocked a slow component of the composite EPSC (Fig. 5B) and 100 μM D,L-APV alone blocked NMDAR-mediated test currents in other experiments (to 4.6 ± 1.0% of baseline, n = 5). Similarly, the Ca2+-calmodulin antagonist calmidazolium (50 μM) did not prevent transient depression (n = 3 of 4; Fig. 5A). Next, we blocked NO activation by preincubating slices with a combination of well-established NO antagonists: the NO synthase inhibitor NG-nitro-L-arginine methyl ester (L-NAME, 100 μM) and the NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO, 30 μM). Transient depression occurred normally in the presence of these antagonists (n = 5; Fig. 5A). In another series of experiments we tested whether endocannabinoids released from neighboring neurons might be contributing to transient depression. Addition of the endocannabinoid receptor blocker AM251 (2–5 μM) failed to block transient depression (n = 5; Fig. 5C). AM251 (5 μM) was verified to antagonize the inhibition of evoked IPSCs produced by bath application of the endocannabinnoid agonist WIN 55,212-2 (1 μM; n = 1; data not shown). Together, these data are inconsistent with the hypothesis that Ca2+-dependent release of modulatory factors from neighboring neurons and glia mediates transient depression. Rather, they strengthen the case for a presynaptic mechanism of the transient depression we observe after brief trains of synaptic stimulation.
FIG. 5.

Diffusible messengers nitric oxide (NO) and endocannabinoids do not mediate transient depression. A: NO antagonists do not prevent transient depression. NMDAR antagonists D,L-APV (100 μM) and MK-801 (20 μM) were bath applied to inhibit NMDAR activation during the conditioning train and NO production (open symbols, n = 3). Ca2+-calmodulin antagonist calmidazolium (50 μM) was applied by bath or by preincubation to inhibit NO production (black symbols, n = 3). NO synthase inhibitor NG-nitro-L-arginine methyl ester (L-NAME, 100 μM) and NO scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (carboxy-PTIO, 30 μM) were applied together as a preincubation (gray symbols, n = 5). B: NMDAR antagonists block a slow component of composite EPSCs. Traces are averages of 10 consecutive responses before (black) and after (gray) APV and MK-801 application. Traces are scaled to illustrate the difference in time course. Stimulus intensity was different between the 2 conditions. C: endocannabinoid CB1R antagonist AM251, applied as a preincubation at 2 μM (n = 1) or 5 μM (n = 4), does not prevent transient depression.
Optimal transient depression depends on coincident synaptic activation of Gq-coupled receptors
Because transient depression in our experiments resulted from a decrease in presynaptic glutamate release (Fig. 2), we examined the role of receptors known to depress neurotransmitter release. More specifically, glutamate release at Schaffer collateral axon terminals can be depressed by activation of several types of presynaptic G-protein–coupled receptors, including Group I mGluRs (Baskys and Malenka 1991; Gereau and Conn 1995), mAChRs (Fernandez de Sevilla and Buno 2003; Fernandez de Sevilla et al. 2002; Hounsgaard 1978; Sheridan and Sutor 1990; Valentino and Dingledine 1981), adenosine receptors (Dunwiddie and Hoffer 1980; Scholz and Miller 1992; Schubert and Mitzdorf 1979), and GABABRs (Ault and Nadler 1982; Bowery et al. 1980; Inoue et al. 1985; Isaacson et al. 1993; Lanthorn and Cotman 1981; Olpe et al. 1982). Consistent with previous reports and with the presence of presynaptic Group I mGluRs at Schaffer collateral axon terminals, bath application of the specific Group I mGluR agonist 9-[(1,3-dihydroxy-2-propoxy)methyl] guanine (DHPG, 50 μM) decreased the amplitude and increased the PPR of AMPAR-mediated EPSCs (amplitude to 53.9 ± 4.4% and PPR to 123.9 ± 3.9% of baseline 0–5 min after onset of the DHPG effect, P < 0.001; Fig. 6C) (Baskys and Malenka 1991; Gereau and Conn 1995). We investigated the contribution of presynaptic receptor types to transient depression, either alone or in combination, using receptor agonists and antagonists.
Blockade of individual receptor subtypes impeded but did not entirely prevent conditioning train-elicited transient depression (Fig. 6A). Depression was not blocked during bath application of Group I mGluR antagonists (100 μM LY367385 and 10 μM MPEP, n = 5), the mAChR antagonist atropine (10 μM, n = 3), or the adenosine receptor antagonist DPCPX (0.5–5 μM, n = 7). However, mGluR or mAChR antagonists did make depression more difficult to elicit (subsequently described). In control experiments, DPCPX (5 μM) blocked the depression of glutamatergic transmission produced by a pressure application of 200 μM adenosine (depression to 64.4 ± 3.8% of baseline in control 10–30 s after puff and to 96.9 ± 2.0% with DPCPX, n = 3, P < 0.001; Fig. 6D). GABABRs were blocked in most experiments using CGP55845 (1–2 μM) and depression was observed with or without this antagonist (n = 14; see Fig. 1D).
To evaluate the possibility that Group I mGluRs and mAChRs act together in triggering transient depression, we looked retrospectively at stimulus intensities used during conditioning trains in comparable experiments represented in Fig. 6, A and B. Although neither mGluR nor mAChR activation was absolutely required for transient depression, mGluR or mAChR antagonists increased the stimulus intensity required during the conditioning train to trigger transient depression (14.6 ± 1.8 nC per stimulus during conditioning train with mGluR antagonists, n = 6; 9.6 ± 1.0 nC with mAChR antagonist, n = 6; vs. 5.2 ± 0.4 nC in control conditions, n = 17; P < 0.005; Fig. 7A). Pharmacologic conditions that increased the stimulus requirement during the conditioning train, however, did not require more intense stimulation to elicit test synaptic responses (2.3 ± 0.4 nC per test stimulus with mGluR antagonists, n = 6; 3.1 ± 0.3 nC with mAChR antagonist, n = 6; vs. 3.7 ± 0.4 nC in control conditions; P > 0.15; n = 17). In the course of increasing the conditioning train stimulation intensity during experiments we noted that depression tended to appear at a threshold level of intensity and rapidly approached a magnitude of depression roughly 40% below baseline (data not shown). We thus did not attempt to examine the effect of receptor blockade on depression magnitude. The observation that mGluR or mAChR blockade made depression more difficult to elicit suggested the possibility that evoked glutamate and acetylcholine release might act cooperatively through activation of Gq-protein–coupled receptors to mediate transient depression. Consistent with this possibility, depression was blocked after conditioning trains when both the Group I mGluR antagonists and the mAChR antagonist were present in the recording solution (antagonists described earlier, n = 6; Fig. 6B).
No significant increase in the stimulus intensity used during the conditioning train was observed when DPCPX (5 μM) antagonized the adenosine receptor, a Gi-coupled receptor (6.7 ± 0.8 nC per stimulus during conditioning train, 2.8 ± 0.3 nC per test stimulus; P > 0.3). However, the implication of adenosine receptor activation in presynaptic depression studied at this synapse (Grover and Teyler 1993b; Manzoni et al. 1994; Mitchell et al. 1993; Sekino and Koyama 1992) led us to test whether transient depression was prevented by a combination of DPCPX with either mGluR or mAChR antagonists. Neither of these combinations prevented transient depression (n = 3 for DPCPX plus mGluR antagonists; n = 3 of 4 for DPCPX plus atropine; Fig. 6B), and there was no significant increase in conditioning train stimulus intensity compared with mGluR or mAChR antagonists alone (P > 0.1). Stimulus intensity data from these combinations were thus pooled with data from mGluR or mAChR blockade for plotting in Fig. 7A. Experiments using receptor antagonists suggest at most a small, insufficient role for Gi-coupled adenosine receptors in transient depression under our recording conditions, but leave open the possibility of a greater contribution from this receptor type in vivo or on a faster timescale (see DISCUSSION).
In an effort to confirm that glutamate and acetylcholine work together to trigger transient depression, we used the acetylcholinesterase inhibitor eserine to enhance the action of acetylcholine during conditioning trains. In experiments where transient depression was not observed after conditioning trains at baseline, we found that bath application of eserine (5 μM) facilitated the occurrence of depression (n = 3; Fig. 7B). The effect of eserine was reversed by atropine (10 μM) in each experiment. Conditioning train stimulus intensities used during these experiments were similar to those used in control experiments in Fig. 7A (5.3 ± 0.7 nC). These findings, taken together with data presented in Figs. 6 and 7A, support our conclusion that the combined action of evoked glutamate and acetylcholine release in stratum radiatum depresses glutamate release from Schaffer collateral synapses. The data raise the possibility that spatial and temporal convergence of glutamatergic and cholinergic inputs to CA1 may help to shape hippocampal network activity in vivo (see schematic, Fig. 8).
FIG. 8.

Schematic of convergent afferents to CA1. Our data support a model in which cholinergic and glutamatergic afferents converge in area CA1 to produce a transient presynaptic depression and postsynaptic Ca2+ waves. Stratum radiatum stimulation elicits release of acetylcholine (ACh) by axons from basal forebrain nuclei or from hippocampal cholinergic interneurons. The same stimuli evoke glutamate release from Schaffer collateral axons originating in CA3 and perhaps from axons from basal forebrain nuclei (Allen et al. 2006; Colom et al. 2005). Released ACh and glutamate act in unison on presynaptic and postsynaptic Gq-coupled receptors.
DISCUSSION
We have studied a synaptically elicited transient depression of glutamatergic neurotransmission at Schaffer collateral synapses in area CA1 using acute slice electrophysiology and Ca2+ fluorescence imaging. This depression was induced by a brief conditioning train of electrical stimulation in stratum radiatum and lasted 2–3 min. NMDAR- and composite EPSCs were similarly depressed and depression coincided with an increase in the paired-pulse ratio (PPR), consistent with a presynaptic locus of depression. Eliciting transient depression was most easily achieved when both mGluRs and mAChRs were activated, suggesting a cooperative interaction involving a common G-protein subtype. Although transient depression correlates with postsynaptic Ca2+ release from internal stores mediated by these same receptor types, it does not require increases in postsynaptic [Ca2+]i or other forms of postsynaptic or network signaling that we tested. The involvement of two neurotransmitters— elicited with modest synaptic activation—in triggering transient depression is consistent with numerous behavioral, systems, and theoretical reports suggesting a functional importance for convergent glutamatergic and cholinergic afferents to area CA1 (Hasselmo 1999; Hasselmo and Schnell 1994).
Although it is notoriously difficult to be certain whether changes in synaptic transmission are expressed presynaptically and/or postsynaptically, our data, in toto, are strongly consistent with a decrease in glutamate release from presynaptic terminals. First, the amplitude and time course of the depression were remarkably similar for two species of ionotropic glutamate receptors, consistent with an overall decrease of glutamate available to postsynaptic receptors. Second, there was a transient increase in the PPR with a time course that closely matched the time course of the transient depression, consistent with a decrease in the probability of release of glutamate from the axon terminal. Slight deviations between the time courses of PPR increase and amplitude depression likely arise from factors other than Pr that affect PPR measurements (e.g., AMPAR desensitization and differential sensitivity of silent synapses to depression) (Cabezas and Buno 2006; Colquhoun et al. 1992; Fernandez de Sevilla et al. 2002). Finally, no changes were observed in postsynaptic cells during transient depression (e.g., Rin and Vm) and a multitude of experimental manipulations known to block plasticity at postsynaptic sites or the release of retrograde messengers failed to prevent transient depression.
Previous studies reported altered NMDAR function resulting from Gq-coupled receptor activation and increases in [Ca2+]i (Grishin et al. 2004; Markram and Segal 1990). In the present study, we observed a Gq-coupled receptor-mediated depression of NMDAR and composite EPSCs that correlated with a robust increase in postsynaptic [Ca2+]i arising from internal Ca2+ release. We demonstrate, however, that these two phenomena are not causally related in our experiments because blocking increases in [Ca2+]i with high concentrations of BAPTA (50 mM), antagonizing postsynaptic G-proteins, or depleting intracellular Ca2+ stores failed to block transient depression. These previous studies, through direct activation of postsynaptic NMDARs by pressure application of agonists, may have unmasked a distinct postsynaptic effect of internal Ca2+ release on NMDARs by disengaging the presynapse. Also, the use of mGluR and mAChR agonist applications in these studies may have provided a stronger and more widespread stimulus of Gq-coupled receptors on the recorded cell than that achieved during our conditioning trains. The findings from these previous studies and the findings reported here suggest the possibility of interplay between presynaptic and postsynaptic effects of metabotropic receptor activation in the control of network activity in CA1, and the potential importance of factors that independently regulate these effects.
Others reported a depression of glutamate release from Schaffer collateral nerve terminals after activation of presumed presynaptic G-protein–coupled receptors. Some showed that repeated trains of stimulation in the stratum oriens/alveus depress glutamate release through activation of presynaptic M1 acetylcholine receptors (Fernandez de Sevilla and Buno 2003; Fernandez de Sevilla et al. 2002). The present study builds on this work by describing a role for acetylcholine released during a single train and its cooperation with synaptically released glutamate. Other studies describe depression that depends partly on presynaptic adenosine receptor activation after conditioning stimulation (Grover and Teyler 1993b; Manzoni et al. 1994; Sekino and Koyama 1992) and one reported that adenosine release was dependent on NMDAR activation (Manzoni et al. 1994). In contrast, the transient depression we studied occurs despite blockade of NMDARs or adenosine receptors. A likely explanation for this discrepancy is that we are studying the adenosine-receptor–independent component of the depression they observed, and that the physiologic effects of adenosine on the presynapse occur on a shorter timescale than the effects of Gq-coupled receptor activation (Grover and Teyler 1993b; Mitchell et al. 1993). A greater or longer-lasting role for adenosine in previous studies may depend on a greater level of adenosine release achieved through stronger and spatially more extensive stimulation, or other differences in experimental conditions (Manzoni et al. 1994; Sekino and Koyama 1992). Similar to Manzoni et al., we found that transient depression was difficult to elicit after long-term blockade of NMDARs (antagonists present for 1–4 h; n = 4; unpublished observations). This preliminary finding may indicate some role—perhaps a permissive one— of tonic or ongoing NMDAR activation for transient depression and may warrant further study.
We favor a mechanism of transient depression wherein intrinsic glutamatergic signaling interacts with extrinsic cholinergic signaling and the signals converge at the level of Gq activation in the presynaptic terminal. This potential mechanism would be consistent with numerous studies demonstrating the importance of cholinergic input from the basal forebrain in hippocampal function (Aigner 1995; Dragoi et al. 1999; Hasselmo 1999), and with the capacity of G-protein–coupled receptors to converge on the same G-protein class and/or the same target molecule (Hille 1992). While our data indicate that evoked, rather than tonic, release of glutamate and acetylcholine triggers transient depression, they are not conclusive about the source of acetylcholine afferent input. More specifically, the source of acetylcholine axons might be cholinergic interneurons intrinsic to the hippocampus (Frotscher et al. 2000) or extrinsic inputs originating in the medial septum or diagonal band of Broca (Frotscher and Leranth 1985; Lewis and Shute 1967). Finally, although we find that depression is presynaptic and can occur when certain types of signaling are antagonized in the recorded neuron and in neighboring cell types, we cannot entirely exclude the possibility that metabotropic receptors on neighboring pyramidal cells or other cell types may mediate this depression through a more complex mechanism.
Functional significance
Our study reveals that the effects of glutamatergic and cholinergic signals in CA1 combine to depress EPSCs by attenuating neurotransmitter release. Cholinergic inputs from the basal forebrain have long been known to influence the ability of the hippocampal formation to participate in some forms of memory by affecting hippocampal network properties, hippocampal theta rhythm, and the induction of LTP (Bland et al. 1999; Louie and Wilson 2001; Otto et al. 1991; Pavlides et al. 1988; Winson and Abzug 1978). The transient depression we have studied may affect LTP induction by limiting ionotropic and metabotropic glutamatergic activation, thereby limiting increases in [Ca2+]i normally mediated by influx through NMDA channels and mGluR-activated internal Ca2+ release (Morris et al. 1986; Yeckel et al. 1999). The possibility of cooperative action between acetylcholine and glutamate raises the possibility of depression at specific subsets of synapses with certain activity patterns or anatomical arrangements. Furthermore, were transient depression to be coincident with the potentially more general excitatory postsynaptic effects of acetylcholine (Cole and Nicoll 1983; Colino and Halliwell 1993; Dodd et al. 1981; Halliwell 1990), the combined effect could be to alter the distribution of synaptic weights along the dendrite, postsynaptic computations, and the induction of long-term synaptic plasticity at distant synapses as well.
Acknowledgments
We thank D. Hertle, A. Sleeper, J. Fitzpatrick, A. Hagenston, and S. Krueger for a critical reading of the manuscript; T. Koos for useful discussions; F. Sigworth for advice on analytical strategies; and J. Fitzpatrick for writing the data collection software.
GRANTS
This work was supported by the Hellman Family Fund, the Whitehall Foundation, and National Institute of Mental Health Grant RO1 MH-067830.
References
- Abraham WC, Bear MF. Metaplasticity—the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Aigner TG. Pharmacology of memory: cholinergic–glutamatergic interactions. Curr Opin Neurobiol. 1995;5:155–160. doi: 10.1016/0959-4388(95)80021-2. [DOI] [PubMed] [Google Scholar]
- Allen TG, Abogadie FC, Brown DA. Simultaneous release of glutamate and acetylcholine from single magnocellular “cholinergic” basal forebrain neurons. J Neurosci. 2006;26:1588–1595. doi: 10.1523/JNEUROSCI.3979-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Martin ED, Perea G, Arellano JI, Buno W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ault B, Nadler JV. Baclofen selectively inhibits transmission at synapses made by axons of CA3 pyramidal cells in the hippocampal slice. J Pharmacol Exp Ther. 1982;223:291–297. [PubMed] [Google Scholar]
- Barrionuevo G, Brown TH. Associative long-term potentiation in hippocampal slices. Proc Natl Acad Sci USA. 1983;80:7347–7351. doi: 10.1073/pnas.80.23.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskys A, Malenka RC. Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol. 1991;444:687–701. doi: 10.1113/jphysiol.1991.sp018901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Milh M, Morozov YM, Ben-Ari Y, Freund TF, Gozlan H. Altering cannabinoid signaling during development disrupts neuronal activity. Proc Natl Acad Sci USA. 2005;102:9388–9393. doi: 10.1073/pnas.0409641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland BH, Oddie SD, Colom LV. Mechanisms of neural synchrony in the septohippocampal pathways underlying hippocampal theta generation. J Neurosci. 1999;19:3223–3237. doi: 10.1523/JNEUROSCI.19-08-03223.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton CL, Irving AJ, Southam E, Potier B, Garthwaite J, Collingridge GL. The nitric oxide–cyclic GMP pathway and synaptic depression in rat hippocampal slices. Eur J Neurosci. 1994;6:1528–1535. doi: 10.1111/j.1460-9568.1994.tb00543.x. [DOI] [PubMed] [Google Scholar]
- Bowery NG, Hill DR, Hudson AL, Doble A, Middlemiss DN, Shaw J, Turnbull M. (−)Baclofen decreases neurotransmitter release in the mammalian CNS by an action at a novel GABA receptor. Nature. 1980;283:92–94. doi: 10.1038/283092a0. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas C, Buno W. Distinct transmitter release properties determine differences in short-term plasticity at functional and silent synapses. J Neurophysiol. 2006;95:3024–3034. doi: 10.1152/jn.00739.2005. [DOI] [PubMed] [Google Scholar]
- Christie BR, Eliot LS, Ito K, Miyakawa H, Johnston D. Different Ca2+ channels in soma and dendrites of hippocampal pyramidal neurons mediate spike-induced Ca2+ influx. J Neurophysiol. 1995;73:2553–2557. doi: 10.1152/jn.1995.73.6.2553. [DOI] [PubMed] [Google Scholar]
- Cole AE, Nicoll RA. Acetylcholine mediates a slow synaptic potential in hippocampal pyramidal cells. Science. 1983;221:1299–1301. doi: 10.1126/science.6612345. [DOI] [PubMed] [Google Scholar]
- Colino A, Halliwell JV. Carbachol potentiates Q current and activates a calcium-dependent non-specific conductance in rat hippocampus in vitro. Eur J Neurosci. 1993;5:1198–1209. doi: 10.1111/j.1460-9568.1993.tb00974.x. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Herron CE, Lester RA. Synaptic activation of N-methyl-D-aspartate receptors in the Schaffer collateral-commissural pathway of rat hippocampus. J Physiol. 1988;399:283–300. doi: 10.1113/jphysiol.1988.sp017080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom LV, Castaneda MT, Reyna T, Hernandez S, Garrido-Sanabria E. Characterization of medial septal glutamatergic neurons and their projection to the hippocampus. Synapse. 2005;58:151–164. doi: 10.1002/syn.20184. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Jonas P, Sakmann B. Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hippocampal slices. J Physiol. 1992;458:261–287. doi: 10.1113/jphysiol.1992.sp019417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd J, Dingledine R, Kelly JS. The excitatory action of acetylcholine on hippocampal neurones of the guinea pig and rat maintained in vitro. Brain Res. 1981;207:109–127. doi: 10.1016/0006-8993(81)90682-x. [DOI] [PubMed] [Google Scholar]
- Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, Zieglgansberger W, Lutz B, Rammes G. Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci. 2006;26:5794–5799. doi: 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragoi G, Carpi D, Recce M, Csicsvari J, Buzsáki G. Interactions between hippocampus and medial septum during sharp waves and theta oscillation in the behaving rat. J Neurosci. 1999;19:6191–6199. doi: 10.1523/JNEUROSCI.19-14-06191.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Hoffer BJ. Adenine nucleotides and synaptic transmission in the in vitro rat hippocampus. Br J Pharmacol. 1980;69:59–68. doi: 10.1111/j.1476-5381.1980.tb10883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez de Sevilla D, Buno W. Presynaptic inhibition of Schaffer collateral synapses by stimulation of hippocampal cholinergic afferent fibres. Eur J Neurosci. 2003;17:555–558. doi: 10.1046/j.1460-9568.2003.02490.x. [DOI] [PubMed] [Google Scholar]
- Fernandez de Sevilla D, Cabezas C, de Prada AN, Sanchez-Jimenez A, Buno W. Selective muscarinic regulation of functional glutamatergic Schaffer collateral synapses in rat CA1 pyramidal neurons. J Physiol. 2002;545:51–63. doi: 10.1113/jphysiol.2002.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Leranth C. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. J Comp Neurol. 1985;239:237–246. doi: 10.1002/cne.902390210. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Vida I, Bender R. Evidence for the existence of non-GABAergic, cholinergic interneurons in the rodent hippocampus. Neuroscience. 2000;96:27–31. doi: 10.1016/s0306-4522(99)00525-4. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature. 1988;336:385–388. doi: 10.1038/336385a0. [DOI] [PubMed] [Google Scholar]
- Gereau RWt, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- Grishin AA, Gee CE, Gerber U, Benquet P. Differential calcium-dependent modulation of NMDA currents in CA1 and CA3 hippocampal pyramidal cells. J Neurosci. 2004;24:350–355. doi: 10.1523/JNEUROSCI.4933-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Presynaptic mechanism for heterosynaptic, posttetanic depression in area CA1 of rat hippocampus. Synapse. 1993a;15:149–157. doi: 10.1002/syn.890150207. [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ. Role of adenosine in heterosynaptic, posttetanic depression in area CA1 of hippocampus. Neurosci Lett. 1993b;154:39–42. doi: 10.1016/0304-3940(93)90166-i. [DOI] [PubMed] [Google Scholar]
- Halliwell JV. Physiological mechanisms of cholinergic action in the hippocampus. Prog Brain Res. 1990;84:255–272. doi: 10.1016/s0079-6123(08)60910-3. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. Neuromodulation: acetylcholine and memory consolidation. Trends Cogn Sci. 1999;3:351–359. doi: 10.1016/s1364-6613(99)01365-0. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Schnell E. Laminar selectivity of the cholinergic suppression of synaptic transmission in rat hippocampal region CA1— computational modeling and brain slice physiology. J Neurosci. 1994;14:3898–3914. doi: 10.1523/JNEUROSCI.14-06-03898.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestrin S, Nicoll RA, Perkel DJ, Sah P. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recording from rat hippocampal slices. J Physiol. 1990;422:203–225. doi: 10.1113/jphysiol.1990.sp017980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. G protein-coupled mechanisms and nervous signaling. Neuron. 1992;9:187–195. doi: 10.1016/0896-6273(92)90158-a. [DOI] [PubMed] [Google Scholar]
- Hounsgaard J. Presynaptic inhibitory action of acetylcholine in area CA1 of the hippocampus. Exp Neurol. 1978;62:787–797. doi: 10.1016/0014-4886(78)90284-4. [DOI] [PubMed] [Google Scholar]
- Inoue M, Matsuo T, Ogata N. Characterization of pre- and postsynaptic actions of (−)-baclofen in the guinea-pig hippocampus in vitro. Br J Pharmacol. 1985;84:843–851. doi: 10.1111/j.1476-5381.1985.tb17378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Kapur A, Yeckel M, Johnston D. Hippocampal mossy fiber activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci. 2001;21:9608–9618. doi: 10.1523/JNEUROSCI.21-24-09608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha SA, MacDonald JF. Signaling molecules and receptor transduction cascades that regulate NMDA receptor-mediated synaptic transmission. Int Rev Neurobiol. 2003;54:51–106. doi: 10.1016/s0074-7742(03)54003-x. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde signaling by endocannabinoids. Curr Opin Neurobiol. 2002;12:324–330. doi: 10.1016/s0959-4388(02)00328-8. [DOI] [PubMed] [Google Scholar]
- Lanthorn TH, Cotman CW. Baclofen selectively inhibits excitatory synaptic transmission in the hippocampus. Brain Res. 1981;225:171–178. doi: 10.1016/0006-8993(81)90326-7. [DOI] [PubMed] [Google Scholar]
- Ledo A, Frade J, Barbosa RM, Laranjinha J. Nitric oxide in brain: diffusion, targets and concentration dynamics in hippocampal subregions. Mol Aspects Med. 2004;25:75–89. doi: 10.1016/j.mam.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Lewis PR, Shute CC. The cholinergic limbic system: projections to hippocampal formation, medial cortex, nuclei of the ascending cholinergic reticular system, and the subfornical organ and supra-optic crest. Brain. 1967;90:521–540. doi: 10.1093/brain/90.3.521. [DOI] [PubMed] [Google Scholar]
- Louie K, Wilson MA. Temporally structured replay of awake hippocampal ensemble activity during rapid eye movement sleep. Neuron. 2001;29:145–156. doi: 10.1016/s0896-6273(01)00186-6. [DOI] [PubMed] [Google Scholar]
- Lowenstein CJ, Dinerman JL, Snyder SH. Nitric oxide: a physiologic messenger. Ann Intern Med. 1994;120:227–237. doi: 10.7326/0003-4819-120-3-199402010-00009. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Manabe T, Nicoll RA. Release of adenosine by activation of NMDA receptors in the hippocampus. Science. 1994;265:2098–2101. doi: 10.1126/science.7916485. [DOI] [PubMed] [Google Scholar]
- Markram H, Segal M. Acetylcholine potentiates responses to N-methyl-D-aspartate in the rat hippocampus. Neurosci Lett. 1990;113:62–65. doi: 10.1016/0304-3940(90)90495-u. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RGM. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Mitchell JB, Lupica CR, Dunwiddie TV. Activity-dependent release of endogenous adenosine modulates synaptic responses in the rat hippocampus. J Neurosci. 1993;13:3439–3447. doi: 10.1523/JNEUROSCI.13-08-03439.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RGM, Andersen E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Olpe HR, Baudry M, Fagni L, Lynch G. The blocking action of baclofen on excitatory transmission in the rat hippocampal slice. J Neurosci. 1982;2:698–703. doi: 10.1523/JNEUROSCI.02-06-00698.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, Eichenbaum H, Weiner SI, Wible CG. Learning-related patterns of CA1 spike trains parallel stimulation parameters optimal for inducing hippocampal long-term potentiation. Hippocampus. 1991;1:181–192. doi: 10.1002/hipo.450010206. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Greenstein YJ, Grudman M, Winson J. Long-term potentiation in the dentate gyrus is induced preferentially on the positive phase of theta rhythm. Brain Res. 1988;439:383–387. doi: 10.1016/0006-8993(88)91499-0. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. Calcium concentration dynamics produced by synaptic activation of CA1 hippocampal pyramidal cells. J Neurosci. 1992;12:4202–4223. doi: 10.1523/JNEUROSCI.12-11-04202.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Inhibition of quantal transmitter release in the absence of calcium influx by a G protein-linked adenosine receptor at hippocampal synapses. Neuron. 1992;8:1139–1150. doi: 10.1016/0896-6273(92)90134-y. [DOI] [PubMed] [Google Scholar]
- Schubert P, Mitzdorf U. Analysis and quantitative evaluation of the depressive effect of adenosine on evoked potentials in hippocampal slices. Brain Res. 1979;172:186–190. doi: 10.1016/0006-8993(79)90910-7. [DOI] [PubMed] [Google Scholar]
- Sekino Y, Koyama I. Selective inhibition of homosynaptic depression in a tetanized pathway by an adenosine A1 blocker in the CA1 region of rat hippocampal slice. Neurosci Lett. 1992;148:109–113. doi: 10.1016/0304-3940(92)90816-p. [DOI] [PubMed] [Google Scholar]
- Sheridan RD, Sutor B. Presynaptic M1 muscarinic cholinoceptors mediate inhibition of excitatory synaptic transmission in the hippocampus in vitro. Neurosci Lett. 1990;108:273–278. doi: 10.1016/0304-3940(90)90653-q. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Dingledine R. Presynaptic inhibitory effect of acetylcholine in the hippocampus. J Neurosci. 1981;1:784–792. doi: 10.1523/JNEUROSCI.01-07-00784.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Winson J, Abzug C. Neuronal transmission through hippocampal pathways dependent on behavior. J Neurophysiol. 1978;41:716–732. doi: 10.1152/jn.1978.41.3.716. [DOI] [PubMed] [Google Scholar]
- Yamakura T, Shimoji K. Subunit- and site-specific pharmacology of the NMDA receptor channel. Prog Neurobiol. 1999;59:279–298. doi: 10.1016/s0301-0082(99)00007-6. [DOI] [PubMed] [Google Scholar]
- Yeckel MF, Berger TW. Spatial distribution of potentiated synapses in hippocampus: dependence on cellular mechanisms and network properties. J Neurosci. 1998;18:438–450. doi: 10.1523/JNEUROSCI.18-01-00438.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]