

Abstract
Background & Aims
Helicobacter pylori infection disrupts the balance between gastric epithelial cell proliferation and apoptosis, which is likely to lower the threshold for the development of gastric adenocarcinoma. H. pylori infection is associated with EGF receptor (EGFR) activation through metalloproteinase-dependent release of EGFR ligands in gastric epithelial cells. Since EGFR signaling regulates cell survival, we investigated whether activation of EGFR following H. pylori infection promotes gastric epithelial survival.
Methods
Mouse conditionally immortalized stomach epithelial cells (ImSt) and AGS cells, as well as wild-type and kinase-defective EGFR (EGFRwa2) mice, were infected with the H. pylori cag+ strain 7.13. Apoptosis, caspase activity, EGFR activation (phosphorylation) and EGFR downstream targets were analyzed.
Results
Inhibiting EGFR kinase activity or decreasing EGFR expression significantly increased H. pylori-induced apoptosis in ImSt. Blocking H. pylori-induced EGFR activation with a heparin-binding (HB)-EGF neutralizing antibody or abrogating a disintegrin and matrix metalloproteinase-17 (ADAM-17) expression increased apoptosis of H. pylori-infected AGS and ImSt, respectively. Conversely, pretreatment of ImSt with HB-EGF completely blocked H. pylori-induced apoptosis. H. pylori infection stimulated gastric epithelial cell apoptosis in EGFRwa2, but not in wild-type mice. Furthermore, H. pylori-induced EGFR phosphorylation stimulated PI3K-depnedent activation of the anti-apoptotic factor Akt, increased expression of the anti-apoptotic factor Bcl-2, and decreased expression of the pro-apoptotic factor Bax.
Conclusions
EGFR activation by H. pylori infection has an anti-apoptotic effect in gastric epithelial cells that appears to involve Akt signaling and Bcl family members. These findings provide important insights into the mechanisms of H. pylori-associated tumorigenesis.
Introduction
Helicobacter pylori, a Gram-negative bacterium that selectively colonizes the gastric mucosa, increases the risk of developing gastric adenocarcinoma and gastric non-Hodgkin’s lymphoma in humans and animal models.1, 2 Host responses that mediate H. pylori-associated disease include disruption of the balance between gastric epithelial cell proliferation and apoptosis, which can manifest as increased proliferation and either increased or decreased apoptosis at different stages of infection.3, 4 Increased proliferation that is not balanced by compensatory increases in apoptosis in advanced stage of infection may heighten the risk for development of gastric neoplasia.5 Thus, understanding the mechanism through which H. pylori regulates cell fate is critical for establishing therapeutic approaches to potentially prevent H. pylori infection-induced gastric tumorigenesis.
Both in vivo and in vitro studies have demonstrated that several H. pylori virulence factors stimulate gastric epithelial apoptosis, such as products of the cag pathogenicity island,4, 6 a secreted vacuolating toxin (VacA),7 and lipopolysaccharide.8 The cellular mechanisms by which H. pylori induces apoptosis include activation of Fas9 and cytokine receptors, such as tumor necrosis factor (TNF) receptor10 and induction of spermine oxidase (SMO) activity,11 which lead to Caspase activation and apoptosis in gastric epithelial cells.
H. pylori has also been reported to be associated with attenuated apoptosis following infection,12 and chronically colonized humans and Mongolian gerbils harboring H. pyloricag+ strains develop increased gastric epithelial cell proliferation without an associated increase in apoptosis, suggesting that increased proliferation in the absence of a concomitant increase in apoptosis may contribute to increased risk for gastric cancer with cag+ strains.13–15 However, the mechanism through which H. pylori exerts anti-apoptotic effects remains unclear.
Recent reports show that H. pylori induces up-regulation of EGF receptor (EGFR) expression16 and EGFR activation by heparin-binding (HB)-EGF release from gastric epithelial cells.17, 18 EGFR is a member of the ErbB family which consists of four tyrosine kinase receptors, EGFR (ErbB1), and ErbB2-4. These four receptors are essential for modulating cell survival, proliferation, and differentiation in many tissue types.19
However, there is no report that has identified the role of EGFR activation in the pathogenesis of H. pylori infection. Therefore, the purpose of this study was to define gastric epithelial cellular responses regulated by EGFR activation following H. pylori infection. Our studies using in vitro cell culture and in vivo animal models show that transactivation of EGFR by H. pylori protects gastric epithelial cells from apoptosis and that this anti-apoptotic response is mediated by PI3K-dependent activation of Akt and Bcl family members. Thus, our studies demonstrate that activation of EGFR may represent a previously unrecognized molecular regulatory step in determining the effects of H. pylori on regulation of gastric epithelial cell homeostasis. Since EGFR has been implicated in a number of epithelial cancers and serves as a target for cancer therapy,20, 21 results in this report provide a novel focus for further investigations into the mechanisms of H. pylori-associated tumorigenesis
Materials and Methods
Information regarding inhibitors, antibodies, reagents, cell treatment, gastric epithelial cell isolation, cellular lysate preparations, immunohistochemistry, and apoptosis assays are provided in the Supplemental Data.
H. pylori culture
The H. pylori cag+ rodent adapted strain 7.13 was grown in Brucella broth with 5% FBS for 24 h.22 Bacteria were identified as H. pylori by urease and oxidase activity and Gram’s stain morphology. H. pylori were washed with PBS and cell culture medium before addition to cells.
Cell culture, retroviral transduction, and transient transfection of siRNA
Mouse conditionally immortalized stomach epithelial cells (ImSt) were isolated from the gastric epithelium of transgenic mice with a temperature-sensitive mutation of the simian virus 40 (SV40) large tumor antigen gene (tsA58) fused to the promoter of the mouse H-2Kb class I gene (H-2Kb-tsA58 mice).23 A disintegrin and matrix metalloproteinase-17 (ADAM-17)−/−ImSt were isolated from the gastric epithelium of ADAM-17 null heterozygous mice crossed with the H-2Kb-tsA58 mice.24
ImSt were maintained in RPMI 1640 medium containing 10% FBS, 5 U/ml of murine IFN-γ and 100 U/ml penicillin and streptomycin on collagen-coated plates and grown under permissive conditions at 33°C with 5% CO2. Prior to all experiments, cells were transferred to 37°C (non-permissive) conditions and cultured in medium containing 0.5% FBS, without IFN-γ or antibiotics for 16–18 hours.
ADAM-17−/− ImSt reconstituted to express wild-type (wt) ADAM-17 or catalytically-inactive E406A mutant ADAM-1725 were prepared as described before.26 Briefly, C-terminally HA-tagged murine wt and E406A ADAM17 were cloned into the pBM-IRES-PURO retroviral vector. All cDNA constructs were verified by DNA sequencing. For retroviral transduction, ADAM17−/− ImSt were seeded into tissue culture flasks and cultured for 24 h before incubation with virus stock for 12 h in the presence of 4 μg/ml polybrene. Cells were then grown for 48 h in normal cell culture medium before passage into medium containing 5 μg/ml puromycin.
ImSt were transiently transfected with either non-targeting siRNA or mouse EGFR SMARTpool siRNA at 70% confluence using Lipofectamine 2000.27 After 24 h of transfection, cells were cultured in medium containing 0.5% FBS for 16–18 hours before treatment.
AGS cells were grown in RPMI 1640 medium containing 10% FBS and 100 U/ml penicillin and streptomycin at 37°C with 5% CO2. Prior to all experiments, cells were cultured in medium containing 0.5% FBS without antibiotics for 16–18 h.
Mice and H. pylori infection and gastric tissue preparations
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at Vanderbilt University. C57BL/6wa2 mice with a naturally occurring EGFR kinase defective mutation (Val to Gly mutation at residue 743 in the kinase domain)28 were obtained from Dr. David Threadgill (University of North Carolina, Chapel Hill). PCR primers specific for the EGFR sequence containing the point mutation were used for genotyping (sequences available upon request). Mice (8–10 weeks old, 25–30g) were anesthetized and then infected orally with H. pylori (109 cfu) or broth only in a total volume of 500 μl for 24 h.29
Gastric tissue was fixed in 10% neutral-buffered formalin and paraffin-embedded before sectioning. The gastric mucosa was scraped into homogenization buffer and tissue was lysed.30
Apoptosis assays
Apoptosis in cell lines was detected using ApopTag In Situ Apoptosis Detection Kits (TUNEL).31, 32 For Annexin V-FITC staining, attached cells were dissociated using Accutase and double stained with Annexin V-FITC and propidium iodide.33 The percentage of apoptotic cell populations was measured by flow cytometry.11
Apoptosis was detected in gastric tissue sections using ApopTag™ In Situ Oligo Ligation (ISOL) Kit, and observed by DIC microscopy.31, 32 Apoptotic cells were quantified by counting the absolute number of positive stained cells in at least 500 gastric glands.
Flow cytometric analysis of active caspase-3 and Bcl-2
ImSt were fixed with 0.1% paraformaldehyde and incubated with a rabbit anti-active caspase-3 antibody and a mouse anti-Bcl-2 antibody, and were further stained using FITC-conjugated anti-rabbit and APC-conjugated anti-mouse antibodies, respectively.33 The percentage of active caspase-3 and/or Bcl-2 positive staining cell populations was measured by flow cytometry.
Statistical analysis
Statistical significance in each study was determined by one-way ANOVA followed by Newman-Keuls analysis for multiple comparisons using Prism 5.0 (GraphPad Software, Inc.). A p-value < 0.05 was defined as statistically significant. All data presented are representative of at least three repeat experiments and are presented as mean±S.E.M.
Results
Inhibition of H. pylori-stimulated EGFR kinase activity and protein expression increases apoptosis in ImSt
H. pylori induces up-regulation of EGF receptor (EGFR) expression16 and EGFR transactivation18 in gastric epithelial cells. Phosphorylation of Y1068 on EGFR mediates activation of the ERK/MAP and results in recruitment of the p85 subunit of PI3K to the EGF receptor, leading to activation of Akt.34 One significant role of EGFR activation is to promote cell survival. Therefore, we studied the effect of inhibiting EGFR tyrosine kinase activity on H. pylori-stimulated apoptosis in ImSt. H. pylori were co-cultured with ImSt in the presence or absence of the EGFR tyrosine kinase inhibitor, AG1478, and EGFR Y1068 phosphorylation and apoptosis were analyzed. As expected, H. pylori-induced EGFR activation was inhibited by the EGFR tyrosine kinase inhibitor (Figure 1A). However, the expected increase in apoptosis initiated by H. pylori was further increased by blocking EGFR tyrosine kinase activity, as detected by Annexin V staining and flow cytometry (Figure 1B and C) and TUNEL assay (Figure 1D and E).
Figure 1. EGFR kinase activity is required for surv ival of gastric epithelial cells infected with H. pylori.

ImSt were infected with the wt H. pyloricagA+ strain 7.13 (bacteria:cells = 100:1) for 90 min (A) or 24 h (B–E) pretreated for 30 min with AG1478 (150 nM) or not pretreated. A: EGFR Y1068 phosphorylation was detected by Western blot analysis of cellular lysates using an anti-EGFR-phospho (P)-Y1068 antibody. An anti-EGFR antibody was used as a loading control. Band density was quantified using ImageJ software. The relative density was calculated by comparing density of EGFR-P-Y1068 to the density of EGFR. The relative density in the control group (no AG1478 and no H. pylori) was set as 1.0. The relative densities in the treated groups were compared to that of control. B and C: Cells were stained with Annexin V-FITC and PI, and flow cytometry was performed. The percentage of cells in each quadrant is shown (C). The upper right quadrant represents late apoptosis, and the lower right quadrant represents early apoptosis. D and E: Cells were fixed for TUNEL assay, and apoptotic nuclei were labeled with FITC (green) and total nuclei were labeled with DAPI (blue). FITC and DAPI labeled images were taken from the same field. Apoptosis was determined by counting at least 500 cells. The percentage of cells undergoing apoptosis is shown (E). Data in this and subsequent figures are representative of at least three separate experiments. * p < 0.05 vs cells alone, ** and *** p < 0.001 vs cells alone, § and §§ p < 0.001 vs cells infected with H. pylori, + p < 0.001 vs cells treated with AG1478.
To investigate a potential role for EGFR in suppressing H. pylori-induced apoptosis, we used siRNA directed against EGFR to reduce EGFR expression in ImSt (Figure 2A). Consistent with the results of experiments in which EGFR kinase activity was inhibited, loss of EGFR expression significantly enhanced H. pylori-stimulated apoptosis, compared to non-targeting control siRNA transfection (Figure 2B). Taken together, these results demonstrate that EGFR expression and EGFR tyrosine kinase activity enhance survival of gastric epithelial cells infected by H. pylori.
Figure 2. Suppression of EGFR expression promotes apoptosis in H. pylori-infected gastric epithelial cells.
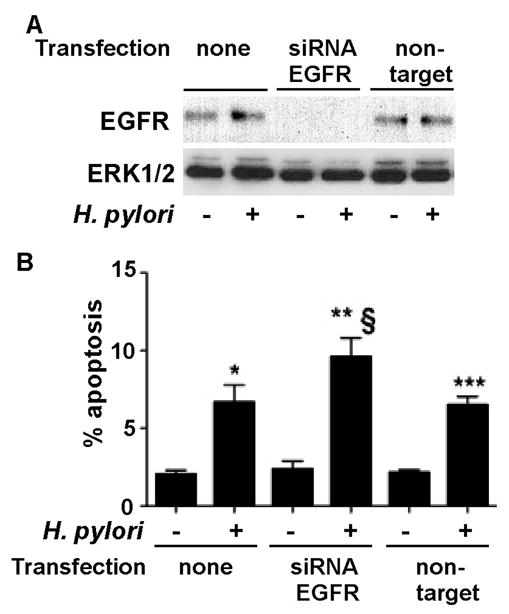
ImSt tansfected with EGFR siRNA or non-targeting siRNA were infected with H. pylori for 24 h. A: EGFR expression levels were analyzed by Western blot analysis of cellular lysates using an anti-EGFR antibody. An anti-ERK1/2 antibody was used as a loading control. B. Apoptosis was detected using TUNEL assay as described in Figure 1. Apoptosis was determined by counting at least 500 cells. The percentage of cells undergoing apoptosis is shown. *, **, and ***p < 0.001 vs uninfected cells in each cell line, § p < 0.001 vs non-transfected cells infected with H. pylori.
Loss of EGFR kinase activity enhances H. pylori-induced gastric epithelial cell apoptosis in vivo
Our in vitro data indicate that EGFR activation plays an anti-apoptotic role in H. pylori-infected cells. To determine whether these effects could be extended in vivo, wild type (wt) or EGFR kinase defective (EGFRwa2) mice on a C57BL/6 background were infected with H. pylori. Mice were sacrificed 5 h or 24 h after infection and tissue was fixed and prepared for immunohistochemistry or mucosal scrapings were prepared for Western blot analysis. The presence of H. pylori in both wt and EGFRwa2 mice was confirmed by immunostaining (Figure 3A). H. pylori-induced EGFR Y1068 phosphorylation was detected in gastric mucosal lysates isolated from wt, but not in EGFRwa2 mice, despite similar levels of bacterial infection (Figure 3B). Importantly, immunostaining showed that H. pylori-stimulated EGFR Y1068 phosphorylation was present in wt, but was absent in EGFRwa2 mice (Figure 3C), which is consistent with our in vitro findings using ImSt (Figure 1A).
Figure 3. Loss of EGFR kinase activity promotes gastric epithelial cell apoptosis in mice infected with H. pylori.

Wt and EGFR wa2 (EGFR kinase defective) mice on a C57BL/6 background were infected with H. pylori (109 cfu/mouse) for 5 or 24 h. Paraffin-embedded stomach tissues (A, C–D) and gastric mucosal lysates (B) were prepared. A. Immunostaining with anti-H. pylori antiserum was performed to detect the presence of H. pylori (arrowheads). B: EGFR Y1068 phosphorylation was detected by Western blot analysis of gastric mucosal lysates as described in Figure 1. C: EGFR Y1068 phosphorylation was detected using immunostaining with anti-EGFR Tyr1068 and FITC-labeled secondary antibody. Green represents positive labeling (L: Luminal side). D: Apoptosis was detected by ISOL staining and active caspase-3 immunostaining using an anti-active caspase-3 antibody. Apoptotic nuclei were labeled with peroxidase and developed with DAB and observed using DIC microscopy. Brown nuclei (arrows) represent ISOL or activated caspase-3 positive staining cells. n = 5–7 mice for each condition.
To determine if EGFR mediates H. pylori regulation of cell survival in vivo, we performed ISOL and anti-active caspase-3 staining to detect apoptosis. H. pylori infection for 24 h did not stimulate apoptosis in the gastric epithelium of wt mice. However, apoptosis was induced by H. pylori infection in EGFRwa2 mice (Figure 3D). Thus, these data indicate that survival of gastric epithelial cells in mucosa infected by H. pylori requires functional EGFR in vivo.
We next examined the relationship between EGFR-regulated apoptosis and proliferation in H. pylori-infected mice. Interestingly, apoptotic cells were limited to the epithelial cell layer of the lower one third of the gastric glands (Figure 3D). However, proliferating gastric epithelial cells were localized to the upper one third of the gastric glands, and no difference in gastric epithelial cell proliferation rates was detected in wt mice after 24 h of H. pylori infection compared to non-infected mice (SD Figure 1). Therefore, no overlap was present between proliferative and apoptotic cell populations. Given the evidence that both gastric epithelial cells with H. pylori-activated EGFR and apoptotic gastric epithelial cells localized to the lower one-third of gastric glands (Figure 3C and D) and no proliferation was stimulated by H. pylori in this zone, these data suggest that H. pylori-activated EGFR after 24 h infection exerts an anti-apoptotic, but not a proliferative role, in gastric epithelial cells.
Metalloproteinase activity and EGFR ligand release are required for suppressing H. pylori-induced gastric epithelial cell apoptosis through activation of EGFR
Activation of Src family protein kinases, leading to metalloproteinase-dependent ligand release, serves as one of the mechanisms by which EGFR is transactivated.35 Previous studies have reported that H. pylori activated EGFR is mediated by stimulation of HB-EGF release from gastric epithelial cells,17, 18 since ADAM-17 can regulate HB-EGF cleavage and ligand binding,36, 37 we determined whether ADAM-17 is involved in H. pylori-regulated EGFR transactivation and apoptosis. In ADAM-17−/− ImSt, H. pylori failed to activate EGFR (Figure 4A) and stimulated increased apoptosis (Figure 4B–C). Infection of wt ADAM-17, but not the catalytically-inactive E406A mutant ADAM-17 restored EGFR activation (Figure 4A) and reduced apoptosis induced by H. pylori (Figure 4B–C), indicating a requirement for ADAM-17 in the H. pylori-induced EGFR activation-dependent cell survival phenotype.
Figure 4. ADAM-17 mediates H. pylori-induced activation of EGFR and is required for an anti-apoptotic effects.

Cells were infected with H. pylori for 90 min (A) or 24 h (B and C). EGFR Y1068 phosphorylation was analyzed by Western blot analysis of cellular lysates (as described in Figure 1), activation of Akt using anti-phospho (P)-Ser 473 Akt, ERK1/2 MAPK activation using anti-phospho (P)-Thr183/Tyr185 ERK1/2, and p38 using anti-phospho (P)-Tyr180/182 p38 antibodies. B–C: Cells were fixed for TUNEL assay and apoptotic nuclei were labeled with peroxidase and developed with DAB and observed using DIC microscopy. Brown staining represents apoptotic nuclei (B). Apoptotic cells were enumerated by counting at least 500 cells. The percentage of cells undergoing apoptosis is shown (C). *, **, *** and ****p < 0.001 vs uninfected cells in each cell line. § and # p < 0.001 vs ImSt and wt ADAM-17 transfected cells infected with H. pylori.
Since HB-EGF is a ligand generated by ADAM-17 to activate EGFR38 and H. pylori stimulates production of HB-EGF, we next determined whether inhibition of HB-EGF altered H. pylori-stimulated EGFR activation and apoptosis. Pretreatment of ImSt with HB-EGF neutralizing antibody blocked H. pylori activation of EGFR (Figure 5A) and increased apoptosis (Figure 5B). Importantly, pretreatment of AGS cells with HB-EGF completely blocked H. pylori-stimulated apoptosis (Figure 5B), suggesting that HB-EGF is not only required for EGFR activation, but also for gastric epithelial cell survival in response to H. pylori infection.
Figure 5. H. pylori -induced gastric epithelial cell apoptosis is enhanced by blocking HB-EGF binding toEGFR, but is prevented by HB-EGF pretreatment.

AGS cells were pretreated for 30 min with an anti-HB-EGF neutralizing antibody (3 μg/ml) or HB-EGF (10 ng/ml), as indicated, and then infected with H. pylori for 90 min (A) or 24 h (B) A: Cellular lysates were analyzed by Western blot analysis to detect EGFR, Akt, ERK1/2 MAPK and p38 activation as described in Figures 1 and 4. B. Apoptosis was detected using TUNEL assay as described in Figure 4. Apoptotic cells were enumerated by counting at least 500 cells. The percentage of cells undergoing apoptosis is shown. * p < 0.001 vs cells alone, ** p < 0.001 vs cells treated with HB-EGF antibody only. § p < 0.001 vs cells infected with H. pylori.
Akt and Bcl-2 family members are targets of EGFR activation that prevent H. pylori-stimulated apoptosis in gastric epithelial cells
One of the major downstream signaling targets of EGFR is PI3K. Akt is the principal mediator of PI3K-regulated signaling pathways for promoting cell survival. Akt is also an anti-apoptotic target for EGFR transactivation by TNF.27 Therefore, we tested whether EGFR is required for activation of Akt by H. pylori. H. pylori-induced AKT and EGFR activation was blocked in ADAM-17−/−ImSt, and these responses were reconstituted in cells expressing wt, but not catalytically-inactive E406A ADAM-17−/− (Figure 4A). Furthermore, HB-EGF neutralizing antibody inhibited Akt and EGFR activation by H. pylori (Figure 5A). H. pylori infection-induced Akt activation in ImSt was inhibited by blocking EGFR or PI3K kinase activities using an EGFR kinase inhibitor AG1478 or a PI3K kinase inhibitor wortmannin, respectively (Figure 6A). Importantly, pretreatment of ImSt with wortmannin enhanced H. pylori-induced apoptosis (Figure 6B).
Figure 6. Akt is a target of EGFR for survival of gastric epithelial cells infected with H. pylori.

ImSt were infected with H. pylori 90 min (A) or 24 h (B) in the presence or absence of 30 min pretreatment with AG1478 (150 nM), a PI3K inhibitor, wortmannin (100 nM), or MEK inhibitor, PD98059 (10 μM). A: Cellular lysates were collected for detecting EGFR, Akt and ERK1/2 MAPK activation by Western blot analysis as described in Figures 1 and 4. B: Cells were fixed for TUNEL assay as described in Figure 4. Apoptosis was determined by counting at least 500 cells. The percentage of cells undergoing apoptosis is shown. *, **, and *** p < 0.001 vs uninfected cells in each inhibitor treatment group. § p < 0.001 vs ImSt infected with H. pylori.
Since Akt has direct effects on the apoptotic machinery through regulating expression of several factors, including a pro-apoptotic protein, Bax, and an anti-apoptotic protein, Bcl-2,39 we studied expression of the Bcl family member Bcl-2 in gastric epithelial cells. Bcl-2 and active caspase-3, markers for apoptosis, were assessed in the same cells by flow cytometry. Blocking EGFR kinase activity by AG1478 decreased the population of ImSt with H. pylori-stimulated Bcl-2 expression. However, the total population of cells with active caspase-3 was enhanced by AG1478 (Figure 7A–B), due to a large increase in Bcl-2−/active caspase-3+ cells (left upper quadrant, Figure 7A)
Figure 7. Role of EGFR in regulating anti-apoptotic and pro-apoptotic proteins in gastric epithelial cells.

A and B: ImSt were pretreated for 30 min with a EGFR kinase inhibitor, AG1478 (150 nM) or not pretreated, and then infected with H. pylori 24 h. Cells were fixed and stained using antibodies against active caspase-3 and Bcl-2 and flow cytometry was performed. Representative dot plots (A) show the presence of a population of cells that are both low Bcl-2 and active caspase-3 (left lower quadrant), high Bcl-2 and low active caspase-3 (right lower quadrant), low Bcl-2 and high active caspase-3 (left upper quadrant), and high Bcl-2 and active caspase-3 (right upper quadrant). Percentage of Bcl-2 or active caspase-3 cells are shown (B). C and D: Mice were infected with H. pylori (109 cfu/mouse) for 24 h. C: Gastric mucosal lysates were prepared for Western Blot analysis to detect relative Bax and Bcl-2 protein expression levels using anti-Bax and anti-Bcl-2 antibodies, respectively, and EGFR phosphorylation and actin levels were determined as described in Figure 1. D: The relative densities of Bax and Bcl-2 protein bands on Western blots were compared to determine the protein expression ratio of Bax to Bcl-2 in each group. The ratio in broth treated mice was set as 100%, and fold change of the ratios in H. pylori-infected mice were compared to the broth treated group. N = 5–7 mice for each condition. * p < 0.05 vs cells alone, ** p < 0.001 vs cells alone, *** p < 0.001 vs cells treated with AG1478, § p < 0.001 vs cells infected with H. pylori. @ p < 0.001 vs broth treatment in wt mice. # p < 0.001 vs broth treatment in EGFRwa2 mice.
We also investigated Bcl-2 and Bax expression in the gastric mucosa of mice infected with H. pylori. Infection with H. pylori decreased Bax expression and increased Bcl-2 expression in wt mice, but these effects were not observed in the EGFRwa2 mice (Figure 7C). In fact, the ratio of Bax:Bcl-2 was reversed in EGFRwa2 mice, compared to wt mice (Figure 7D), which represents an increased apoptotic response in the EGFRwa2 mice upon H. pylori infection. This regulatory effect of H. pylori on Bcl-2 and Bax was confirmed in gastric epithelial cells isolated from the stomachs of mice infected with H. pylori (SD Figure 2). Thus, these results suggest that Bax and Bcl-2 are downstream targets of EGFR transactivation that promote an anti-apoptotic response in H. pylori-infected gastric epithelial cells.
Discussion
In this study, we have demonstrated that activation of EGFR is a key regulatory step in promoting an anti-apoptotic response in gastric epithelial cells infected with H. pylori in vitro and in vivo. Furthermore, our data provide evidence that H. pylori-induced EGFR activation results in activation of Akt/PI3K kinase activity and regulation of Bcl family members which promote gastric epithelial cell survival. These novel findings indicate that activation of EGFR may be a previously unrecognized “molecular switch” determining the survival response of H. pylori-infected gastric epithelial cells. We speculate that EGFR activation may function to inhibit apoptosis caused by activation of Fas9 and cytokine receptors10 and SMO activation that leads to generation of H2O2, which results in cytochrome c release and caspase-3 activation11, 33 (Figure 8).
Figure 8.
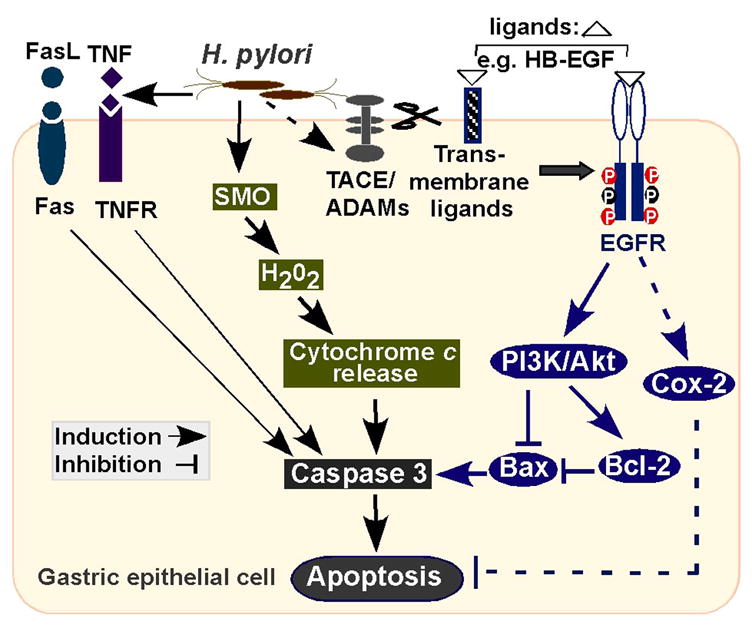
A schematic model of mechanisms for determining the fate of gastric epithelial cells infected with H. pylori.
EGFR is a member of the ErbB family which consists of four tyrosine kinase receptors, EGFR (ErbB1), and ErbB2-4. These receptors promote cell survival, proliferation, migration, and differentiation in many tissue types.19 EGFR and other ErbB family members can be activated by direct interaction with EGF-like ligands or transactivated by various extracellular stimuli, such as TNF,27 which initiate formation of homo- and/or hetero-dimers and increased kinase activity.40, 41 Therefore, we recognize that other ErbBs may also be potential targets of H. pylori infection. In fact, H. pylori stimulates ErbB3 tyrosine phosphorylation, but not ErbB2 or 4 in ImSt (data not shown), thus, H. pylori may increase EGFR/ErbB3 heterodimer formation. Since ErbBs have been shown to couple EGFR to the PI3K/Akt pathway in gefitinib-sensitive non-small cell lung cancer cell lines,42, EGFR activation by H. pylori may be amplified by heterodimer formation with ErbB3. These studies are currently underway in our laboratory.
Given a requirement for metalloproteinase activity in H. pylori-initiated HB-EGF release for EGFR transactivation, ADAM-17 is an ideal candidate enzyme for regulation of this pathway.18, 43 Our data show that ADAM-17 is required for preventing H. pylori-induced apoptosis (Figure 4). ADAM-17 is the first ADAM to have a defined physiological substrate, which is the precursor transmembrane form of TNF. ADAM-17 is required for the release of soluble TNF and at least 3 EGFR ligands, HB-EGF, amphiregulin (AR), transforming growth factor (TGF)-α.36, 44 Previous studies have shown that H. pylori stimulates AR release from gastric mucosal cells.45 Thus AR and TNF release may be another ligand for H. pylori activation of EGFR. TNF production is increased in a number of gastrointestinal disorders, including H. pylori-induced gastritis,46 and TNF transactivates EGFR to promote cell survival in colon epithelial cells. 27 Although ADAM-17 is ubiquitously present in the human gastrointestinal tract, its expression and catalytic activity are increased in ulcerative colitis,47 and it regulates TNF levels in rodent models of colitis.48 However, while ADAM-17 is a target of drug development for inflammatory conditions,49 the disorganized and inflamed nature of the gastrointestinal tract seen in ADAM-17-deficient mice suggests this metalloproteinase may also play important roles in gut epithelial homeostasis, perhaps through regulation of EGFR ligands.36, 44, 50, 51 Therefore, a better understanding of the function of ADAM-17 during H. pylori-induced gastric epithelial injury may provide insights into its potential role in gastric adenocarcinoma.
Elucidating the down-stream targets of EGFR activation by H. pylori is important for understanding the cellular responses induced by H. pylori. We show that H. pylori-initiated EGFR activation is required for activation of an anti-apoptotic signal, Akt/PI3K. In fact, since the submission of this current manuscript another report has now demonstrated specific reduction of Akt via siRNA increases apoptosis in response to H. pylori infection of human gastric epithelial cells (Nagy et al., Journal of Infectious Diseases, In press). However, inhibition of EGFR does not block H. pylori-induced pro-apoptotic signals, such as p38 (Figures 4 and 5), or JNK activation.18 This evidence supports a model whereby pro-apoptotic signals become physiologically predominant in cells that do not express EGFR or lose EGFR kinase activity. We show that H. pylori-initiated EGFR activation is required for Akt/PI3K and ERK1/2 MAPK activation in gastric epithelial cells (Figures 4, 5). Interestingly, although H. pylori-stimulated ERK1/2 MAPK activation in ImSt requires EGFR (Figures 4A and 5A and18), inhibition of ERK1/2 MAPK activation does not enhance H. pylori-induced apoptosis (Figure 6B). These data suggest that Akt is a potential target of EGFR activation for preventing H. pylori-stimulated apoptosis in gastric epithelial cells. However, another group reported a role of H. pylori-induced ERK1/2 MAPK activation in prevention of apoptosis.52 This difference in the role of H. pylori-activated ERK1/2 MAPK may depend on host and/or bacterial strain differences.
In addition to PI3K-dependent Akt activation up-regulation of Bcl-2 and down-regulation of Bax expression (Figure 7), cyclooxygenase 2 (COX-2) may serve as another downstream target of EGFR activation by H. pylori that can induce anti-apoptotic responses in gastric epithelial cells. The COX-2 level in gastric epithelial cells is up-regulated by H. pylori infection,53, 54 and COX-2 inhibitors increase H. pylori-induced apoptosis.55 We have also found that transactivation of EGFR is required for H. pylori up-regulation of COX-2 expression (unpublished observation).
In summary, our study demonstrates that H. pylori-stimulated EGFR activation exerts an anti-apoptotic role in gastric epithelial cells in vitro and in vivo through its downstream targets, Akt and Bcl2 family members. In vivo chronically infected humans and Mongolian gerbils harboring cag+ strains have increased gastric epithelial cell proliferation without an associated increase in apoptosis, suggesting that increased proliferation in the absence of a concomitant increase in apoptosis may contribute to increased risk for gastric cancer.14, 15 Additionally, the lack of gastric epithelial cell apoptosis with acute H. pylori infection in vivo that we have attributed to EGFR transactivation may contribute to the ability of H. pylori to colonize, as there may be less neutrophil infiltration that would otherwise be recruited to engulf cellular debris and may serve to defend against H. pylori infection.56 Important questions regarding the role of EGFR transactivation in the progression from gastric infection to neoplasia and the interaction of this pathway with the recently reported links between EGFR activation and macrophage recruitment57 and activation58 need to be addressed to fully understand the preventive and therapeutic implications of the findings on gastric carcinoma development.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by NIH grants DK56008 for DBP, DK053620 and Office of Medical Research, Department of Veterans Affairs for KTW, DK58587, DK73902, and CA77955 for RMP, AI 068009, AI39657, and Department of Veterans Affairs for TLC, DK63363 and a Crohn’s and Colitis Foundation of America grant for PJD, DK58404 for Vanderbilt University Digestive Disease Research Center, and a NCI program project grant CA116087.
Abbreviations
- ADAM-17/TACE
A disintegrin and matrix metalloproteinase-17/tumor necrosis factor a converting enzyme
- EGFR
EGF receptor
- HB-EGF
heparin-binding-EGF
- ISOL
in situ oligo ligation
- ImSt
immortalized stomach epithelial cells
Footnotes
No conflicts of interest exist.
Author contribution: FY and RC contributed to the collection, analysis and interpretation of date and manuscript preparation. HC, UK, SSH, PJD, and MKW contributed to the collection, analysis, and interpretation of date. RMP, TLC, KTW, and DBP contributed to the analysis and interpretation of date and manuscript preparation.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 2.Peek RM, Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–48. doi: 10.1002/path.1868. [DOI] [PubMed] [Google Scholar]
- 3.Crabtree JE, Court M, Aboshkiwa MA, et al. Gastric mucosal cytokine and epithelial cell responses to Helicobacter pylori infection in Mongolian gerbils. J Pathol. 2004;202:197–207. doi: 10.1002/path.1498. [DOI] [PubMed] [Google Scholar]
- 4.Moss SF, Sordillo EM, Abdalla AM, et al. Increased gastric epithelial cell apoptosis associated with colonization with cagA+Helicobacter pylori strains. Cancer Res. 2001;61:1406–11. [PubMed] [Google Scholar]
- 5.Ernst PB, Crowe SE, Reyes VE. How does Helicobacter pylori cause mucosal damage? The inflammatory response. Gastroenterology. 1997;113:S35–42. doi: 10.1016/s0016-5085(97)80009-1. [DOI] [PubMed] [Google Scholar]
- 6.Gupta RA, Polk DB, Krishna U, et al. Activation of peroxisome proliferator-activated receptor γ suppresses nuclear factor κB-mediated apoptosis induced by Helicobacter pylori in gastric epithelial cells. J Biol Chem. 2001;276:31059–31066. doi: 10.1074/jbc.M104141200. [DOI] [PubMed] [Google Scholar]
- 7.Cover TL, Krishna US, Israel DA, et al. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951–7. [PubMed] [Google Scholar]
- 8.Kawahara T, Teshima S, Kuwano Y, et al. Helicobacter pylori lipopolysaccharide induces apoptosis of cultured guinea pig gastric mucosal cells. Am J Physiol Gastrointest Liver Physiol. 2001;281:G726–34. doi: 10.1152/ajpgi.2001.281.3.G726. [DOI] [PubMed] [Google Scholar]
- 9.Rudi J, Kuck D, Strand S, et al. Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori-induced gastric epithelial apoptosis. J Clin Invest. 1998;102:1506–14. doi: 10.1172/JCI2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibata J, Goto H, Arisawa T, et al. Regulation of tumour necrosis factor (TNF) induced apoptosis by soluble TNF receptors in Helicobacter pylori infection. Gut. 1999;45:24–31. doi: 10.1136/gut.45.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H, Chaturvedi R, Cheng Y, et al. Spermine oxidation induced by Helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Res. 2004;64:8521–5. doi: 10.1158/0008-5472.CAN-04-3511. [DOI] [PubMed] [Google Scholar]
- 12.Shirin H, Sordillo EM, Kolevska TK, et al. Chronic Helicobacter pylori infection induces an apoptosis-resistant phenotype associated with decreased expression of p27(kip1) Infect Immun. 2000;68:5321–8. doi: 10.1128/iai.68.9.5321-5328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mimuro H, Suzuki T, Nagai S, et al. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe. 2007;2:250–63. doi: 10.1016/j.chom.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Peek RM, Jr, Moss SF, Tham KT, et al. Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst. 1997;89:863–8. doi: 10.1093/jnci/89.12.863. [DOI] [PubMed] [Google Scholar]
- 15.Peek RM, Jr, Wirth HP, Moss SF, et al. Helicobacter pylori alters gastric epithelial cell cycle events and gastrin secretion in Mongolian gerbils. Gastroenterology. 2000;118:48–59. doi: 10.1016/s0016-5085(00)70413-6. [DOI] [PubMed] [Google Scholar]
- 16.Keates S, Keates AC, Katchar K, et al. Helicobacter pylori induces up-regulation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Infect Dis. 2007;196:95–103. doi: 10.1086/518440. [DOI] [PubMed] [Google Scholar]
- 17.Dickson JH, Grabowska A, El-Zaatari MA, et al. Helicobacter pylori can induce heparin-binding epidermal growth factor expression via gastrin and its receptor. Cancer Res. 2006;66:7524–31. doi: 10.1158/0008-5472.CAN-05-3246. [DOI] [PubMed] [Google Scholar]
- 18.Keates S, Sougioultzis S, Keates AC, et al. cag+Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Biol Chem. 2001;276:48127–48134. doi: 10.1074/jbc.M107630200. [DOI] [PubMed] [Google Scholar]
- 19.Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–16. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 20.O’Dwyer PJ, Benson AB., 3rd Epidermal growth factor receptor-targeted therapy in colorectal cancer. Semin Oncol. 2002;29:10–7. doi: 10.1053/sonc.2002.35643. [DOI] [PubMed] [Google Scholar]
- 21.Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of Cancer. J Clin Oncol. 2003;21:2787–2799. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]
- 22.Franco AT, Israel DA, Washington MK, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. 2005;102:10646–51. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitehead RH, VanEeden PE, Noble MD, et al. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci, USA. 1993;90:587–591. doi: 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garton KJ, Gough PJ, Philalay J, et al. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-α-converting enzyme (ADAM 17) J Biol Chem. 2003;278:37459–37464. doi: 10.1074/jbc.M305877200. [DOI] [PubMed] [Google Scholar]
- 25.Garton KJ, Gough PJ, Blobel CP, et al. Tumor necrosis factor-α-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276:37993–8001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- 26.Sanderson MP, Erickson SN, Gough PJ, et al. ADAM10 mediates ectodomain shedding of the betacellulin precursor activated by p-aminophenylmercuric acetate and extracellular calcium influx. J Biol Chem. 2005;280:1826–37. doi: 10.1074/jbc.M408804200. [DOI] [PubMed] [Google Scholar]
- 27.Yamaoka T, Yan F, Cao H, et al. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci USA. 2008;105:11772–11777. doi: 10.1073/pnas.0801463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luetteke NC, Phillips HK, Qiu TH, et al. The mouse waved-2 phenotype results from a point mutation in the EGF receptor tyrosine kinase. Genes & Dev. 1994;8:399–413. doi: 10.1101/gad.8.4.399. [DOI] [PubMed] [Google Scholar]
- 29.Fox JG, Wang TC, Rogers AB, et al. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology. 2003;124:1879–90. doi: 10.1016/s0016-5085(03)00406-2. [DOI] [PubMed] [Google Scholar]
- 30.Polk DB. Ontogenic regulation of PLCγ1 activity and expression in the rat small intestine. Gastroenterology. 1994;107:109–116. doi: 10.1016/0016-5085(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 31.Yan F, Cao H, Cover TL, et al. Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology. 2007;132:562–75. doi: 10.1053/j.gastro.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan F, John SK, Wilson G, et al. Kinase suppressor of Ras protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. J Clin Invest. 2004;114:1272–80. doi: 10.1172/JCI21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaturvedi R, Cheng Y, Asim M, et al. Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J Biol Chem. 2004;279:40161–73. doi: 10.1074/jbc.M401370200. [DOI] [PubMed] [Google Scholar]
- 34.Rodrigues GA, Falasca M, Zhang Z, et al. A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol Cell Biol. 2000;20:1448–1459. doi: 10.1128/mcb.20.4.1448-1459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fischer OM, Hart S, Gschwind A, et al. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203–8. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 36.Sunnarborg SW, Hinkle CL, Stevenson M, et al. Tumor necrosis factor-α converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J Biol Chem. 2002;277:12838–12845. doi: 10.1074/jbc.M112050200. [DOI] [PubMed] [Google Scholar]
- 37.Lee DC, Sunnarborg SW, Hinkle CL, et al. TACE/ADAM17 processing of EGFR ligands indicates a role as a physiological convertase. Ann N Y Acad Sci. 2003;995:22–38. doi: 10.1111/j.1749-6632.2003.tb03207.x. [DOI] [PubMed] [Google Scholar]
- 38.Hinkle CL, Sunnarborg SW, Loiselle D, et al. Selective roles for tumor necrosis factor α-converting enzyme/ADAM17 in the shedding of the epidermal growth factor receptor ligand family: the juxtamembrane stalk determines cleavage efficiency. J Biol Chem. 2004;279:24179–88. doi: 10.1074/jbc.M312141200. Epub 2004 Apr 5. [DOI] [PubMed] [Google Scholar]
- 39.Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–82. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 40.Citri A, Skaria KB, Yarden Y. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003;284:54–65. doi: 10.1016/s0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 41.Carpenter G. ErbB-4: mechanism of action and biology. Exp Cell Res. 2003;284:66–77. doi: 10.1016/s0014-4827(02)00100-3. [DOI] [PubMed] [Google Scholar]
- 42.Engelman JA, Janne PA, Mermel C, et al. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102:3788–93. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wallasch C, Crabtree JE, Bevec D, et al. Helicobacter pylori-stimulated EGF receptor transactivation requires metalloprotease cleavage of HB-EGF. Biochem Biophys Res Commun. 2002;295:695–701. doi: 10.1016/s0006-291x(02)00740-4. [DOI] [PubMed] [Google Scholar]
- 44.Peschon JJ, Slack JL, Reddy P, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 45.Romano M, Ricci V, Di Popolo A, et al. Helicobacter pylori upregulates expression of epidermal growth factor-related peptides, but inhibits their proliferative effect in MKN 28 gastric mucosal cells. J Clin Invest. 1998;101:1604–13. doi: 10.1172/JCI1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindholm C, Quiding-Jarbrink M, Lonroth H, et al. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–71. doi: 10.1128/iai.66.12.5964-5971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brynskov J, Foegh P, Pedersen G, et al. Tumour necrosis factor α converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut. 2002;51:37–43. doi: 10.1136/gut.51.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colon AL, Menchen LA, Hurtado O, et al. Implication of TNF-α convertase (TACE/ADAM17) in inducible nitric oxide synthase expression and inflammation in an experimental model of colitis. Cytokine. 2001;16:220–6. doi: 10.1006/cyto.2001.0969. [DOI] [PubMed] [Google Scholar]
- 49.Black RA. Tumor necrosis factor-α converting enzyme. Int J Biochem Cell Biol. 2002;34:1–5. doi: 10.1016/s1357-2725(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 50.Rio C, Buxbaum JD, Peschon JJ, et al. Tumor necrosis factor-α-converting enzyme is required for cleavage of ErbB4/HER4. J Biol Chem. 2000;275:10379–87. doi: 10.1074/jbc.275.14.10379. [DOI] [PubMed] [Google Scholar]
- 51.van Deventer SJH. A place for TACE. Gut. 2002;51:5–6. doi: 10.1136/gut.51.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi IJ, Kim JS, Kim JM, et al. Effect of inhibition of extracellular signal-regulated kinase 1 and 2 pathway on apoptosis and bcl-2 expression in Helicobacter pylori-infected AGS cells. Infect Immun. 2003;71:830–7. doi: 10.1128/IAI.71.2.830-837.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu S, Ramanujam KS, Wong A, et al. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology. 1999;116:1319–29. doi: 10.1016/s0016-5085(99)70496-8. [DOI] [PubMed] [Google Scholar]
- 54.Romano M, Ricci V, Memoli A, et al. Helicobacter pylori up-regulates cyclooxygenase-2 mRNA expression and prostaglandin E2 synthesis in MKN 28 gastric mucosal cells in vitro. J Biol Chem. 1998;273:28560–3. doi: 10.1074/jbc.273.44.28560. [DOI] [PubMed] [Google Scholar]
- 55.Kim TI, Lee YC, Lee KH, et al. Effects of nonsteroidal anti-inflammatory drugs on Helicobacter pylori-infected gastric mucosae of mice: apoptosis, cell proliferation, and inflammatory activity. Infect Immun. 2001;69:5056–63. doi: 10.1128/IAI.69.8.5056-5063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133:288–308. doi: 10.1053/j.gastro.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 57.Sasaki T, Nakamura T, Rebhun RB, et al. Modification of the primary tumor microenvironment by transforming growth factor α-epidermal growth factor receptor signaling promotes metastasis in an orthotopic colon cancer model. Am J Pathol. 2008;173:205–16. doi: 10.2353/ajpath.2008.071147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oguma K, Oshima H, Aoki M, et al. Activated macrophages promote Wnt signalling through tumour necrosis factor-α in gastric tumour cells. Embo J. 2008;27:1671–81. doi: 10.1038/emboj.2008.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.