

Abstract
Tetrodotoxin (TTX) is the quintessential ligand of voltage-gated sodium channels (NaVs). Like TTX, μ-conotoxin peptides are pore blockers, and both toxins have helped to define the properties of neurotoxin receptor Site 1 of NaVs. Here, we report unexpected results showing that the recently discovered μ-conotoxin KIIIA and TTX can simultaneously bind to Site 1 and act in concert. Results with saturating concentrations of peptide applied to voltage-clamped Xenopus oocytes expressing brain NaV1.2, and single-channel recordings from brain channels in lipid bilayers, show that KIIIA or its analog, KIIIA[K7A], block partially, with a residual current that can be completely blocked by TTX. In addition, the kinetics of block by TTX and peptide are each affected by the prior presence of the other toxin. For example, bound peptide slows subsequent binding of TTX (an antagonistic interaction) and slows TTX dissociation when both toxins are bound (a synergistic effect on block). The overall functional consequence resulting from the combined action of the toxins depends on the quantitative balance between these opposing actions. The results lead us to postulate that in the bi-liganded NaV complex, TTX is bound between the peptide and the selectivity filter. These observations refine our view of Site 1 and open new possibilities in NaV pharmacology.
Keywords: conotoxin, contratoxin, NaV1.2, oocyte, sodium channel, site 1, syntoxin, tetrodotoxin, voltage clamp
Voltage-gated sodium channels (NaVs) are responsible for the upstroke of action potentials and play a critical role in electrical signaling in the nervous system. There has been an ongoing interest in ligands to probe the structure and function of these channels. Nine mammalian isoforms of the main pore-forming α-subunit of the channel have been characterized,1 and ligands that can discriminate among them are useful not only for basic research but also for the development of drugs for diseases such as epilepsy and for pain management.2, 3 Antagonists of NaVs such as tetrodotoxin (TTX) and μ-conotoxins are pore blockers and have been instrumental in the study of these channels.2, 4 TTX is a small guanidinium alkaloid (Mr = 319), whereas μ-conotoxins are positively-charged peptides of 16 to 25 AA residues sharing a common three disulfide-bridge framework. TTX blocks six of the nine NaV isoforms with IC50s in the nM range, including NaV1.4 (from skeletal muscle) and NaV1.2 (from brain). In contrast, the best-studied μ-conotoxin, GIIIA, is >100-fold more potent in blocking NaV1.4 (IC50 = 19 nM in oocyte experiments) than other isoforms, such as NaV1.2 (IC50 ≈ 2.7 μM).5 Competition-binding studies show that TTX, saxitoxin (STX, another guanidinium alkaloid), and GIIIA all bind to neurotoxin receptor Site 1 of NaV1.4 at the mouth of the permeation pathway for sodium ions.2, 4, 6, 7
A residue critical for the functional activity of GIIIA is Arg13.8 Thus for example, the mutation R13A not only lowers the affinity for NaV1.4 by >80-fold,9 but also lowers efficacy of block, insofar as the bound mutant peptide incompletely occludes the channel, as manifested by a residual current when single channels are analyzed.10
We recently discovered a subset of μ-conotoxins that blocks neuronal NaVs as well as, or better than, NaV1.4.11, 12 The sequence of one of these, KIIIA, is shown below along with that of GIIIA (here, “O” is hydroxyproline, the dashes are spacers to align the Cys's in the two peptides, and * indicates a C-terminal amide).
| GIIIA | RDCCTOOKKCKDRQCKOQ-RCCA* |
| KIIIA | --CCN----CSSKWCRDHSRCC* |
KIIIA had the following intriguing properties that prompted us to examine its interaction with NaV channels in greater detail. 1, Single-channel analysis revealed that when KIIIA bound to rat brain sodium channels, most of the unitary current was blocked, but a small residual current remained.13 The mutant KIIIA[K7A] permits an even larger residual current to flow (JRM & RJF, unpublished; cf. Fig. 5 below). 2, The homolog of GIIIA's Arg13 in KIIIA is Lys7 (see underlined residues in sequences above). In contrast to GIIIA, single residue replacements of an alanine-walk identified other residues in the C-terminal half of KIIIA that contributed more strongly to high affinity binding than did Lys7.13 3, In contrast to GIIIA, KIIIA showed much stronger binding to NaV1.2 than to NaV1.4.13 4, KIIIA has 6 fewer residues in its N-terminal half than GIIIA, suggesting that it may rely on a different complement of interactions to determine targeting specificity and affinity than does GIIIA.
Figure 5.
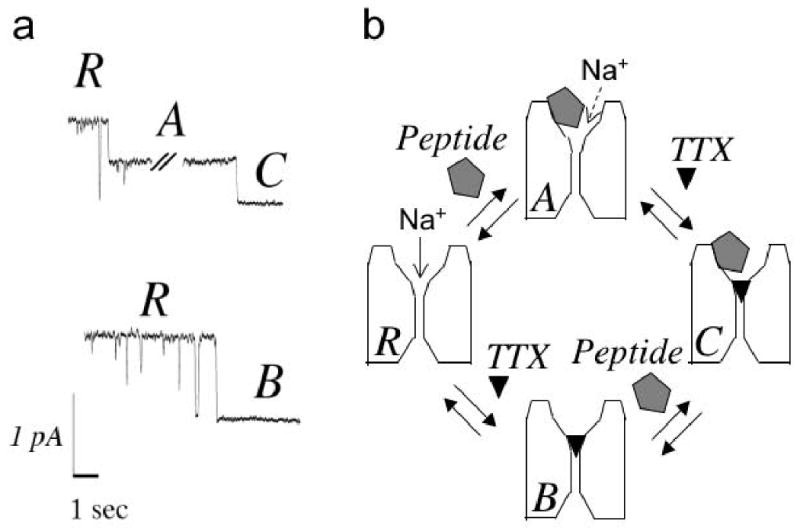
Single-channel records (a) and proposed reaction scheme (b). The conductance states “R”, “A”, “B”, and “C” in (a) correspond to the 4 configurations in (b). All putative transitions except “B” to “C” are illustrated in (a); the “B” to “C” transition would not result in a change in the current level due to complete block by TTX. (a) Recordings from a single rat brain sodium channel incorporated into a planar bilayer membrane were obtained as described in Methods. Upper trace: Begins in the “R” level (open level, unblocked) in the presence of KIIIA[K7A] (2 μM), and then steps to the partially blocked level “A” indicating binding of the peptide. After KIIIA[K7A] binding, TTX concentration was increased to 10 μM, and ∼1 minute later, a transition was seen to a fully blocked level “C”, which we interpret as the bi-liganded state (NaV•TTX•KIIIA[K7A]). Lower trace, channel block in the presence of TTX (50 nM), without peptide, clearly defines the unblocked open level and fully blocked/closed level “B”, which shows the same conductance as the bi-liganded state “C” shown in the upper trace. (b) Reaction scheme consistent with both single-channel and macroscopic results. “R” represents ligand-free (fully functional) channel; “A”, peptide-blocked channel (where residual INa is indicated by dashed arrow); “B”, TTX-blocked channel; and “C”, the ternary (bi-liganded NaV) complex.
To further investigate these distinctive features of KIIIA and KIIIA[K7A], we used these peptides together with TTX and examined the dose-dependence and kinetics of the block of sodium current of NaV1.2, expressed in Xenopus oocytes, during and after exposure to individual toxins and toxin mixtures. The results indicate that either peptide, KIIIA or KIIIA[K7A], and the alkaloid, TTX, can simultaneously bind to the channel. This conclusion is supported by single-channel recordings of brain NaV in planar lipid bilayers. Finally, the mutation F385C in NaV1.2, which is near the selectivity filter and renders the channel resistant to TTX, had a small effect on the sensitivity to KIIIA. The overall results are consistent with peptide and TTX being able to form a ternary complex with Site 1 of NaV1.2 and suggest new possibilities in sodium channel pharmacology that could be exploited to substantially expand the repertoire of ligands with which to experimentally probe, or therapeutically target, voltage-gated sodium channels. We introduce the terms “syntoxin” and “contratoxin” that encapsulate two disparate conceptual frameworks engendered by our results.
Results
A residual sodium current persists at saturating concentrations of KIIIA or KIIIA[K7A]
At high peptide concentrations, the dose-response curves of KIIIA and its analog, KIIIA [K7A], were flat, even though 100% block was not achieved (Fig. 1). Experimental determination of the IC50's of these peptides was problematic because of the slow rates of block by the peptides at concentrations near their expected IC50's. However, Kd's of the two peptides were calculated from their kinetics (Table 1), and these Kd's were well below the concentrations tested in Figure 1, where the curves were flat. The residual sodium current (rINa) with KIIIA was 5% of the control current, and rINa with KIIIA[K7A] was 23% of control. A similar range of residual, unblocked currents has been observed with conotoxin GIIIA derivatives in both whole-cell14 and single-channel studies.10 The frequency and durations of the partially blocked states are generally consistent with the kinetics of toxin wash in and washout observed in whole-cell recordings. We designate the peptide-bound states of the channel (with residual current) as NaV•KIIIA or NaV•KIIIA[K7A], and the TTX-bound state as NaV•TTX.
Figure 1.
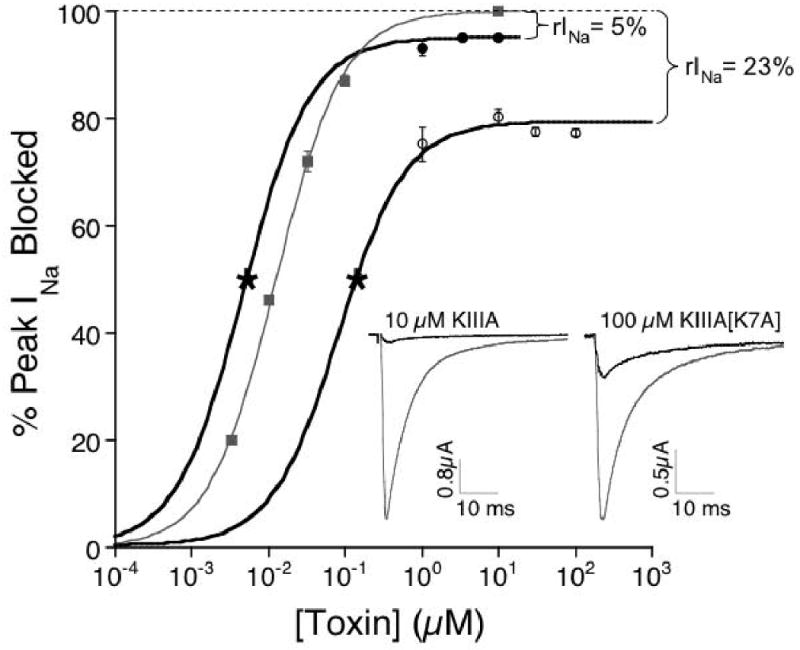
Steady state dose-response curves reveal a residual Na current (rINa) at saturating peptide concentrations. Data (KIIIA, closed circles; KIIIA[K7A], open circles; TTX, closed squares) were obtained with oocytes expressing NaV1.2 as described in Methods. Small kon prevented acquisition of steady-state data at [peptide] in the vicinity of Kd, represented by asterisk, obtained from koff/kon (Table 1). Solid lines represent fit of data, including asterisks, to the Langmuir isotherm, Y = 100 • plateau/[1 + (IC50/[toxin])]. Plateau (i.e., Y-axis value at saturating [toxin]) was 95 ± 3% for KIIIA and 77 ± 1% for KIIIA[K7A] and 100% for TTX. Insets, representative current recordings in the absence (control, gray traces) and presence (black traces) of saturating concentrations of KIIIA (10 μM, left) and KIIIA[K7A] (100 μM, right).
Table 1. Kinetica and equilibrium constants of μ-conotoxins, TTX and toxin-combinations on NaV1.2.
| Peptide or complexb | IC50 (nM) |
kon (μM-1•min-1) |
koff (min-1) |
Kd (nM) |
|---|---|---|---|---|
| KIIIA | N.D.c | 0.3±0.03 | 0.0016±0.0016d | 5.3±5 |
| KIIIA[K7A] | N.D.c | 0.13±0.006 | 0.015±0.005 | 115±38 |
| TTX | 12.6 (11.7-13.6) | 58±5.3 | 2.2±0.3 | 37.9±6 |
| NaV•KIIIA•TTX | 0.017±0.002 | |||
| NaV•TTX•KIIIA | 0.02±0.005 | |||
| NaV•KIIIA[K7A]•TTX | 0.024±0.004 | |||
| NaV•TTX•KIIIA[K7A] | 0.025±0.003 |
Kinetic data were obtained as described in Methods, unless otherwise indicated.
The order of the components in a complex indicates the order in which the components were added: for NaV•KIIIA•TTX, the channel was exposed to KIIIA first, then to a mixture of KIIIA and TTX (see Fig. 2f); for NaV•TTX•KIIIA, the order was reversed (see Fig. 2g) (the koff's were the same within experimental error, suggesting the same ternary complex formed)—likewise for NaV•KIIIA[K7A]•TTX (see Fig. 2c) and NaV•TTX•KIIIA[K7A] (see Fig. 2d).
When [peptide] near the expected IC50 was tested, the reaction was too slow to reach steady-state within the time-frame of the experiment.
Estimated from level of recovery after 20 min and assuming exponential time course. Values in parentheses are 95% confidence interval, and ± value indicates S.D. (n ≥ 3). kon of KIIIA is as previously reported11, all other constants were determined in experiments of the present report.
TTX blocks the residual current through peptide-bound channels and accelerates peptide dissociation, indicating that bi-liganded complexes can form
The rINa with either peptide could be completely blocked by TTX (Figs. 2c and 2f). Furthermore, the rate of functional recovery following toxin-washout differed depending on whether the exposure was to TTX alone, peptide alone, or to both peptide and TTX (Fig. 2). The order of ligand-addition (i.e., peptide-first or TTX-first) did not seem to affect the recovery rate (Figs. 2c vs 2d, 2f vs 2g, and Table 1). Thus, these kinetic data, as well as those presented below, argue that bi-liganded complexes can form, which we will refer to as NaV•TTX•KIIIA and NaV•TTX•KIIIA[K7A].
Figure 2.
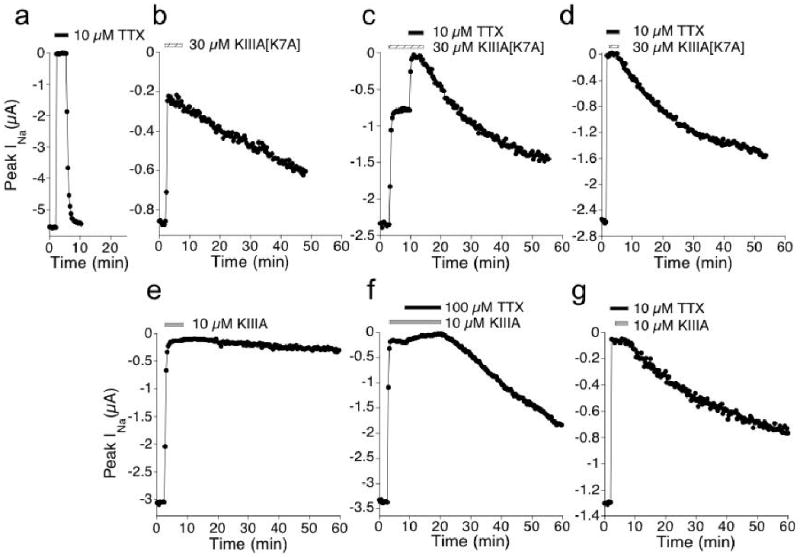
TTX blocks residual INa that persists in saturating concentrations of KIIIA[K7A] or KIIIA and accelerates the rate of peptide-washout. Example plots of peak INa recorded from oocytes during exposure to: TTX (a); KIIIA[K7A] (b); KIIIA[K7A] followed by KIIIA[K7A]+TTX (c); TTX followed by TTX+KIIIA[K7A] (d); KIIIA (e); KIIIA followed by KIIIA+TTX (f); and TTX followed by TTX+KIIIA (g). Different oocyte was used in each of panels b to g, where previous exposure to 10 μM TTX obliterated INa (not shown, but as in a). Presence of 10 or 100 μM TTX is indicated by black bar, 30 μM KIIIA[K7A] by hatched bar, and 10 μM KIIIA by gray bar. In all cases, except e, recovery of INa during toxin-washout could be fit by a single exponential time course (koff values are listed in Table 1). Test pulses were applied at 20 s intervals (see Methods).
TTX blocks rINa orders of magnitude more slowly than it blocks control INa
Relatively high [TTX] was used in the experiments illustrated in Figure 2 because the block of rINa was slow. The rate of block of rINa was examined over a range [TTX]. The block of rINa by TTX followed an exponential time course (not illustrated) to yield kobs, and plots of kobs for the block of rINa as a function of [TTX] are linear (Fig. 3). It is evident from the slopes of the linear regression curves that the apparent kon values for TTX block of NaV•KIIIA[K7A] and NaV•KIIIA were reduced relative to the kon for TTX block of NaV by factors of 10-2 and 5×10-5, respectively.
Figure 3.
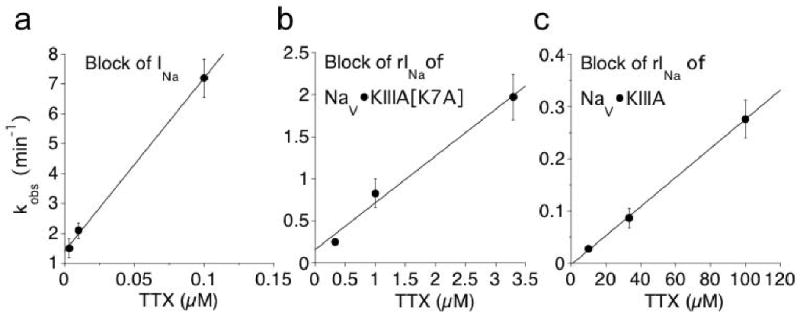
Rate of TTX-block of control INa (a), residual INa in 30 μM KIIIA[K7A] (b), and in 10 μM KIIIA(c) differ radically from each other (note different time scales). Time course of block of rINa by various [TTX], each fit to a single exponential (not illustrated), provided the observed rate constants (kobs). Plots show kobs for different [TTX]. Relative timing of the exposure to toxin was essentially as in Fig. 2a for NaV, Fig. 2c for NaV•KIIIA[K7A], and Fig. 2f for NaV•KIIIA. Linear regression slope, providing apparent kon, was 58 ± 5.3 μM-1min-1 (a), 0.56 ± 0.08 μM-1min-1 (b), and 0.003 ± 0.0004 μM-1min-1 (c).
Binding of KIIIA and KIIIA[K7A] to NaV•TTX appear to occur at rates similar to their binding to unliganded-NaV
To examine the rate of peptide binding to NaV already bound with TTX, we exploited the disparity in the rates of recovery from block of NaV•TTX vs NaV•TTX•peptide; i.e., following toxin-washout, the block produced by either peptide persisted much longer than the block by TTX alone. The time course of recovery during toxin washout following exposure first to a saturating [TTX] (10 μM), then to 10 μM TTX plus 1 μM peptide, was well fit by a double exponential (Fig. 4b). The fast component had a rate constant consistent with that of NaV•TTX and the slow component had a rate constant consistent with that of NaV•TTX•KIIIA (or NaVTTX•KIIIA[K7A]). The span of the slower component, when plotted as function of time-of-exposure to peptide, was fit by a single exponential time course (closed circles in Figs. 4c and d) to yield the kobs for the binding of 1 μM peptide to NaV•TTX. For comparison, also shown is the block of NaV by 1 μM of each peptide alone (open circles in Figs. 4c and 4d); the presence of TTX reduced the kobs of each peptide by only about 25%.
Figure 4.
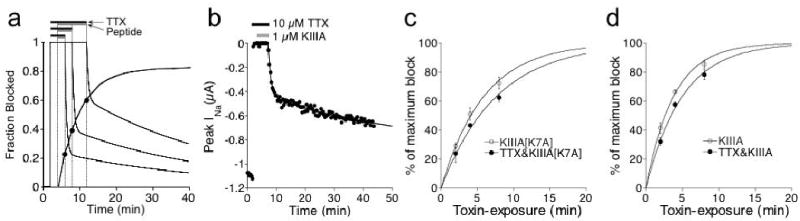
Apparent on-rate of peptide binding in the presence of TTX was only slightly slower than the rate of block by peptide in the absence of TTX. Level of peptide bound to NaV1.2 in the face of 10 μM TTX was determined by the amount of persistent-block following washout of both toxins. (a) Protocol used to determine level of persistent-block by peptide (i.e., formation of NaV•TTX•peptide). Results from series of three hypothetical experiments are superimposed: oocyte was exposed to TTX for 2 min. followed by exposure to TTX + peptide for 2, 4, or 8 min before washing out both toxins (black bars represent presence of TTX, and gray bars, TTX + peptide). In each case, the washout curve was fit to a double exponential, of which the span and koff of the faster and slower components represent the recovery from block of NaV•TTX and NaV•TTX•peptide, respectively. The closed circles represent the span of the slower component and correspond to the closed circles in c and d. (b) Representative results showing peak INa during exposure to, and following washout of, TTX and KIIIA (black and gray bars, respectively). (c, d) Time course for the formation of NaV • TTX•peptide (closed circles), estimated by the persistent-block produced by 1 μM peptide as described in a for different durations of simultaneous-exposure to TTX+peptide (cf., closed circles in a), compared with the formation of NaV • peptide (open circles) simply measured by block produced by 1 μM peptide alone. Data were fit to single exponential curves (solid lines). kobs for formation of NaV • KIIIA[K7A] and NaV • TTX • KIIIA[K7A] was 0.17 ± 0.006 and 0.13 ± 0.006 min-1, respectively (c). kobs for formation of NaV • KIIIA and NaV •TTX • KIIIA was 0.27 ± 0.01 and 0.20 ± 0.008 min-1, respectively (d).
Single-channel recordings of rat brain NaV in lipid bilayers show that rINa with KIIIA[K7A] can be blocked by TTX
In principle, single channel recordings should easily distinguish between conducting states resulting from partial block by a peptide toxin and complete block by TTX, as has been demonstrated for NaV1.4 channels in the simultaneous presence a saxitoxin derivative (dcSTX), and GIIIA[R13Q].15 For those toxins, blocking events originate only from the unblocked, fully open state, regardless of whether they reflect complete block by dcSTX or partial block by GIIIA[R13Q]. This, along with kinetic analysis, indicated a simple competition between these toxins for the channel vestibule. The macroscopic current analysis of NaV1.2 above suggests a more complex interaction for the smaller KIIIA peptides and TTX, in which TTX can apparently by-pass the bound peptide, albeit with slow kinetics, and completely block single-channel current. Thus, transitions to a fully blocked state are expected from the peptide-bound, partially blocked state to the non-conducting level. The long bound times of the KIIIA peptides allowed us to observe complete blocking events by adding a high concentration of TTX to the bath, after observing discrete onset of block by the peptide. Such transitions can be recorded from brain Na channels in the presence of KIIIA[K7A] (Fig 5a). Similar events have been recorded from skeletal muscle channels (not illustrated), and these observations offer direct evidence that TTX binds to and blocks the peptide-bound channel.
KIIIA blocks the TTX-resistant mutant NaV1.2[F385C] almost as well as the wild-type channel
The single point mutation of the Phe at position 385 of NaV1.2 to a Cys, resulting in NaV1.2[F385C], renders the channel insensitive to TTX concentrations up to 10 μM.16 We constructed the mutant clone and tested TTX concentrations up to 100 μM and found the IC50 to be 39 μM (Table 2). Thus, the mutation decreased TTX's affinity 3,000-fold. At these concentrations the rate of block was too fast for our experimental system to determine kon. The koff of TTX for the mutant channel was likewise too fast to be determined. In contrast, the mutation had a much smaller effect on KIIIA's activity: kon was relatively unchanged, and koff was about doubled, resulting in a mere doubling of the Kd (Table 2). Intriguingly, as also indicated in Table 2, the residual current at saturating concentrations of KIIIA was two-fold larger for the mutant channel than for the wild type channel. The weak block of the mutant channel by TTX precluded experiments to see whether TTX blocked the residual current.
Table 2. Comparison of block of wild-type NaV1.2 versus NaV1.2[F385C] by TTX and KIIIA.
| Ligand | Variable | WT NaV1.2 | NaV1.2[F385C] |
|---|---|---|---|
| TTX | IC50 (μM) | 0.013 (0.012-14)a | 39 (34-44)c |
| KIIIA | kon (μM-1•min-1) | 0.3±0.03a | 0.5±0.06 |
| KIIIA | koff (min-1) | 0.0016±0.0016a | 0.0047±0.0016d |
| KIIIA | Kd (nM) | 5±5a | 9±3 |
| KIIIA | rINa (% of control) | 5±3b | 10±2 |
From Table 1.
From Fig. 1.
IC50 was determined using [TTX] up to 100 μM (at which 72 ± 3% block was observed) and fitting data to the Langmuir isotherm (see Fig. 1 legend) assuming 100% block at saturating [TTX].
Estimated from level of recovery after 20 min and assuming exponential time course. Values in parentheses are 95% confidence interval, and ± value indicates S.D. (n ≥ 3).
Discussion
Formation of the ternary, NaV•TTX•peptide, complex
Dose-response curves clearly show that an rINa persists at saturating peptide concentrations (Fig. 1). Residual INa with the peptides was anticipated from single-channel recordings of rat brain channels incorporated into lipid bilayers, where a small rINa was observed with the KIIIA-bound channel13 and an even larger rINa can be observed with KIIIA[K7A] (Fig. 5a and JRM & RJF, unpublished). However, the observation that TTX can block the (macroscopic) rINa was not anticipated (Figs. 2c and 2f). The latter observation and the kinetic data reviewed below lead to the conclusion that the two toxins bind to the channels with different affinities and kinetics, and that one molecule of each toxin can bind simultaneously to a single channel to form a bi-liganded complex, NaV•TTX•peptide (Fig. 5b).
Consistent with this conclusion are observations on single channels incorporated into planar lipid bilayer membranes (Fig 5a). These experiments are challenging, given the intrinsically slow kinetics of the peptide interaction with rNaV1.213 and the vast slowing of the TTX interaction when peptide is bound. However, all the conductance states and transitions, which are expected for a scheme in which either or both toxins can bind to the channel were observed: unblocked – state R; KIIIA[K7A]-bound, partially blocked – state A; TTX blocked, non-conducting – state B; and a long duration, non-conducting state entered from state A – the bi-liganded state C.
Implications of the observed off-kinetics
The respective off-rates of KIIIA and KIIIA[K7A] from the mono-liganded NaV•peptide complex differ from each other by a factor of 10; however, presence of TTX appeared to accelerate the off-rate of KIIIA by about 10-fold but that of KIIIA[K7A] only about 1.7-fold (Table 1). Two factors may contribute to TTX's stronger interactions with KIIIA. First, one would expect an electrostatic repulsion between the positively charged TTX and K7 of KIIIA. Second, even in the absence of TTX, KIIIA[K7A] inherently lacks the normal coupling expected between K7 and negative residues on the channel. When TTX is bound, it would be expected act as a shield against the native KIIIA toxin's K7-channel interactions, leading to off rates close to those of KIIIA[K7A].
It can be inferred that peptide reduces TTX's off-rate; however, measurements of the off-rates of TTX from NaV•TTX•peptide in the continuous presence of peptide were not performed, because such experiments require larger quantities of peptide than were available.
The same ternary complex appears to be formed regardless of the order of ligand-addition, insofar as the rate of functional recovery following toxin washout from the ternary complex was essentially the same whether peptide or TTX was added first (Fig. 2 and Table 1).
Implications of the observed on-kinetics
The presence of bound TTX reduced the apparent kon of both peptides modestly and equally (by ∼ 25%, Fig. 4) suggesting that TTX does not substantially shield sites on the channel that determine the on-rate of peptide. In stark contrast, the presence of bound peptide drastically reduced the apparent kon of TTX, with KIIIA interfering with TTX binding much more strongly than did KIIIA[K7A] (Fig. 3). In either case however, when TTX does bind, it blocks the rINa of both NaV•KIIIA and NaV•KIIIA[K7A] completely, as in the control INa (Fig. 2). This is consistent with the view that TTX is situated in the same fashion on NaV in both NaV•TTX and NaV•TTX•peptide complexes. These results would be naturally accommodated if the peptide bound to the channel in a manner that still allowed TTX to access its normal binding site, with residue K7 of KIIIA playing an important role in the ease with which TTX can do so.
Implications of interactions between TTX and peptide for biochemical competition-binding studies
As noted above, pre-bound TTX accelerates the off-rate of subsequently-bound peptide, while pre-bound peptide slows the on-rate of subsequently-bound TTX. Thus, in conventional biochemical competition-binding experiments peptide and TTX could appear bind to the channel in a simple competitive fashion unless the experiments were conducted under conditions where 1) binding kinetics were examined over a broad time range, and/or 2) both ligands were labeled, each with a distinctive reporter.
Reaction mechanisms proposed for the formation of the ternary complex
A scheme that takes into account all of our findings is illustrated in a cartoon that also incorporates the possibility that TTX is located between the bound peptide and the channel's narrow selectivity filter, in the ternary complex (Fig 5b). In this scenario, when the peptide is bound first, TTX can “sneak by” and traverse between its non-bound and bound states, but at a considerably slower rate in each direction than it does in the absence of the peptide; thus during toxin-washout, the rate limiting factor for recovery of function of the ternary complex is the rate at which the peptide comes off. The presence of TTX reduces the peptide's affinity, so activity is recovered more rapidly from NaV•TTX•peptide than from NaV•peptide. The scheme is also supported by the observation that the F385C mutation of NaV1.2, which drastically reduces affinity for TTX, minimally affected KIIIA's activity. At the same time, the mutation doubled the residual INa seen at saturating [KIIIA] (Table 2). This behavior with the mutant channel is consistent with KIIIA binding close to the selectivity filter.
Our results support the view that a neurotoxin receptor site may be considered as a macrosite composed of an amalgamation of microsites.17 In the present case of (macro)Site 1 of NaV1.2, TTX occupies one set of microsites, the peptide another set of microsites, and a third set of microsites might exist that is accessible to both toxins when each binds in the absence of the other. We cannot rule out the possibility that KIIIA is binding at a location distinct from, but allosterically linked to, the site where TTX binds. However, our working assumption of a single multivalent macrosite seems more parsimonious.
Is our conclusion that Site 1, at least that of NaV1.2, can accommodate both TTX and KIIIA consistent with current structural models of Site 1? Recent experiments with NaV1.4 indicate that TTX binds via a cation-pi interaction, where the cationic guanidinium group of TTX associates with the aromatic ring of the Tyr residue 401 (homologous to Phe 385 of NaV1.2) that is next to Asp of the selectivity filter in domain I of the channel.18 The sensitivity of NaV1.4 to TTX is drastically decreased by the Y401C mutation (IC50 increased 43- to >3125-fold);19, 20 yet that mutation reduces GIIIA's affinity only 2- to 4-fold.20, 21 These observations are echoed by our results with KIIIA and NaV1.2[F385C], and together they suggest that, in contrast to TTX, μ-conotoxins generally dock at a more superficial site in the outer vestibule of the channel,2, 22 raising the possibility that KIIIA might allow TTX, albeit with the observed reduced probability and association rate, to pass by the peptide and bind at or near its normal site deeper in the vestibule. This contrasts with the case of the STX analog, dcSTX, and the bulkier conotoxin derivative, GIIIA(R13Q), for which binding is mutually exclusive on NaV1.4.15
TTX and KIIIA act as “syntoxins”
Given that our present data indicate that peptide (KIIIA or KIIIA[K7A]) and TTX can cohabit Site 1, with functional consequences different from those when each toxin acts alone, they might be thought of as “syntoxins”. The expression, syntoxin, encapsulates the notion that two toxins can act in concert. In the case of KIIIA (or KIIIA[K7A]) and TTX, the concerted action has mixed synergistic and antagonistic effects: presence of peptide slows both dissociation and association of TTX (the former action enhances, while the latter diminishes, TTX affinity); reciprocally, the presence of TTX accelerates peptide dissociation.
It will be interesting to further test for syntoxin interactions using different ligands and receptors. Possible ligands include other μ-conotoxins, such as SIIIA, which has 6 more residues than KIIIA;11 other guanidinium alkaloids, including STX, which has 94% the molecular mass of TTX but with two, instead of only one, guanidinium groups; possible targets, include NaV1.4, whose binding with GIIIA has received extensive scrutiny (see ref. 22 for a recent report). Positive findings in this endeavor would suggest new possibilities in sodium channel pharmacology. A syntoxin pair, in context, may be considered as a self-assembling, two-part toxin, where one member has a motif of a guanidinium alkaloid, and the other member has one of a μ-conopeptide. Pairs of members with each motif could be readily tested, in a mix-and-match fashion, for activity against different NaV subtypes. Syntoxin pairs, themselves, might be exploited to substantially expand the repertoire of pharmacological actions targeted at specific subtypes of voltage-gated sodium channels. Furthermore, those pairs yielding the highest affinities might be subsequently engineered into covalently linked, bivalent ligands with the expectation that some could achieve even greater affinity than the corresponding non-linked pair (see e.g., ref. 23).
A modified μ-conotoxin with a large residual INa could serve as a “contratoxin”
Quite apart from the syntoxin properties of KIIIA and KIIIA[K7A], our results show that the peptide's structure can be engineered to attenuate its efficacy; that is, to block a smaller faction of the single-channel current when it binds. Thus, a ligand with high affinity and low (or ideally no) efficacy could be used as a “contratoxin.” For example, in the case of sodium channels, a ligand with high affinity for Site 1 but minimal ability to block the pore, could act as a contratoxin against other toxins that target Site 1, such as TTX and functionally active μ-conotoxins. Note that the action of a contratoxin is distinct from that of an antitoxin, which is an antibody that neutralizes a toxin by binding to the toxin; in contrast, a contratoxin prevents a toxin's action by binding to the toxin's receptor. A contratoxin could not only serve as an antidote to TTX, but (depending on its specificity for different NaVs) could also be used in conjunction with TTX to sharpen the latter's specificity.
Methods
Wild type and mutant NaV1.2 clones
The cDNA for rat NaV1.2 was kindly provided by Alan Goldin. The point mutant, NaV1.2[F385C], where Phe 385 is mutated to Cys, was prepared from the wild type clone as follows. Forward and reverse primers containing the mutation were designed with the following sequences: forward, 5′ctcatgactcaagactgttgggaaaacctttatc3′; reverse, 5′gataaaggttttcccaacagtcttgagtcatgag3′. The mutagenic primers were extended and incorporated by PCR using Finnzyme Phusion™ High-Fidelity DNA polymerase (New England Biolabs, Ipswich, MA). The methylated, non-mutated parental cDNA was digested with Dpn I. The mutated DNA was transformed into DH5α competent cells, and positive colonies were selected based on tetracycline-resistance. Plasmid was isolated using the Qiaprep Spin Miniprep kit (Qiagen, Valencia, CA) and sequenced to ascertain the incorporation of the desired mutation. Capped cRNA for NaV1.2[F385C] was made using the mMessage mMachine in vitro transcription kit (Ambion, Austin, TX), following linearization of the plasmid and subsequent transcription with T7 RNA polymerase. The final product was purified using the Qiagen RNeasy kit.
Oocyte Electrophysiology
Oocytes expressing mutant and wild type NaV1.2 were prepared and two-electrode voltage clamped essentially as previously described.13 Briefly, oocytes were placed in a 30 μL chamber containing ND96 and two electrode voltage clamped with a holding potential of −80 mV. To activate Na channels, the membrane potential was stepped to −10 mV for 50 ms every 20 sec. The oocytes were exposed to toxin in static baths to conserve material. During toxin-washout, the oocyte chamber was perfused at 1.5 ml/min for 20 sec, then at 0.5 ml/min thereafter. Recordings were done at room temperature (∼21 °C).
Single-channel Recordings
Membrane vesicles were isolated from adult rat fore-brains as described previously.24 Membrane vesicles were incubated with batrachotoxin (1 μM in ethanol) then pipetted directly over a horizontal bilayer (4PE:1PC) in symmetric solutions (200mM NaCl, 10mM MOPS, 0.1mM EDTA, pH 7.0) and incorporation was detected as a step increase in current (∼1 pA) at 60 mV. Orientation of the channel was deduced as an increase in closure frequency at −70 mV and 2 μM KIIIA[K7A] toxin with 50 nM TTX was then perfused into the “extracellular” chamber (2 × 400 μL). Steady-state recordings at +40 mV were collected; upon block by the peptide, TTX (10 μM) was added.
Toxins
μ-Conopeptides were synthesized as previously described13. TTX was obtained from Alomone Labs.
Data Analysis
Off-rate constants (koff's) were determined from single-exponential fits of peak sodium current following toxin washout, unless otherwise indicated. Observed rate constants (kobs's) were obtained by single exponential fits of peak sodium current following toxin addition, and on-rate constants (kon) were obtained from slopes of kobs versus [toxin].25
Acknowledgments
We thank Alan Goldin, UCLA, for providing the NaV1.2 clone and Aleksandra Walewska for synthesizing conotoxins. This work was supported by National Institutes of Health grants GM 48677 and NS055845. Work in the French laboratory was supported by the Heart & Stroke Foundation of Alberta, NWT & Nunavut.
Abbreviations
- GIIIA
μ-conotoxin GIIIA
- INa
sodium current
- KIIIA
μ-conotoxin KIIIA
- KIIIA[K7A]
μ-conotoxin KIIIA[K7A]
- NaV
voltage-gated sodium channel
- NaV•KIIIA
the binary complex of NaV with bound KIIIA
- NaV•KIIIA[K7A]
the binary complex of NaV with bound KIIIA[K7A]
- NaV•TTX
the binary complex of NaV with bound TTX
- NaV•TTX•KIIIA
the ternary complex of NaV with bound TTX and KIIIA
- NaV•TTX•KIIIA[K7A]
the ternary complex of NaV with bound TTX and KIIIA[K7A]
- rINa
residual sodium current
References
- 1.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacological reviews. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 2.French RJ, Terlau H. Sodium channel toxins--receptor targeting and therapeutic potential. Curr Med Chem. 2004;11:3053–64. doi: 10.2174/0929867043363866. [DOI] [PubMed] [Google Scholar]
- 3.Wood JN, Boorman JP, Okuse K, Baker MD. Voltage-gated sodium channels and pain pathways. Journal of neurobiology. 2004;61:55–71. doi: 10.1002/neu.20094. [DOI] [PubMed] [Google Scholar]
- 4.Cestele S, Catterall WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000;82:883–92. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- 5.Safo P, Rosenbaum T, Shcherbatko A, Choi DY, Han E, Toledo-Aral JJ, Olivera BM, Brehm P, Mandel G. Distinction among neuronal subtypes of voltage-activated sodium channels by mu-conotoxin PIIIA. J Neurosci. 2000;20:76–80. doi: 10.1523/JNEUROSCI.20-01-00076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Sabi A, McArthur J, Ostroumov V, French RJ. Marine toxins that target voltage-gated sodium channels. Mar Drugs. 2006;4:157–92. [Google Scholar]
- 7.Catterall WA. Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu Rev Pharmacol Toxicol. 1980;20:15–43. doi: 10.1146/annurev.pa.20.040180.000311. [DOI] [PubMed] [Google Scholar]
- 8.Sato K, Ishida Y, Wakamatsu K, Kato R, Honda H, Ohizumi Y, Nakamura H, Ohya M, Lancelin JM, Kohda D, et al. Active site of mu-conotoxin GIIIA, a peptide blocker of muscle sodium channels. The Journal of biological chemistry. 1991;266:16989–91. [PubMed] [Google Scholar]
- 9.Becker S, Prusak-Sochaczewski E, Zamponi G, Beck-Sickinger AG, Gordon RD, French RJ. Action of derivatives of mu-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry. 1992;31:8229–38. doi: 10.1021/bi00150a016. [DOI] [PubMed] [Google Scholar]
- 10.Hui K, Lipkind G, Fozzard HA, French RJ. Electrostatic and steric contributions to block of the skeletal muscle sodium channel by mu-conotoxin. The Journal of general physiology. 2002;119:45–54. doi: 10.1085/jgp.119.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bulaj G, West PJ, Garrett JE, Watkins M, Zhang MM, Norton RS, Smith BJ, Yoshikami D, Olivera BM. Novel conotoxins from Conus striatus and Conus kinoshitai selectively block TTX-resistant sodium channels. Biochemistry. 2005;44:7259–65. doi: 10.1021/bi0473408. [DOI] [PubMed] [Google Scholar]
- 12.Zhang MM, Fiedler B, Green BR, Catlin P, Watkins M, Garrett JE, Smith BJ, Yoshikami D, Olivera BM, Bulaj G. Structural and functional diversities among mu-conotoxins targeting TTX-resistant sodium channels. Biochemistry. 2006;45:3723–32. doi: 10.1021/bi052162j. [DOI] [PubMed] [Google Scholar]
- 13.Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A, Terlau H, McArthur JR, French RJ, Gulyas J, Rivier JE, Smith BJ, Norton RS, Olivera BM, Yoshikami D, Bulaj G. Structure/function characterization of micro-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. The Journal of biological chemistry. 2007;282:30699–706. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- 14.French RJ, Horn R. Shifts of macroscopic current activation in partially blocked sodium channels Interaction between the voltage sensor and a mu-conotoxin. In: Latorre R, Saez JC, editors. From Ion Channels to Cell-to-Cell Conversations. New York: Plenum Press; 1997. pp. 67–89. [Google Scholar]
- 15.French RJ, Prusak-Sochaczewski E, Zamponi GW, Becker S, Kularatna AS, Horn R. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron. 1996;16:407–13. doi: 10.1016/s0896-6273(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 16.Heinemann SH, Terlau H, Imoto K. Molecular basis for pharmacological differences between brain and cardiac sodium channels. Pflugers Arch. 1992;422:90–2. doi: 10.1007/BF00381519. [DOI] [PubMed] [Google Scholar]
- 17.Olivera BM, Rivier J, Scott JK, Hillyard DR, Cruz LJ. Conotoxins. The Journal of biological chemistry. 1991;266:22067–70. [PubMed] [Google Scholar]
- 18.Santarelli VP, Eastwood AL, Dougherty DA, Horn R, Ahern CA. A cation-pi interaction discriminates among sodium channels that are either sensitive or resistant to tetrodotoxin block. The Journal of biological chemistry. 2007;282:8044–51. doi: 10.1074/jbc.M611334200. [DOI] [PubMed] [Google Scholar]
- 19.Backx PH, Yue DT, Lawrence JH, Marban E, Tomaselli GF. Science. Vol. 257. New York, NY: 1992. Molecular localization of an ion-binding site within the pore of mammalian sodium channels; pp. 248–51. [DOI] [PubMed] [Google Scholar]
- 20.Chen LQ, Chahine M, Kallen RG, Barchi RL, Horn R. Chimeric study of sodium channels from rat skeletal and cardiac muscle. FEBS letters. 1992;309:253–7. doi: 10.1016/0014-5793(92)80783-d. [DOI] [PubMed] [Google Scholar]
- 21.Li RA, Tsushima RG, Kallen RG, Backx PH. Pore residues critical for mu-CTX binding to rat skeletal muscle Na+ channels revealed by cysteine mutagenesis. Biophysical journal. 1997;73:1874–84. doi: 10.1016/S0006-3495(97)78218-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choudhary G, Aliste MP, Tieleman DP, French RJ, Dudley SC., Jr Docking of mu-conotoxin GIIIA in the sodium channel outer vestibule. Channels. 2007;1:344–52. doi: 10.4161/chan.5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao J, Lahiri J, Isaacs L, Weis RM, Whitesides GM. Science. Vol. 280. New York, NY: 1998. A trivalent system from vancomycin.D-ala-D-Ala with higher affinity than avidin.biotin; pp. 708–11. [DOI] [PubMed] [Google Scholar]
- 24.Krueger BK, Worley JF, 3rd, French RJ. Block of sodium channels in planar lipid bilayers by guanidium toxins and calcium. Are the mechanisms of voltage dependence the same? Annals of the New York Academy of Sciences. 1986;479:257–68. doi: 10.1111/j.1749-6632.1986.tb15574.x. [DOI] [PubMed] [Google Scholar]
- 25.West PJ, Bulaj G, Garrett JE, Olivera BM, Yoshikami D. Mu-conotoxin SmIIIA, a potent inhibitor of tetrodotoxin-resistant sodium channels in amphibian sympathetic and sensory neurons. Biochemistry. 2002;41:15388–93. doi: 10.1021/bi0265628. [DOI] [PubMed] [Google Scholar]