

Abstract
Autophagic and endocytic pathways are tightly regulated membrane rearrangement processes that are crucial for homeostasis, development and disease. Autophagic cargo is delivered from autophagosomes to lysosomes for degradation through a complex process that topologically resembles endosomal maturation. Here, we report that a Beclin1-binding autophagic tumour suppressor, UVRAG, interacts with the class C Vps complex, a key component of the endosomal fusion machinery. This interaction stimulates Rab7 GTPase activity and autophagosome fusion with late endosomes/lysosomes, thereby enhancing delivery and degradation of autophagic cargo. Furthermore, the UVRAG-class-C-Vps complex accelerates endosome–endosome fusion, resulting in rapid degradation of endocytic cargo. Remarkably, autophagosome/endosome maturation mediated by the UVRAG-class-C-Vps complex is genetically separable from UVRAG–Beclin1-mediated autophagosome formation. This result indicates that UVRAG functions as a multivalent trafficking effector that regulates not only two important steps of autophagy — autophagosome formation and maturation — but also endosomal fusion, which concomitantly promotes transport of autophagic and endocytic cargo to the degradative compartments.
Autophagy is a tightly regulated membrane rearrangement process that ensures lysosome-dependent bulk degradation of cytosolic proteins or organelles, and is highly conserved in eukaryotic cells, as seen with the endocytic pathway1. In response to environmental stresses, portions of cytoplasmic constituents are engulfed by a unique membrane structure, the phagophore, as it elongates to form a double- or multiple-membrane-bound compartment called the autophagosome. Newly synthesized autophagosomes then undergo extensive remodelling to acquire degradative capabilities. The remodelling process, also known as autophagosomal maturation, involves sequential fusion of autophagosomes with endocytic vesicles (early and late endosomes) and lysosomes, producing degradative autolysosomes. The sequestered material is then degraded into building blocks for synthesis of macromolecules and energy production1-3.
Although the autophagic pathway has been studied extensively, little is known about the molecular mechanism underlying the conversion of autophagosome to degradative autolysosome2. It is generally thought that autophagosomal maturation probably has similar features as the progression of endosomes to lysosomes, a complex process that involves a number of vesicle-trafficking components4-9. In fact, inhibition of lysosomal fusion by depletion of lysosomal membrane proteins or lysosomal enzymes inhibits both autophagic and endocytic degradation10-12. Furthermore, morphologic convergence between autophagic and endocytic pathways has been frequently observed at the ‘early and late endosomes’ node, as well as lysosomes in the cell13-15. Thus, autophagy, particularly the late stages of autophagic maturation, may interconnect with the endocytic pathway by sharing similar machinery for the concomitant lysosome fusion process.
A prerequisite for vesicle fusion is vesicle tethering. The tethering events at the yeast endosome/vacuole (equivalent to mammalian lysosomes) have been thoroughly studied and shown to require the class C vacuolar protein sorting (Vps) complex16-19. The core class C Vps complex (hereafter referred to as C-Vps), including Vps11, Vps16, Vps18 and Vps33 exists into two configurations: the HOPS complex (for homotypic vacuole fusion and protein sorting), which contains two additional subunits (Vps39/Vam6 and Vps41), acts at the vacuole20,21, whereas the class C core vacuole/endosome tethering (CORVET) complex has Vps3 (human Vps39 (hVps39) homologue) and Vps8 (Vps41 homologue) instead, and functions at the endosome17. Both complexes interact with Ypt–Rab GTPase and are thought to couple Rab activation and SNAREs (soluble N-ethylmaleimide-sensitive fusion protein attachment receptors) assembly during fusion17,22. Although the specific function of each individual subunit remains to be established, Vps39 has been shown to confer GTPase exchange factor (GEF) activity to Ypt7p, the yeast Rab7 orthologue. In mammalian cells, hVps39 regulates the recruitment/activation of Rab7 onto the Rab5-labelled early endosomes, a process called Rab conversion23,24. Although the role of C-Vps in mammals is unclear, mutations in the Drosophila melanogaster homologues of C-Vps components induce defects in lysosomal protein transport as well as autophagosome maturation25,26, suggesting that the role of C-Vps may be conserved in multicellular organisms.
Previously we reported the identification of a UV-irradiation-resistance-associated gene (UVRAG) as a Beclin1-interacting protein27. UVRAG association with Beclin1 enhances phophatidylinositol-3-OH kinase class III (PI(3)KC3) activity and induces autophagosome formation27. In our continuing effort to characterize the functions of UVRAG, we report here that UVRAG interacts with the C-Vps tethering complex. Independently of Beclin1, UVRAG interaction with C-Vps stimulates autophagosome maturation and endosomal fusion, thereby enhancing both autophagic and endocytic protein degradation. Thus, our study provides a mechanism for regulation of UVRAG-mediated autophagy, and it extends the function of UVRAG from an autophagy effector to an important factor in multiple membrane-trafficking events.
RESULTS
UVRAG interacts with C-Vps in vivo
Full-length UVRAG is predicted to contain an amino-terminal prolinerich (PR) region (residues 1–41) that mediates Bif-1 interaction, followed by a putative Ca2+-dependent phospholipid binding C2 domain (residues 42–147), a central Beclin1-binding coiled-coil domain (CCD, residues 200–269), and a carboxy-terminal region (residues 270–699) of unidentified function27,28. In an attempt to identify protein(s) other than Beclin1 and Bif-1 that are important for the biological roles of UVRAG, we expressed the distinct C-terminal region of UVRAG (residues 270–699) as a GST fusion protein in 293T cells. Two cellular polypeptides with relative molecular weights of 92,000 and 103,000 (Mr 92K and 103K) that specifically bound to GST–UVRAG270–699, but not to GST alone, were affinity purified and identified by mass spectrometry as the C-Vps proteins Vps16 (92K) and Vps11 (103K) (Supplementary Information, Fig. S1a). Vps16 and Vps11 have been shown to interact physically with Vps18 and Vps33 to form C-Vps, which is required for homotypic fusion at yeast vacuoles18,19,23,29. To confirm their interactions with UVRAG, 293T cells were transfected with Flag–UVRAG and HA-tagged Vps16, Vps18, Vps11, Vps33 or Vps39. Co-immunoprecipitation showed efficient interactions of UVRAG with Vps16, Vps18, Vps11, Vps33 or Vps39 (Fig. 1a). Analogous results were obtained in the reciprocal co-immunoprecipitation (Supplementary Information, Fig. S1b). The interaction between endogenous UVRAG and Vps16, Vps11 and Vps18 was also readily detected (Fig. 1b). Despite their apparent interactions, increasing amounts of UVRAG and Vps33 expression did not affect their interaction with Vps16 at detectable levels (Supplementary Information, Fig. S1). These findings indicate that UVRAG efficiently associates with C-Vps without affecting its subunit stoichiometry in living cells.
Figure 1. Interaction and colocalization of UVRAG with C-Vps.
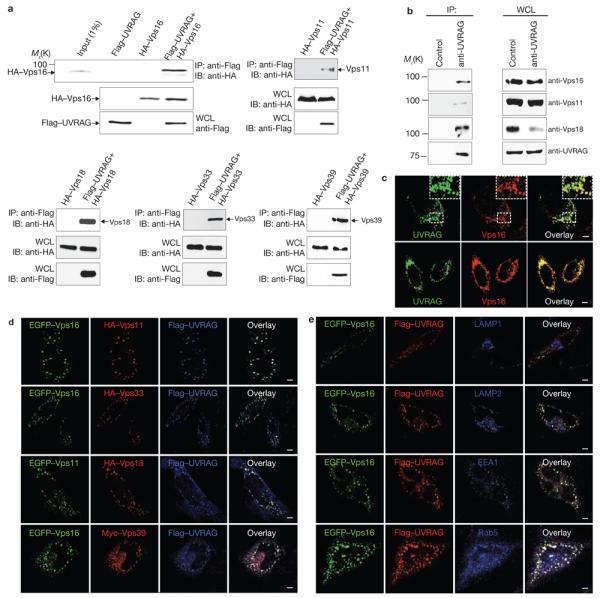
(a) UVRAG interaction with C-Vps. 293T cells were co-transfected with Flag–UVRAG and HA–Vps16, HA–Vps11, HA–Vps18, HA–Vps33 or HA–Vps39. Whole-cell lysates (WCLs) were used for immunoprecipitation (IP) with an anti-Flag antibody, followed by immunoblotting (IB) with an anti-HA antibody. 1% WCL was used as the input. (b) Interaction between endogenous UVRAG and C-Vps subunits. WCLs of 293T cells were used for IP with control serum (control) or an anti-UVRAG antibody, followed by IB with the indicated antibodies. The right panel shows endogenous protein expression. (c) Confocal microscopy analysis of the colocalization of endogenous UVRAG and Vps16 or Vps18 in HeLa cells. Insets highlight colocalization. (d) Confocal microscopy analysis of the colocalization of UVRAG with C-Vps subunits in HeLa cells transfected with Flag–UVRAG, and epitope-tagged C-Vps subunits as indicated. (e) Colocalization of UVRAG–C-Vps in early endosomes. HeLa cells were co-transfected with EGFP–Vps16 and Flag–UVRAG, stained with antibodies to LAMP1, LAMP2, EEA1 and Rab5, followed by confocal microscopy. All images are representative of at least three independent experiments. Scale bars, 5 μm. The raw data for a and b are shown in Supplementary Information, Fig. S6
Subcellular localization of UVRAG and C-Vps in early endosomes
We next investigated the subcellular localization of C-Vps proteins and UVRAG. Epitope-tagged Vps16, Vps18, Vps11 and Vps33 showed punctate staining and extensive colocalization with EEA1 and p40(phox)PX–GFP, a probe for phosphatidylinositol-3-phosphate (PtdIns(3)P)-enriched early endosomes30, but not with the late endosome or lysosome markers LAMP1, LAMP2, cathepsin D and CD63 (Supplementary Information, Fig. S2a, b). Significant colocalizations were also observed for the epitope-tagged C-Vps subunits with each other, having cytoplasmic punctate patterns reminiscent of endosome staining (Supplementary Information, Fig. S2c). Endogenous and epitope-tagged UVRAG were consistently observed outlining EEA1+ and Rab5+ early endosomal compartments, but not with the late endosomal markers (Supplementary Information, Fig. S2). Notably, deletion of the N-terminal phospholipid-binding C2 domain (UVRAGΔN(1–147)) abolished the endosomal distribution of UVRAG, suggesting that the C2 region is probably required for directing UVRAG endosomal localization (Supplementary Information, Fig. S2f).
We next evaluated the spatial correlation of UVRAG and C-Vps proteins in HeLa cells. Endogenous and epitope-tagged UVRAG colocalized with Vps16 or Vps18 in a perinuclear region (Fig. 1c, d). Furthermore, the punctate staining of UVRAG and C-Vps proteins did not co-distribute with LAMP1 and LAMP2 but colocalized extensively with EEA1+ and Rab5+ early endosomes (Fig. 1e). These results indicate that UVRAG associates with C-Vps primarily in early endosomal compartments.
Domains of UVRAG associate with C-Vps
To map the specific domains of UVRAG required for C-Vps interaction, a series of UVRAG deletion mutants was constructed for co-immunoprecipitation (Fig. 2e). It showed that either the C2 (UVRAGC2) or the C-terminal domain (UVRAG270-CT) alone was sufficient to bind Vps16, whereas the CCD (UVRAGCCD) and PR region were not (Fig. 2a, e and data not shown). Moreover, a small region of the C-terminal domain encompassing residues 270–442 (UVRAG270–442), but not residues 443–583 (UVRAG443–583) and residues 584–699 (UVRAG584–699), seemed to sufficiently interact with Vps16 (Fig. 2c, e). Similar results were obtained for these mutants with other C-Vps subunits (data not shown). Consistently, we observed near-complete colocalization of UVRAGC2 or UVRAG270-CT mutant with Vps16, whereas UVRAGCCD failed to colocalize with Vps16 (Fig. 2b).
Figure 2. Mapping of Vps16 binding domains of UVRAG.

(a) The N-terminal C2 domain or the C-terminal region of UVRAG interacts individually with Vps16. 293T cells were co-transfected with HA–Vps16, together with vector, Flag–UVRAG or Flag–UVRAG mutants. WCLs were immunoprecipitated with anti-Flag followed by immunoblotting with an anti-HA antibody. (b) HeLa cells were transfected with EGFP–Vps16 together with Flag–UVRAG mutants as indicated and stained with anti-Flag (red), followed by confocal microscopy. Scale bars, 5 μm. (c) Interactions of the UVRAG C-terminal truncated mutants with Vps16. At 48 h post-transfection with HA–Vps16 and Flag–UVRAG C-terminal mutants, 293T WCLs were immunoprecipitated with anti-Flag, followed by immunoblotting with anti-HA. (d) UVRAG C2 domain or C-terminal region deletion abolishes Vps16 binding. At 48 h post-transfection with HA–Vps16 together with Flag–UVRAG or its mutants, 293T WCLs were immunoprecipitated with anti-Flag followed by immunoblotting with an anti-HA antibody. (e) Schematic representation of UVRAG and its deletion mutants. ++, strong binding; +, weak binding; −, no binding. The raw data for a, c and d are shown in Supplementary Information, Fig. S6
To further define UVRAG interaction with Vps16, UVRAG mutants containing deletions of C2, CCD or the entire C-terminal region were constructed for binding studies. The UVRAGΔCCD mutant bound Vps16 as efficiently as wild-type UVRAG, whereas UVRAGΔN(1–147) and UVRAGΔ270-CT showed substantially reduced Vps16 binding (Fig. 2d). This indicates that although the C2 or C-terminal region of UVRAG binds Vps16 sufficiently, the presence of the central Beclin1-binding CCD domain suppresses their Vps16 binding activity (Fig. 2d). However, Beclin1 overexpression did not negatively affect the UVRAG–C-Vps interaction (Supplementary Information, Fig. S1e). Furthermore, both UVRAGΔN(1–147) and UVRAGΔ270-CT mutants retained efficient Beclin1-binding capability (data not shown). Thus, the weak Vps16 binding of UVRAGΔN(1–147) and UVRAGΔ270-CT mutants was probably not due to either their Beclin1 interaction or misfolding. Nevertheless, these results demonstrate that UVRAG contains two separate regions for Vps16 binding that are genetically distinct from Beclin1 interaction.
UVRAG enhances association of C-Vps with autophagosomes
Although UVRAG is part of the Beclin1–PI(3)KC3 complex for autophagy induction27, its direct association with autophagosomes has not yet been addressed. To this end, we expressed Flag–UVRAG, in combination with GFP–LC3, which specifically labels autophagic membranes31, in HeLa cells. Rapamycin treatment caused a rapid shift in GFP–LC3 staining from a diffuse cytosolic/nuclear staining to a punctate pattern outlining autophagic vacuoles in which UVRAG colocalized substantially (Supplementary Information, Fig. S3a). To analyse whether UVRAG affected the distribution of C-Vps when autophagy was induced, HA–Vps16 and GFP–LC3 were expressed in HeLa. Vector (HeLa.Vec) and HeLa.UVRAG cells (Fig. 3a). More than 70% of the LC3-labelled autophagosomes colocalized with HA–Vps16 in HeLa.UVRAG cells, whereas only partial colocalization was observed in HeLa.Vec cells (Fig. 3a). Similar results were obtained in HCT116 cells, in which UVRAG expression is much lower due to a mono-allelic frameshift mutation32 (data not shown). In addition, small interference RNA (siRNA)-mediated depletion of UVRAG caused a significant reduction in the colocalization of GFP–LC3 with Vps16, suggesting that UVRAG expression effectively facilitates the recruitment of Vps16 onto autophagosome membranes (Supplementary Information, Fig. S3b). However, Vps16 depletion in HCT116 and HeLa cells stably expressing vector or UVRAG showed no discernable effect on UVRAG-mediated autophagosome formation (Fig. 3b). These data indicate that UVRAG efficiently recruits C-Vps to LC3+ autophagosomes; but C-Vps does not appear to act on UVRAG-mediated autophagosome formation.
Figure 3. UVRAG recruits C-Vps protein to the GFP–LC3-labelled autophagosomes.
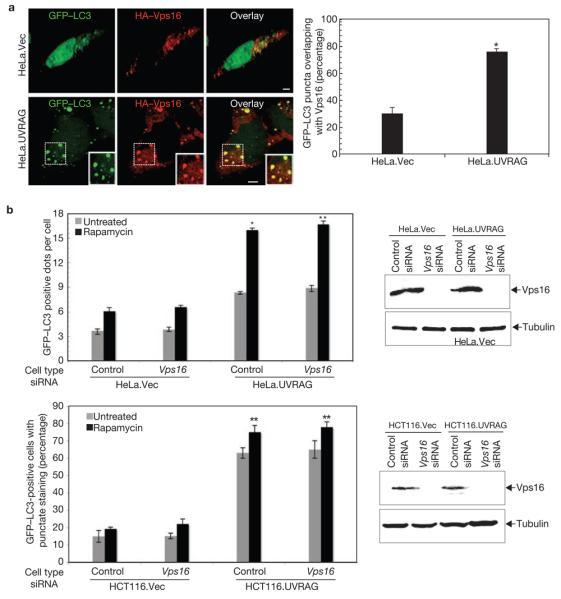
(a) HeLa.Vec and HeLa.UVRAG cells transfected with HA–Vps16 and GFP–LC3 were treated with 2 μM rapamycin for 2 h, and processed for confocal microscopy (left panel; scale bars, 5 μm). The percentage of GFP–LC3 punctae positive for HA–Vps16 staining was quantified (right panel; data are mean ± s.e.m., n = 60, *P < 0.01). (b) Effects of Vps16 on UVRAG-mediated autophagosome formation. Light microscopic quantification of autophagosomes in HeLa.Vec and HeLa.UVRAG cells (upper left panel) or in HCT116.Vec and HCT116.UVRAG cells (bottom left panel) when transfected with GFP–LC3 together with control siRNA or Vps16 siRNA (data are mean ± s.e.m., n = 60 for HeLa cells; n = 450 for HCT116 cells, *P < 0.01; **P < 0.001). Immunoblotting of Vps16 and tubulin are shown in the right panel.
Autophagosome maturation in UVRAG-expressing cells
Given that C-Vps in yeast acts as a tethering factor in vesicle fusion33, and that mutations in Drosophila homologues of C-Vps subunits induce autophagosome maturation defects25,26,34,35, we initially assessed the effects of UVRAG on autophagosome maturation by measuring the colocalization efficiency of GFP–LC3-labelled autophagosomes with LAMP1-stained late endosomes/lysosomes (Fig. 4a). The percentage of LAMP1+/LC3+ co-stained autophagosomes was significantly higher in HeLa.UVRAG than in HeLa.Vec cells, and was further enhanced by autophagy induction (Fig. 4a). However, treatment with bafilomycin A1, a specific inhibitor of vacuolar H+-ATPase, blocked the UVRAG-induced autophagosome maturation (Fig. 4a). Similar effects were observed when CD63 late endosomal protein was used as an autophagosome maturation marker (Fig. 4b). As detected in HeLa cells, the fusion efficiency of GFP–LC3 with LAMP1+ or acidotropic LysoTracker Red+ late endosomes/lysosomes was approximately doubled in HCT116.UVRAG, compared with HCT116.Vector (HCT116.Vec) cells (Supplementary Information, Fig. S3c). Additionally, LC3+ vesicles showed higher proteolytic activity in UVRAG-expressing cells when compared with the control, as illustrated by enhanced staining with DQ-Red bovine serum albumin (BSA) whose fluorescence dequenches upon proteolysis (Supplementary Information, Fig. S3d). Furthermore, the proteolysis rate of long-lived proteins in HeLa.UVRAG and HCT116.UVRAG cells during starvation was significantly accelerated, compared with that in control cells, but was markedly suppressed by bafilomycin A1 (Fig. 4c; data not shown). Together, these data indicate that UVRAG expression enhances autophagosome maturation or autophagic flux.
Figure 4. Autophagosome maturation in UVRAG-expressing HeLa cells.

(a) UVRAG enhances the colocalization efficiency of GFP–LC3 with LAMP1. HeLa.Vec and HeLa.UVRAG cells transfected with GFP–LC3 were either untreated or treated with rapamycin (2 μM) in the absence or presence of bafilomycin A1 (0.1 μM) and stained for LAMP1. Insets highlight the colocalization. The percentage of LAMP1-positive autophagosomes was calculated in each setting (data are mean ± s.e.m., n = 200; 5 independent experiments, **P < 0.01; *P < 0.05). (b) UVRAG enhances the acquisition of CD63 by autophagosomes. The GFP–LC3-transfected HeLa.Vec and HeLa.UVRAG cells were treated as described in a and stained for CD63 and the percentage of CD63-positive autophagosomes was calculated (data are mean ± s.e.m., n = 200, **P < 0.01; *P < 0.05). (c) UVRAG promotes autophagic degradation of long-lived proteins. HeLa.Vec and HeLa.UVRAG cells were incubated for 16 h with l-3H-Leu (1 μCi ml−1). The degradation of long-lived proteins was measured at the indicated time points in complete medium (NC), EBSS alone (Starv.), or EBSS + 0.1 μM bafilomycin A1 (Starv.+BalfA1). Results (data are mean ± s.e.m. of triplicates) are representative of 2 independent experiments. Scale bars, 5 μm.
The roles of Beclin1 and C-Vps in UVRAG-mediated autophagosome maturation
As UVRAG interacts directly with Beclin1 through its CCD domain to induce autophagy, we evaluated whether Beclin1 interaction influenced UVRAG-mediated autophagosome maturation. HeLa and HCT116 cells expressing wild-type UVRAG, UVRAGΔCCD or UVRAGCCD mutant were examined for autophagosome maturation level by measuring the colocalization efficiency of LC3+ vesicles with LAMP1+ late endosomes/lysosomes. Consistent with our previous findings27, the UVRAGΔCCD mutant, which lost Beclin1 binding, failed to induce autophagosome formation (Fig. 5a; Supplementary Information, Fig. S4). In contrast, the UVRAGΔCCD mutant, which is able to interact with C-Vps, promoted autophagosome maturation as efficiently as wild-type UVRAG (Fig. 5a; Supplementary Information, Fig. S4). In addition, the UVRAGCCD mutant did not bind C-Vps and failed to induce autophagosome maturation (Fig. 5a; Supplementary Information, Fig. S4). Moreover, siRNA-mediated knockdown of Beclin1 led to a marked reduction of UVRAG-mediated autophagosome formation, but had a minimal effect on its autophagosome maturation activity (Supplementary Information, Fig. S3e). These results indicate that Beclin1 interaction and expression are not necessary for UVRAG-mediated autophagosome maturation.
Figure 5. UVRAG-mediated autophagosome maturation is dependent on C-Vps, but not Beclin1.

(a) Colocalization of GFP–LC3 and LAMP1. HeLa cells expressing wild-type or mutant UVRAG were transfected with GFP–LC3 and incubated under normal conditions or treated with rapamycin (2 μM). Autophagosome fusion with LAMP+ structures was analysed by confocal microscopy (left panel; rapamycin-treated) and quantified (right upper panel; data are mean ± s.e.m., n = 100). Arrows and insets show the LAMP1+ autophagosomes. The number of GFP–LC3-positive dots per cell was counted using a fluorescence microscope (right bottom panel; data are mean ± s.e.m., n = 60, *P < 0.05; **P < 0.0001). (b) Vps16 or Vps18 knockdown inhibits UVRAG-mediated autophagosome maturation. HeLa.Vec (left) and HeLa.UVRAG (right) cells were transfected with GFP–LC3 together with control siRNA, Vps16 siRNA, or Vps18 siRNA, followed by confocal microscopy. Arrows in the top panel denote the autophagosomes with LAMP1 staining. Immunoblotting of Vps16, or Vps18 and tubulin are shown in the middle panel. The percentage of LAMP1+ autophagosomes was calculated (data are mean ± s.e.m., n = 50, *P < 0.05). Scale bars, 5 μm. The raw data of the immunoblots are shown in Supplementary Information, Fig. S6.
We next determined the role of C-Vps in UVRAG-mediated autophagosome maturation. Treatment of HeLa cells with vps16 siRNA, but not with scrambled siRNA, resulted in suppression of autophagosome maturation (>30%, Fig. 5b). A nearly identical phenotype was observed when vps18 was depleted (Fig. 5b). To further illustrate the functional significance of the UVRAG–Vps16 interaction in UVRAG-mediated autophagosome maturation, we constructed a UVRAG mutant with the specific deletion of its C2 region (residues 42–147, UVRAGΔC2(42–147)), which substantially lost Vps16 binding but retained Beclin1 binding (Fig. 2e, data not shown; ref.36). Although the UVRAGΔC2(42–147) mutant retained autophagosome formation activity, its ability to promote autophagosome maturation was severely impaired, compared with wild-type UVRAG, suggesting that efficient binding to C-Vps is necessary for UVRAG to fully facilitate autophagosome maturation (Supplementary Information, Fig. S4 and data not shown). Collectively, these results indicate that UVRAG facilitates autophagosome maturation through its interaction with C-Vps, which is independent of Beclin1.
Autophagosome recruitment and activation of Rab7 by UVRAG
The completion of autophagosome maturation requires the small GTPase, Rab7 (refs 9, 36). To further understand how UVRAG–C-Vps interaction promotes autophagosome maturation, we evaluated the intracellular distribution and GTPase activity of Rab7 when UVRAG was expressed. Both GFP–Rab7 and RFP–LC3 showed punctate staining in the cytoplasm; however, UVRAG-expressing cells showed an approximately 2-fold increase in colocalization of GFP–Rab7 and RFP–LC3+ autophagosomes, compared with the control; this further increased up to 70% when autophagy was induced (Fig. 6a). A most noticeable feature in cells expressing UVRAG was that GFP–Rab7 was often present as large, ring-shaped vesicles surrounding the RFP–LC3+ autophagosomes . The ‘swollen’ morphology of GFP–Rab7+ vesicles observed in UVRAG-expressing cells was similar to that observed in cells expressing a constitutively active Rab7Q67L mutant (data not shown). Indeed, Rab7 GTPase activity was substantially higher with UVRAG expression than with vector or Vps16-binding deficient UVRAGΔN(1–147) mutant expression (Fig. 6b). Previous studies37,38 indicate that Rab7 activation leads to efficient recruitment of its downstream effector RILP (Rab7-interacting lysosomal protein); the GTP-bound, active form of Rab7 interacts specifically with the Rab7-binding domain (RBD) of RILP. To further substantiate UVRAG-mediated Rab7 activation, a mammalian GST–RILPRBD fusion protein containing the RBD of RILP was tested for Rab7 interaction in HCT116. vec, HCT116.UVRAG cells or HCT116.UVRAGΔN(1–147) cells. It showed that GST–RILPRBD interaction with Rab7 in HCT116.UVRAG cells was significantly higher than in vector-expressing or mutant cells (Fig. 6c). These data indicate that UVRAG–C-Vps interaction promotes efficient autophagosome recruitment and activation of Rab7 GTPase, which may in part facilitate autophagosome maturation.
Figure 6. Rab7 acquisition and activation by UVRAG.

(a) UVRAG expression promotes the recruitment of Rab7 GTPase to autophagosomes. HeLa. Vec and HeLa.UVRAG cells transfected with GFP–Rab7 and RFP–LC3 were maintained either under normal conditions (NC) or treated with rapamycin, followed by confocal microscopy. Insets highlight the Rab7 acquisition of autophagosomes. The percentage of Rab7+-autophagosomes was quantified (right panel; data are mean ± s.e.m., n = 60, ***P < 0.001; **P < 0.01). Scale bars, 5 μm. (b) Rab7 GTPase activity is increased with UVRAG expression. At 48 h post-transfection with GFP–Rab7 together with vector, UVRAG or UVRAGΔN(1–147) vector, 293T WCLs were used for immunoprecipitation with anti-GFP, followed by the Rab7 GTPase activity assay. Left panel: Autoradiographs of the GTP hydrolysis products analysed by TLC. Right panel: Quantification of the percentage of GTP hydrolyzed by Rab7 (data are mean n = 2 independent experiments). (c) Increased Rab7–RILP interaction UVRAG is expressed. HCT116.vector, HCT116.UVRAG and HCT116.UVRAGΔN(1–147) cells were transfected with GFP–Rab7 together with the GST-tagged Rab7 binding domain of RILP (GST–RILPRBD, residues 243–318). WCLs were used for GST pulldown, followed by immunoblotting with an anti-GFP antibody. The raw data of the immunoblots are shown in Supplementary Information, Fig. S6.
UVRAG enhances endocytic trafficking
Given that the autophagic pathway converges on the endocytic pathway at the endosome node for lysosome-dependent degradation13-15, we hypothesized that UVRAG is functionally linked to endocytic processes through its interaction with C-Vps. To test this, we examined the effect of UVRAG expression on epidermal growth factor (EGF)-stimulated endocytic transport to lysosomes in HCT116 cells. When stimulated, comparable amounts of fluorescent EGF were initially bound on the surface of HCT116.vec and HCT116.UVRAG cells (Fig. 7a). At 15 min post-stimulation, EGF was found in small punctate structures dispersed throughout the cytoplasm in HCT116.Vec, whereas in HCT116.UVRAG cells it was present as cytoplasmic aggregates that were extensively co-stained with LAMP1 (Fig. 7a). This difference became more evident at 30 min post-stimulation: essentially, most or all EGF accumulated in the juxtanuclear LAMP1+ lysosomal compartments in HCT116.UVRAG cells (Fig. 7a). Notably, the amount of internalized EGF was similar for vector- and UVRAG-expressing cells (Supplementary Information, Fig. S5a). Consistent with this notion, HCT116.UVRAG cells showed a marked increase in the rate of EGF receptor (EGFR) degradation, compared with HCT116.UVRAGδN(1–147) or HCT116.vec, whereas UVRAG depletion reduced EGFR degradation in HeLa cells (Fig. 7b; Supplementary Information, Fig. S5b). To further comfirm this, we measured the kinetics of endocytic DQ–Green BSA processing in macrophage RAW264.7 cells stably expressing vector or UVRAG. Consistently, UVRAG overexpression accelerated the DQ-Green BSA degradation rate without affecting the total amount of internalized BSA (Fig. 7c; data not shown). Overall, these findings indicate that UVRAG accelerates the intracellular trafficking of endocytic cargo to late endosomes/lysosomes for subsequent degradation, and that this UVRAG activity requires interaction with C-Vps.
Figure 7. UVRAG promotes endocytic trafficking.

(a) EGF-stimulated endocytosis and post-endocytic trafficking. Uptake of Alexa Fluor 488–EGF (green) by HCT116.Vec (left panel) and HCT116.UVRAG (right panel) cells was followed by confocal microscopy. At the indicated times, cells were fixed and stained with antibodies to LAMP1 (red), and Topro-3 (blue, to stain the nucleus). Magnified images of the insets (fourth row) highlight trafficking of EGF towards LAMP1-positive structures. (b) EGFR degradation. HCT116. Vec, HCT116.UVRAG and HCT116.UVRAGΔN(1–147) cells were treated with EGF (200 ng ml−1) at 37 °C for the indicated times. WCLs were subjected to immunoblotting with an anti-EGFR antibody. EGFR at each time point was quantified and indicated as a percentage relative to that at time zero (right panel; data are mean ± s.e.m., n = 3). (c) DQ-green BSA dequenching assay. Representative confocal microscopy images of DQ-BSA proteolysis (left panel) and flow cytometry analysis (right panel) of RAW264.7 cells expressing an empty vector (RAW264.7.Vec; dotted black line), wild-type UVRAG (red line) or UVRAGΔN(1–147) mutant (green line) loaded with 10 μg ml−1 DQ green–BSA for the indicated times. Background (grey line) represents samples without the BSA load. (d) Effect of UVRAG on endosome fusion. RAW264.7.Vec and RAW264.7.UVRAG cells were pulse-labelled with Oregon green 514–avidin and dextran-568 for 10 min and chased for 30 min. Cells were then labelled with biotin–BSA for 10 min, and processed for confocal microscopy (left panel). In the absence of biotin–BSA, Oregon green 514–avidin showed very low fluorescence (first row of confocal image). The percentage of dextran-568-labelled endosomes with visible Oregon green staining (denoted by arrows in the left panel) was calculated from approximately 20 different cells in 2 independent experiments (mean, right panel). Scale bars, 5 μm. The raw data of the immunoblots in b are shown in the Supplementary Information, Fig. S6.
UVRAG promotes endosomal fusion in vitro and in vivo
We next investigated whether the stimulation of endocytic trafficking induced by UVRAG–C-Vps interaction was due to increased endosomal fusion. To test this, we first used a sensitive fluorometric endosome fusion approach in living cells, taking advantage of the irreversible fluorescence enhancement (>15-fold) of Oregon green 514-conjugated avidin when avidin-labelled endosomes fuse with biotin-labelled endosomes in vivo. In this assay, RAW264.7.Vec and RAW264.7.UVRAG cells were pulse-labelled with fluorescent avidin and biotinylated-BSA sequentially. The fluid-phase marker Alexa Fluor 568–dextran was used as an internal control for endocytosis. The ability of biotin-labelled endosomes to fuse with avidin-labelled endosomes was quantified and normalized with the internalized dextran. A greater fraction of endosomes in UVRAG-expressing cells carried bright green fluorescence, compared with the control (Fig. 7d), suggesting that UVRAG expression enhances endosome–endosome fusion in vivo. The in vitro endosome fusion assay using avidin-conjugated alkaline phosphatase (avidin–ALP)-labelled and biotinylated rabbit immunoglobulin (IgG–biotin)-labelled endosomes in an ATP-regenerating versus ATP-depletion system was also conducted. UVRAG expression increased ATP-dependent endosomal fusion activity by approximately 20% (see Supplementary Information, Fig. S5c). These results indicate that UVRAG–C-Vps interaction stimulates endosome–endosome fusion, resulting in rapid endocytic trafficking and degradation of cargo molecules in lysosomes.
DISCUSSION
Here, we have demonstrated that UVRAG, a Beclin1-binding protein, also interacts with C-Vps and that this interaction promotes autophago-some maturation as well as endocytic vesicle trafficking. Several results indicate that the effect mediated by UVRAG–C-Vps interaction is largely separable from that mediated by the UVRAG–Beclin1 interaction. First, the domain of UVRAG required for Vps16 binding is genetically distinct from that required for Beclin1 binding. In fact, Beclin1 does not compete with C-Vps for binding UVRAG. Second, it has been reported that endogenous Beclin1–PI(3)KC3 localizes predominantly to the trans-Golgi network39, whereas C-Vps proteins are largely endosome-associated29,40,41. Third, although UVRAG recruits C-Vps into autophagosomes when autophagy is induced, the recruited C-Vps proteins do not accordingly enhance UVRAG-mediated autophagosome formation, which is dependent primarily on Beclin1 activity27. These observations are also consistent with previous findings that mutations in yeast Vps33 and Vps18 still allow autophagosome formation; but block their fusion with vacuoles42,43. Finally, a UVRAG mutant that was defective in Beclin1 binding but retained C-Vps interaction was able to mediate efficient assembly of the LC3+ and LAMP1+ structures. In contrast, a UVRAG mutant that was unable to bind C-Vps, but was able to bind Beclin1, showed markedly attenuated ability to promote autophagosome maturation. These results indicate that the role of UVRAG–C-Vps interaction is clearly different from UVRAG–Beclin1 interaction in the autophagy pathway.
Our data thus indicate that UVRAG is involved in the two distinct steps of the autophagic pathway (Fig. 8). At an early stage, UVRAG interacts with Beclin1 and this interaction promotes autophagosome formation. During the later stages, through binding to C-Vps, UVRAG promotes autophagosome fusion with endosome/lysosome, resulting in the rapid turnover of autophagic cargo. An important question that arises is why UVRAG uses two separate mechanisms to regulate the autophagy pathway. The sequential transport of autophagic cargo from autophagosomes to late endosomes/lysosomes suggests the necessity of regulatory mechanisms to coordinate the transition between neighbouring membrane compartments. In this context, UVRAG may induce coupling between the Beclin1-positive autophagosomes and C-Vps-positive endosomes/lysosomes to ensure the sequential transport and dynamic equilibrium of autophagic membranes.
Figure 8.

Model of the role of UVRAG as a coordinator of the autophagosomal and endosomal machineries. At an early stage of autophagy, UVRAG targets Beclin1 to facilitate autophagosome formation, whereas at the later stages, UVRAG interacts with C-Vps to promote autophagosome maturation. UVRAG–C-Vps is also involved in the endosome–lysosome transition by activation of Rab7.
Given the activity of C-Vps in endosomal transport, the presence of UVRAG and C-Vps in the same vesicular structures may also be related to the coordinated activity of UVRAG in endocytic trafficking. Indeed, UVRAG in conjunction with C-Vps accelerated the late stage of endocytic trafficking of EGF ligand and the degradation of EGFR. As C-Vps proteins regulate conversion of Rab5 to Rab7 through its GEF activity on Rab724, it is conceivable that UVRAG–C-Vps interaction may stimulate GDP/GTP exchange on Rab7 to promote the transition from Rab5- to -Rab7-positive membrane domains. This notion is supported by our observation that UVRAG expression robustly activates Rab7 GTPase activity and promotes endosomal fusion in vitro and in vivo. It is possible, however, that other effectors that may function synergistically or in parallel with C-Vps are also involved in this UVRAG action. Nevertheless, this activity of UVRAG suggests that the molecular machineries involved in the autophagic and endocytic trafficking are closely interconnected and intimately coordinated (Fig. 8). UVRAG, as a trafficking effector, targets the two independent trafficking complexes, Beclin1–PI(3)KC3 and C-Vps, to fulfill its dual roles in the autophagy and endocytic trafficking pathways.
METHODS
Autophagy analysis
Autophagy was induced by starvation or rapamycin treatment. For starvation, cells were washed three times with PBS and incubated in Hank's solution or Earle's balanced salts solution (EBSS; Gibco-BRL) for 2–4 h at 37 °C. For rapamycin treatment, cells were cultured in DMEM containing 1% FBS and rapamycin (2 μM; Sigma) for 2–4 h.
Autophagic protein degradation assay
Degradation of long-lived proteins was measured according to a standard protocol44. Cells cultured in 6-well plates were labelled for 16–18 h at 37 °C with l-3H Leu (1 μC ml−1; PerkinElmer) in complete DMEM. Unincorporated radioisotope was removed by extensive washing with PBS containing a 10-fold excess of unlabelled Leu. Samples were then incubated at 37 °C either in complete medium or in starvation medium (EBSS) containing 2 mM unlabelled Leu. When required, bafilomycin A1 (0.1 μM) was added. After incubation for 1 h, at which time short-lived proteins were degraded, the medium was replaced with the appropriate fresh medium and incubated for additional time as indicated in Fig. 4c. The culture medium was precipitated with 10% trichloroacetic acid (TCA) at 4 °C for 1 h, and TCA-soluble radioactivity was measured by scintillation counting. Total cellular radioactivity was measured after lysing with 0.1 N NaOH. The rate of l-3H Leu release was calculated as a percentage of the radioactivity in the TCA-soluble supernatant to the total cellular radioactivity for each time point.
RNAi
All siRNAs were produced by Dharmacon Research. An siRNA specific for human Vps16 (5′-AGCCACAUCCUCAUCUGAGACAUCCUU-3′), Vps18 (5′-AGGCUCAUGCACAGCUGAUUGCUGG-3′), Beclin1 (5′-AAGAUCCUGGACCGUGUCACC-3′) or UVRAG (5′-UCACUUGUGUAGUACUGAA-3′), or a scrambled control siRNA (5′-GCGCGCUUUGUAGGAUUCG-3′) was transfected using DharmaFect reagent (Dharmacon) according to the manufacturer's protocol. At 48–72 h post-transfection, the cells were analysed for autophagosome fusion with lysosomes using confocal microscopy.
Rab7 GTPase assay
The GTPase assay was performed in vitro as described45. Briefly, GFP-tagged Rab7 expressed in cells was immunoprecipitated with an anti-GFP (Santa Cruz Biotechnology) antibody, washed successively with lysis buffer, GTP loading buffer (20 mM Tris-HCl at pH 8.0, 2 mM EDTA, 1 mM DTT) and then incubated for 30 min with 0.1 μM α-32P-GTP (PerkinElmer) in 50 μl of loading buffer. Unbound α-32P-GTP was removed by washing the immunoprecipitates three times with loading buffer. The in vitro GTP hydrolysis assay was initiated by resuspending the immunoprecipitates in reaction buffer (20 mM Tris-HCl pH 8.0, 10 mM MgCl2, 1 mM dithiothreitiol), and then incubating at 37 °C for 0.5–1 h. The reactions were terminated by the addition of elution buffer (0.2% SDS, 5 mM EDTA, 5 mM GTP, 5 mM GDP) followed by heating to 65 °C for 2 min. The eluted GTP and GDP were separated on polyethyleneimine (PEI) cellulose sheets (Sorbtech Chromatography) and developed in a chamber equilibrated with 0.75 M KH2PO4 (pH 3.5). Radiolabelled GTP and GDP were detected and quantified with a Fuji Phosphor Imager scanner.
EGF endocytosis and EGFR degradation assay
EGF coupled to Alexa Fluor 488 (Molecular Probes, Invitrogen) was used as an endocytic probe. For EGF uptake, cells plated on 8-well chamber slides were maintained in serum-free DMEM for 14–16 h at 37 °C. Cells were first washed in ice-cold PBS and then incubated on ice in uptake medium (DMEM, 2% BSA, 20 mM HEPES, pH 7.5) containing 5 μg ml−1 Alexa Fluor 488–EGF. After incubation on ice for 1 h, the cells were washed three times in ice-cold PBS to remove unbound ligand. Cells from one well were fixed to give the total amount of bound ligand and the remaining wells were transferred to a 37 °C incubator. At each indicated time point, the cells were fixed and processed for confocal microscopy. For EGFR degradation, cells cultured in 6-well plates were grown to approximately 80% confluency, washed with PBS and serum-starved overnight. EGFR endocytosis was stimulated by addition of EGF (200 ng ml−1; Invitrogen) in DMEM containing 20 mM HEPES and 0.2% BSA. At each time point after EGF stimulation, the cells were lysed in SDS–PAGE sample buffer, and equal amounts of the lysate were analysed by SDS–PAGE and immunoblotted with anti-EGFR antibody (1:100; Santa Cruz Biotechnology) that recognizes total EGFR.
In vivo endosome fusion assay
The in vivo endosome-labelling and fusion assay was performed as described46 with a slight modification. Control and UVRAG-expressing cells grown on 8-well chamber slides were incubated in complete DMEM for 24 h before labelling. For the assay, the cells were washed with PBS and incubated with a combination of Alexa Fluor 568–dextran (1 mg ml−1, as an internal control) and Oregon green 514-avidin (1 mg ml−1) in uptake medium (1 mM CaCl2, 1 mM MgCl2 and 10 mM glucose) for 10 min at 37 °C. The cells were then briefly rinsed with PBS followed by a 30-min chase to label late endosomes. Cells were then pulse-labelled with the second probe, biotin–BSA (16 mg ml−1) for 10 min in the uptake medium. After labelling, the cells were washed briefly in PBS, fixed and processed for confocal microscopy. Biotin binding, which resulted from endosome fusion, caused a significant increase in the fluorescence of Oregon green 514. Fused vesicles (bright green) were counted and expressed as a percentage of the total number of vesicles (containing Alexa Fluor 568–dextran). Vesicle counts were taken from two independent experiments.
Statistical analysis
Statistical analyses were performed using the Student's t test. Values are expressed as mean ± s.e.m. of at least three independent experiments, unless otherwise noted. A P value of <0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
This work was partly supported by U.S. Public Health Service grants CA82057, CA91819, CA31363, CA106156, RR00168 (J.U.J.) and AI 42999, AI069345 (V.D.). C. L. is a Leukemia & Lymphoma Society Fellow. We thank B. Levine, M.J. Hardwick, S. Virgin, S. Field, T. Yoshimori and Y. Ohsumi for providing reagents, S.W. Richardson for helping with the in vitro endosome fusion assay, S. Gygi for mass spectrometry analysis and Michelle Connole for FACS analysis. Finally, we thank all lab members for their support and discussions.
Footnotes
Note: Supplementary Information is available on the Nature Cell Biology website.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eskelinen EL. Maturation of autophagic vacuoles in mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 3.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 4.van der Goot FG, Gruenberg J. Intra-endosomal membrane traffic. Trends Cell Biol. 2006;16:514–521. doi: 10.1016/j.tcb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Zerial M, McBride H. Rab proteins as membrane organizers. Nature Rev. Mol. Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 6.Nara A, et al. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct. Funct. 2002;27:29–37. doi: 10.1247/csf.27.29. [DOI] [PubMed] [Google Scholar]
- 7.Tamai K, et al. Role of Hrs in maturation of autophagosomes in mammalian cells. Biochem. Biophys. Res. Commun. 2007;360:721–727. doi: 10.1016/j.bbrc.2007.06.105. [DOI] [PubMed] [Google Scholar]
- 8.Surpin M, et al. The VTI family of SNARE proteins is necessary for plant viability and mediates different protein transport pathways. Plant Cell. 2003;15:2885–2899. doi: 10.1105/tpc.016121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jager S, et al. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Polo RA, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J. Cell Sci. 2005;118:3091–3102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 11.Rote KV, Rechsteiner M. Degradation of microinjected proteins: effects of lysosomotropic agents and inhibitors of autophagy. J. Cell Physiol. 1983;116:103–110. doi: 10.1002/jcp.1041160116. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto A, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence BP, Brown WJ. Autophagic vacuoles rapidly fuse with pre-existing lysosomes in cultured hepatocytes. J. Cell Sci. 1992;102(Pt 3):515–526. doi: 10.1242/jcs.102.3.515. [DOI] [PubMed] [Google Scholar]
- 14.Liou W, Geuze HJ, Geelen MJ, Slot JW. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J. Cell Biol. 1997;136:61–70. doi: 10.1083/jcb.136.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lucocq J, Walker D. Evidence for fusion between multilamellar endosomes and autophagosomes in HeLa cells. Eur. J. Cell Biol. 1997;72:307–313. [PubMed] [Google Scholar]
- 16.Wickner W, Haas A. Yeast homotypic vacuole fusion: a window on organelle trafficking mechanisms. Annu. Rev. Biochem. 2000;69:247–275. doi: 10.1146/annurev.biochem.69.1.247. [DOI] [PubMed] [Google Scholar]
- 17.Peplowska K, Markgraf DF, Ostrowicz CW, Bange G, Ungermann C. The CORVET tethering complex interacts with the yeast Rab5 homolog Vps21 and is involved in endo-lysosomal biogenesis. Dev. Cell. 2007;12:739–750. doi: 10.1016/j.devcel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Sato TK, Rehling P, Peterson MR, Emr SD. Class C Vps protein complex regulates vacuolar SNARE pairing and is required for vesicle docking/fusion. Mol. Cell. 2000;6:661–671. doi: 10.1016/s1097-2765(00)00064-2. [DOI] [PubMed] [Google Scholar]
- 19.Peterson MR, Emr SD. The class C Vps complex functions at multiple stages of the vacuolar transport pathway. Traffic. 2001;2:476–486. doi: 10.1034/j.1600-0854.2001.20705.x. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura N, Hirata A, Ohsumi Y, Wada Y. Vam2/Vps41p and Vam6/Vps39p are components of a protein complex on the vacuolar membranes and involved in the vacuolar assembly in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1997;272:11344–11349. doi: 10.1074/jbc.272.17.11344. [DOI] [PubMed] [Google Scholar]
- 21.Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc. Natl Acad. Sci. USA. 2000;97:9402–9407. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins KM, Thorngren NL, Fratti RA, Wickner WT. Sec17p and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J. 2005;24:1775–1786. doi: 10.1038/sj.emboj.7600658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wurmser AE, Sato TK, Emr SD. New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J. Cell Biol. 2000;151:551–562. doi: 10.1083/jcb.151.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 25.Pulipparacharuvil S, et al. Drosophila Vps16A is required for trafficking to lysosomes and biogenesis of pigment granules. J. Cell Sci. 2005;118:3663–3673. doi: 10.1242/jcs.02502. [DOI] [PubMed] [Google Scholar]
- 26.Lindmo K, et al. A dual function for Deep orange in programmed autophagy in the Drosophila melanogaster fat body. Exp. Cell Res. 2006;312:2018–2027. doi: 10.1016/j.yexcr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 27.Liang C, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nature Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 28.Takahashi Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nature Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim BY, et al. Identification of mouse Vps16 and biochemical characterization of mammalian class C Vps complex. Biochem. Biophys. Res. Commun. 2003;311:577–582. doi: 10.1016/j.bbrc.2003.10.030. [DOI] [PubMed] [Google Scholar]
- 30.Kanai F, et al. The PX domains of p47phox and p40phox bind to lipid products of PI(3) K. Nature Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 31.Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer cells with microsatellite instability. Oncogene. 2004;23:639–645. doi: 10.1038/sj.onc.1207178. [DOI] [PubMed] [Google Scholar]
- 33.Robinson JS, Graham TR, Emr SD. A putative zinc finger protein, Saccharomyces cerevisiae Vps18p, affects late Golgi functions required for vacuolar protein sorting and efficient alpha-factor prohormone maturation. Mol. Cell Biol. 1991;11:5813–5824. doi: 10.1128/mcb.11.12.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu JF, Fukamachi S, Mitani H, Hori H, Kanamori A. Reduced expression of vps11 causes less pigmentation in medaka, Oryzias latipes. Pigment Cell Res. 2006;19:628–634. doi: 10.1111/j.1600-0749.2006.00346.x. [DOI] [PubMed] [Google Scholar]
- 35.Sevrioukov EA, He JP, Moghrabi N, Sunio A, Kramer H. A role for the deep orange and carnation eye color genes in lysosomal delivery in Drosophila. Mol. Cell. 1999;4:479–486. doi: 10.1016/s1097-2765(00)80199-9. [DOI] [PubMed] [Google Scholar]
- 36.Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 37.Cantalupo G, Alifano P, Roberti V, Bruni CB, Bucci C. Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. EMBO J. 2001;20:683–693. doi: 10.1093/emboj/20.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu M, Wang T, Loh E, Hong W, Song H. Structural basis for recruitment of RILP by small GTPase Rab7. EMBO J. 2005;24:1491–1501. doi: 10.1038/sj.emboj.7600643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–335. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim BY, et al. Molecular characterization of mammalian homologues of class C Vps proteins that interact with syntaxin-7. J. Biol. Chem. 2001;276:29393–29402. doi: 10.1074/jbc.M101778200. [DOI] [PubMed] [Google Scholar]
- 41.Richardson SC, Winistorfer SC, Poupon V, Luzio JP, Piper RC. Mammalian late vacuole protein sorting orthologues participate in early endosomal fusion and interact with the cytoskeleton. Mol. Biol. Cell. 2004;15:1197–1210. doi: 10.1091/mbc.E03-06-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darsow T, Rieder SE, Emr SD. A multispecificity syntaxin homologue, Vam3p, essential for autophagic and biosynthetic protein transport to the vacuole. J. Cell Biol. 1997;138:517–529. doi: 10.1083/jcb.138.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baba M, Takeshige K, Baba N, Ohsumi Y. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J. Cell Biol. 1994;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogier-Denis E, Houri JJ, Bauvy C, Codogno P. Guanine nucleotide exchange on heterotrimeric Gi3 protein controls autophagic sequestration in HT-29 cells. J. Biol Chem. 1996;271:28593–28600. doi: 10.1074/jbc.271.45.28593. [DOI] [PubMed] [Google Scholar]
- 45.Stenmark H, et al. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emans N, Biwersi J, Verkman AS. Imaging of endosome fusion in BHK fibroblasts based on a novel fluorimetric avidin-biotin binding assay. Biophys. J. 69:716–728. doi: 10.1016/S0006-3495(95)79947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.