

Abstract
Background
Tyrosine kinase inhibitors, such as imatinib, are not considered curative for chronic myeloid leukemia – regardless of the significant reduction of disease burden during treatment – since they do not affect the leukemic stem cells. However, the stochastic nature of hematopoiesis and recent clinical observations suggest that this view must be revisited.
Design and Methods
We studied the natural history of a large cohort of virtual patients with chronic myeloid leukemia under tyrosine kinase inhibitor therapy using a computational model of hematopoiesis and chronic myeloid leukemia that takes into account stochastic dynamics within the hematopoietic stem and early progenitor cell pool.
Results
We found that in the overwhelming majority of patients the leukemic stem cell population undergoes extinction before disease diagnosis. Hence leukemic progenitors, susceptible to tyrosine kinase inhibitor attack, are the natural target for chronic myeloid leukemia treatment. Response dynamics predicted by the model closely match data from clinical trials. We further predicted that early diagnosis together with administration of tyrosine kinase inhibitor opens the path to eradication of chronic myeloid leukemia, leading to the wash out of the aberrant progenitor cells, ameliorating the patient’s condition while lowering the risk of blast transformation and drug resistance.
Conclusions
Tyrosine kinase inhibitor therapy can cure chronic myeloid leukemia, although it may have to be prolonged. The depth of response increases with time in the vast majority of patients. These results illustrate the importance of stochastic effects on the dynamics of acquired hematopoietic stem cell disorders and have direct relevance for other hematopoietic stem cell-derived diseases.
Keywords: chronic myeloid leukemia, tyrosine kinase inhibitors, cure, stochastic dynamics
Introduction
Chronic myeloid leukemia (CML) is an acquired hematopoietic stem cell disorder characterized by expression of the BCR-ABL oncoprotein.1–5 Animal models as well as theoretical considerations on the age-specific incidence of CML in human populations suggest that aberrant BCR-ABL expression alone may be enough to explain the chronic phase of the disease.1,5,6 The BCR-ABL oncoprotein interacts with many substrates in the leukemic cell, which ultimately leads to the CML phenotype.7
The introduction of ABL kinase inhibition with imatinib opened a new era in the therapy of CML.2 However, the lack of evidence that this agent has any direct impact on the leukemic stem cell (LSC)8 has led to questions regarding the capacity of imatinib, or the newer tyrosine kinase inhibitors such as dasatinib or nilotinib, to cure CML.9,10 On the other hand, the therapeutic success of tyrosine kinase inhibitors suggests that they efficiently control disease burden in early progenitors and more committed blood cell lineages. In fact, in the absence of acquired resistance to tyrosine kinase inhibition, CML is no longer fatal and the increasing survival of these patients is projected to make the disease one of the most prevalent leukemias. Moreover, there are now reports of patients with CML who, despite stopping tyrosine kinase inhibitor therapy, have remained free of relapse for significant periods of time.11
Previous investigations of CML, including theoretical models,9,12,13 did not take into account the stochastic nature of hematopoiesis.14 Given the small size of the active hematopoietic stem cell pool,15,16 which is not expanded in CML,3 and of which only a very small fraction is constituted by LSC,13,17 stochastic effects should not be overlooked when investigating cell dynamics.14,18 Moreover, the fact that BCR-ABL does not give a fitness advantage to the LSC19 means that expansion of the LSC clone can only occur by neutral drift. In other words, LSC do not benefit and/or are not dependent on BCR-ABL expression, and their expansion is, therefore, independent of oncoprotein expression. Thus, the expansion or elimination of LSC is the same as that of normal hematopoietic stem cells and dependent on chance alone, a feature which is impossible to capture with a deterministic model, in which equal cell division rates imply a constant ratio of LSC and normal hematopoietic stem cell numbers. Here, we argue that LSC should not be considered the main target for CML eradication. Instead, and in accord with the fact that CML is LSC-derived but progenitor cell driven,20 we show how and why progenitor cells, not LSC, are the major cause of problems related to CML. To this end, we developed a model of hematopoiesis which takes explicitly into consideration its stochastic nature and associated effects. In the majority of simulated cases, we found that continued tyrosine kinase inhibitor therapy (assuming it is well tolerated) has the potential to cure CML despite the fact that these agents do not hit LSC. Our results correlate nicely with independent clinical data21 and we employed our model to predict the probability of disease relapse as a function of duration of therapy.
Design and Methods
Normal hematopoiesis
Normal hematopoiesis can be represented by a hierarchical model in dynamic equilibrium in which cells move along the hematopoietic tree as they become increasingly differentiated.22 In a healthy adult, approximately 400 hematopoietic stem cells, which each replicate on average once per year,15,23 are responsible for the daily marrow output of approximately 3.5×1011 cells. As cells differentiate, they reach new levels of the hematopoietic tree, and we associate a specific compartment to each stage of cell differentiation (Figure 1). Cell divisions contribute to differentiation with a probability ɛ and to amplification with a probability of 1-ɛ across the hematopoietic tree.22 When a cell in compartment i divides and the two daughter cells differentiate they move to the next compartment (i+l). Cells in compartment i replicate at a rate ri that increases exponentially together with compartment size (Nj). Adjacent compartments are related by Ni/Ni−l = y = 1.93 and ri/ri−l = r = 1.26. From prior work, we have determined that there are 32 compartments (K=32) in the hematopoietic tree and that ɛ=0.85 for normal hematopoiesis.22 We capture the dynamics of hematopoiesis combining three different approaches including population dynamics in discrete time, age-structured populations and a continuous model when the cell population is large enough. This is similar to cell dynamics in the colonic crypt as described by Johnston et al.24,25 where these approaches are discussed in detail.
Figure 1.
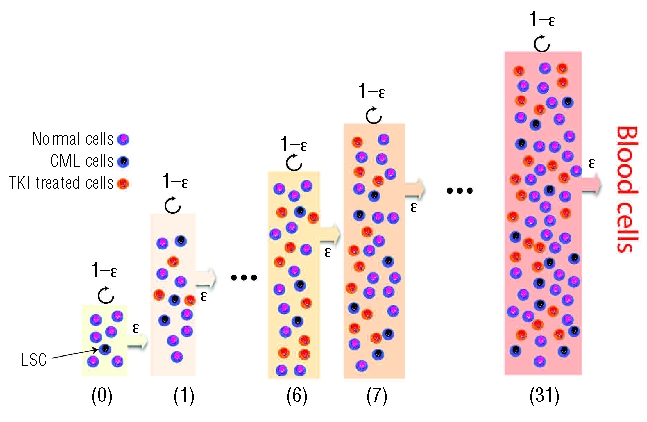
Hematopoiesis and CML dynamics with and without treatment. Illustration of a typical treatment stage of the hierarchical tree model adopted here (further details provided in the Design and Methods section). Normal, CML and tyrosine kinase inhibitor (TKI)-treated cells (imatinib) co-evolve at each stage of differentiation. Cell differentiation occurs with probability ɛ, which depends on cell type, as indicated; otherwise the cell undergoes self-renewal. Because of the relative fitness difference between cell types (inset of Figure 3A), normal cells will out-compete TKI-treated cells (but not CML cells). This leads to the reduction of disease burden achieved with treatment (Figure 5).
Stem cell dynamics
BCR-ABL expression changes the properties of the progeny of LSC, but not the LSC directly.19,26 The active hematopoietic stem cell pool is not expanded in CML,3 hence the evolutionary dynamics of hematopoietic stem cells and LSC can be described by a neutral Moran process in a population of approximately 400 cells.22,23 Disease dynamics typically starts with the appearance of the first LSC and at a given interval of time, one cell is chosen at random for reproduction and subsequently another cell is chosen for export (differentiation) so that the cell population remains constant under an appropriate feedback mechanisms24,25 (Figure 2). When 400 ‘selection-reproduction-export’ events have occurred, 1 year has passed. Export of a LSC starts the expansion of the CML progenitor pool.
Figure 2.
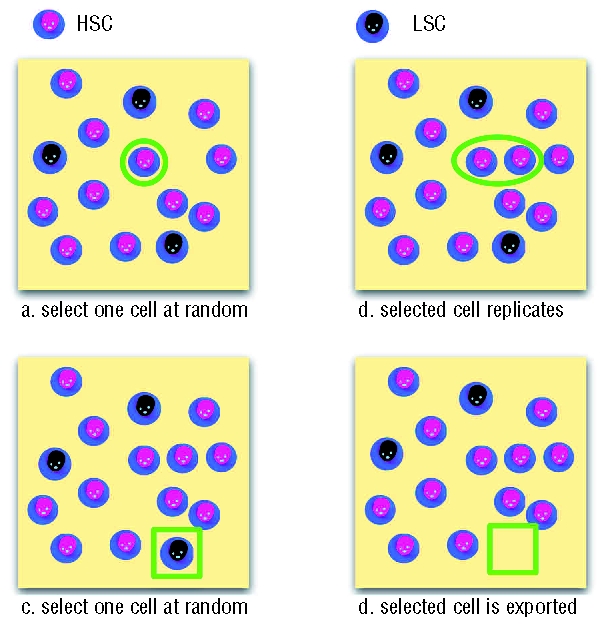
Moran dynamics within the active stem cell pool. Hematopoietic stem cells (HSC) and LSC (identified on top) undergo a stochastic Moran birth-death process which conserves the population size, consistent with the observed lack of expansion of the stem cell pool in CML.3 In the first step (upper panel, a) one of the N cells is chosen at random to replicate (circled cell in a), producing an additional identical cell and increasing the population by one (b). Subsequently one of the N+1 cells is chosen at random to be exported (squared cell in c) – being transformed into one of the downstream cell types bringing the population size back to N (d).
Chronic myeloid leukemia dynamics
The progenitors derived from LSC express BCR-ABL and have a reduced differentiation capacity. Bone marrow expansion concomitant with observations suggests that cells expressing BCR-ABL have a differentiation probability ɛCML = 0.72.13 Besides marrow expansion, this reduced probability of differentiation, compared to normal progenitors (in agreement with what is observed) ultimately also results in an increased hematopoietic output leading to the diagnosis of CML (>1012 cells/day).27
Treatment
From our prior studies, we have estimated that at any time, imatinib therapy affects approximately 5% of the leukemic cell population.13 This percentage will increase when a higher dose of drug is given. Imatinib acts to increase the differentiation capacity of treated cells, leading to a supra-normal value for ɛ1MAT (ɛ1MAT > ɛ0 > ɛCML). Hence, normal cells acquire a relative fitness advantage compared to treated cells (Figure 3).26 Circulating cells have a finite life-time and are continuously being washed out. As a result, the disease burden decreases, as observed clinically.9,12
Figure 3.

Extinction of leukemic stem cells. Pext gives the extinction probability of LSC (fitness rCML≥r0) co-evolving with hematopoietic stem cells (HSC) (fitness r0), while Pneutral provides the limit when rCML=r0. (A) The red line depicts the ratio Pext/Pneutral, which is maximized for neutral drift, as is the case between LSC and HSC in CML19 (inset). The fitter lineage will increase rapidly whenever rCML>r0. This happens for non-stem cells, whose differentiation probabilities ɛ depend on their type (inset; note that larger values of ɛ lead to lower relative fitness) (B) Probability of extinction of the LSC clone as a function of time. At diagnosis (dashed line) approximately 84% of patients have no LSC (further details provided in the Design and Methods section). The probability converges to the theoretical result 1–1/N, where N is the size of the stem cell pool.
Simulations
The hematopoietic tree is modeled here by a sequence of 32 compartments. Since each compartment represents one level of the tree, it maintains information typical for all cells in that stage of differentiation: the number of different cell types (normal, CML and imatinib-treated cells) and the rates of replication for each cell type. To simulate the dynamics we defined very small time intervals τ (compared to the cells’ period of replication 1/Ri), such that in each interval each cell replicates with a probability given by pi=τ Ri. In every step in the simulation we examined every compartment and updated the amounts of each cell type using either a stochastic (i≤K) or a deterministic (i>K) update mechanism: in the stochastic case, every cell in the compartment i replicates with probability pi(1−ɛi) or differentiates with probability pjɛi. In the deterministic case, each cell type changes in time according to the ordinary differential equation:
| Eg. (1) |
where di = (2ɛ −1)ri represents the rate at which cells leave tree level “i” and bi−1 = 2ɛ · ri− 1 represents the rate at which cells originating from tree level “i−l” are injected into level “i”. The stationary state Ni = 0 leads to normal hematopoiesis. The appearance of CML mutations in compartment “i” means we also considered another set of equations besides Eq. (1) formally identical to the first, but for CML cells (NiCML) replacing ɛ with ɛCML < ɛ. In the presence of imatinib, a third set of (formally identical) equations now involving ɛ1MAT must be introduced. Numerical solution of these equations is very efficient and hence this constitutes the preferred method of solution for tree levels that contain a large number of cells (i > K). Below this limit, stochastic effects will have a significant influence on the dynamics of healthy, CML and imatinib-treated cells. At the interface between stochastic and deterministic dynamics, we explicitly took into account that the input of the first deterministic tree level (bi−1 · Ni−1 in Eq. 1) is simply the discrete output of the previous (stochastic) level. As the cell populations increase in size (from compartment 1 onwards), a compartment may be reached in which the number of cells is large enough to render stochastic effects negligible, so that stochastic and deterministic models give the same result. Finding this transition point is very helpful since individual cells need to be tracked only up to that compartment which makes the computations much more efficient and economic with respect to memory storage. Total bone marrow output is still captured by compartment 32.
Results
Stochastic dynamics of leukemic stem cells
We started by investigating the consequences of describing CML dynamics in a stochastic framework. Normal hematopoiesis can be represented by a compartmentalized model (Figure 1) in dynamic equilibrium in which cells move along the hematopoietic tree as they become increasingly differentiated.22 Hematopoietic stem cells lie at the root of the tree, and each branching of the tree may be associated with a compartment in which cells at a given stage of differentiation are accounted for. The implementation details of normal hematopoiesis are explained in the Design and Methods section.
CML disturbs normal hematopoiesis due to expression of BCR-ABL by the leukemic cells. The main phenotypic effect of BCR-ABL expression is to reduce the differentiation capacity of progenitor cells.26 However, it appears that BCR-ABL expression does not give a fitness advantage to the LSC in CML.19 Hence, the active stem cell pool does not grow in CML3 despite the sizable enhancement of marrow cell output which is characteristic of this disease.27 We, therefore, considered that hematopoietic stem cells and LSC, located at the root (i.e. level 0) of the hematopoietic tree (Figure 1), follow a stochastic Moran process28 under neutral drift (Figure 2) with approximately 400 cells that on average replicate each at a rate R0 = 1/year.22 CML dynamics begins when one or more LSC are present in a population of wild-type (normal) hematopoietic stem cells. Export of the first CML cell starts the path leading to disease (in most cases). No new mutations leading to the appearance of another BCR-ABL clone are explicitly considered, since this is a very rare event.29 Bone marrow expansion concomitant with observations suggests that progenitors and differentiated cells expressing BCR-ABL have a higher relative fitness than normal cells (Figure 3A), which ultimately lead to an increase in daily bone marrow output compatible with the diagnosis of CML (>1012 cells/day).27
In Figure 2 we illustrate the typical steps of the Moran process employed to investigate stem cell dynamics. The nature of the Moran process leads to the typical scenarios associated with stochastic evolutionary dynamics: expansion, extinction and latency of the LSC clone.18,28,30 The impact of these stochastic effects14,18 on clone size at the level of the stem cell compartment plays a crucial role and is discussed below.
Leukemic stem cell extinction
We evaluated the natural history of a million virtual patients following their disease over time from the appearance of the first LSC. Figure 3A shows the probability of extinction of the LSC lineage as a function of fitness advantage, illustrating that this probability is maximized precisely in the case of CML in which LSC and hematopoietic stem cells are neutral. Figure 3B displays the increase of the extinction probability as a function of time under neutral evolution. The simulations portray a remarkable and unexpected result in the context of CML: in the vast majority of cases, clonal extinction of LSC occurs. More importantly, by the time the disease is diagnosed (~5 years after the appearance of the first LSC), most patients – 84% – no longer have LSC (Figure 3B). Closer inspection of the results shows that LSC extinction is an early event: after 1 year already 50% of the patients no longer have LSC. For those patients in whom LSC are still present, we observed that the average clone size increases by one LSC per year (R2=0.9999). Moreover, since the current model treats certain parts of the hematopoietic tree stochastically, some patients may never be diagnosed with CML, despite initially carrying LSC (see below).
Stochastic dynamics of progenitor cells
Due to the limited number of most primitive cells within the hematopoietic tree, stochastic effects will play an important role in the dynamics of CML. However, since levels incorporating the more committed cell lineages quickly become very large, stochastic effects may be neglected above a given level K and one can resort to a deterministic description based on ordinary differential equations. As explained in the Design and Methods section, our model is capable of treating the first K levels stochastically, and the subsequent levels deterministically.
In Figure 4A, the average time to diagnosis for different values of K converges to approximately 5.1 years when K → 4, compatible with what has been observed at Hiroshima.31 However, this threshold is not enough to infer the probability that disease diagnosis does not take place once the LSC clone becomes extinct. Indeed, for values of K up to 6, no convergence is achieved for this quantity, from which we learn that at least seven stochastic levels, including the hematopoietic stem cell pool, are required (Figure 4B). Consequently, in all simulations performed in this work, a threshold K=7 was assumed. From Figure 4B, we observe that the number of cases in which no diagnosis is reached is limited to approximately 3% of the cohort of virtual individuals, despite the fact that, in most cases, the LSC clone has become extinguished.
Figure 4.

Stochastic-deterministic dynamics of hematopoietic cells. Starting with a single LSC, the stochastic dynamics of bone marrow cells was followed for a cohort of 106 virtual patients. (A) A plot of the mean time to diagnosis (and its variation as a function of the number of compartments treated stochastically). (B) The probability of no diagnosis is plotted as a function of the number of compartments treated stochastically; in this case, K=7 compartments are the minimum threshold necessary to obtain converged results. This more stringent threshold has been used throughout in assessing the role of stochastic effects in CML dynamics.
Chronic myeloid leukemia dynamics with and without leukemic stem cells
Understanding how CML can be diagnosed in the absence of LSC requires following hematopoietic cells as they traverse the different stages of differentiation linking stem and circulating cells, the process illustrated in Figure 1. Given the limited capacity of self-renewal at every stage (inset of Figure 3A) and also that replication rates increase as cells become more differentiated, propagation of the BCR-ABL oncogene to circulating blood is not immediate, but constrained by the hierarchical dynamics of the hematopoietic tree, leading to almost 5 years between appearance of the first LSC and the diagnosis of CML.31 Crucial to the whole process is the fact that the differentiation probability of non-stem cell lineages depends on whether cells carry the BCR-ABL oncogene (ɛCML) or not (ɛ).32,33 It has been shown in a purely deterministic model13 that in order to explain average CML dynamics, including bone-marrow expansion (a fingerprint of CML), one has ɛ>ɛCML. The effective advantage (inset of Figure 3A) of CML cells due to enhanced self-renewal13,20 leads to their expansion at all stages of differentiation.34 The slow rate of replication of early progenitors, in turn, implies that the wash-out of these cells from early stages of differentiation is a slow process: the appearance of the first LSC sends a ripple down hematopoiesis leading to diagnosis in 97% of the cases, of which only 16% carry LSC at diagnosis. In other words, the same hierarchical architecture that protects the organism from LSC invasion determines a delay in both CML diagnosis and the impact of LSC extinction: CML progenitor cells persist for years, sustaining CML and enabling its diagnosis even in the absence of a LSC clone. These results clearly indicate that the major immediate goal in CML treatment should not be the eradication of LSC but the eradication of the much larger population of leukemic progenitors which drives the disease, increasing the risks of acquired resistance and blast transformation (see below). If one achieves such a goal when LSC no longer feed malignant cells into hematopoiesis, therapies will be mostly curative. We argue that, currently, tyrosine kinase inhibitors are the best strategy to fit this purpose.
Imatinib therapy
The therapeutic effect of imatinib is to increase the differentiation probability of CML progenitor cells ɛCML → ɛIMAT > ɛ > ɛCML,13,32 resulting in a relative fitness advantage of normal cells with respect to treated cells (inset of Figure 3A). Hence, normal cells will partially out-compete treated cells, leading to a progressive reduction in disease burden as shown in Figure 5A, where treatment with imatinib was started right after diagnosis. Consequently, continuous drug administration ensures that normal progenitors dominate hematopoiesis.13,26
Figure 5.

Efficacy of imatinib treatment. Two virtual cohorts, C1 and C2, each with 106 patients, were considered. In all cases, CML dynamics starts from one LSC. Cohort C1 (light green bars) includes only patients with no LSC at diagnosis. Cohort C2 (dark green bars) includes all patients (with and without LSC at diagnosis). (A) Probability of relapse as a function of treatment time. (B) Average log-reduction of disease burden as a function of treatment time. (C) The fraction of C2 patients that reached complete cytogenetic response (CCyR, dark blue) and major molecular response (MMR, cyan). (D) Correlation plot between model prediction of MMR for simulated C2 patients and data for observed MMR for actual patients from Branford et al.36
Duration of imatinib treatment
Up to now, the duration of tyrosine kinase inhibitor therapy required to ‘cure’ CML remains to be determined. In order to address this issue, we followed two populations each with 106 virtual patients who were treated with imatinib for periods varying from 1 up to 9 years after diagnosis of the disease (which takes, on average 5.1 years to reach), treatment being subsequently withdrawn. In both cohorts the simulated patients started out with one LSC. In the first cohort (C1) only those virtual patients who did not have any LSC at diagnosis were retained. As such this population provides an idealized scenario in which the stem cell compartment can no longer feed the disease in any of the simulated patients, in this way providing an upper bound for the results shown in Figure 5. In the second cohort (C2) no restrictions were imposed, and so approximately 16% of the patients in this population had LSC at diagnosis. Once treatment was stopped, the simulations were followed for an additional period of 10 years. We investigated the possible relapse of the disease, associated with the burden at diagnosis. We also examined the reduction in BCR-ABL levels during treatment. Figure 5A shows that the probability of relapse drops quickly with increasing treatment time. Persistence of LSC in a patient always translates into a finite chance of relapse. Hence, since in C2 some patients may still have LSC, the probability of relapse never reaches exactly zero. As the duration of treatment increases from 1 to 9 years, the disease burden is reduced further (Figure 5B), with the probability of relapse falling in parallel (~ 3.1 log reduction at 1 year to ~ 3.8 log reduction at 4 years21). This trend becomes clear when examining Figure 5C. Almost 67% of the virtual patients reach complete cytogenetic response (~3 log reduction in the BCR-ABL/BCR ratio, Figure 5) after 1 year of treatment, compatible with data reported by Druker et al.21 Moreover, at least 2% of these patients achieve major molecular response (≥4 log reduction of disease burden) within 6 months of treatment initiation. Clinically, a greater than 4.5 log reduction in disease burden after 12 months of therapy has been reported.35 For longer treatment periods (Figure 5C), the fractions of patients achieving complete cytogenetic response and major molecular response increase significantly, reaching 94% and 71% respectively after 5 years. On average, complete cytogenetic response is reached after 1 year of treatment, whereas a major molecular response requires 3.5 years or longer periods of treatment. These model predictions are consistent with the independent clinical data from Druker et al.21 and correlate nicely with the experimental data observed by Branford et al.36 (Figure 5D). Overall, our results show that even though there is still a possibility of relapse, in the absence of acquired resistance, continued imatinib therapy is capable of sustained reduction in disease burden down to levels compatible with major molecular response or even lower, in which case relapse probability becomes very low (Figure 5C).
Discussion
The structure and dynamics of hematopoiesis make CML perhaps a unique neoplasm. The small number of hematopoietic stem cells contributing to hematopoiesis and their slow rate of replication explain to a large extent why CML and related disorders are rare.22,37 Arguably, CML is the best studied neoplasm for which the molecular defect leading to many of the disease characteristics is known and introduction of BCR-ABL into hematopoietic stem cells leads to a disease that recapitulates many of the features of the chronic phase of CML in animal models.1,5 Given the fundamental role of BCR-ABL in driving CML, the impact of ABL kinase inhibitor therapy on disease dynamics can be understood. Our work shows that the dire consequences of CML are, in most cases, not a consequence of LSC dynamics, but result from the altered behavior of cells downstream from the stem cell pool, leading to profound changes in progenitor cell dynamics which may subsequently trigger the emergence of blast crisis and resistance to therapy. The enhanced self-renewal imparted by aberrant BCR-ABL expression explains why CML cells undergo a higher number of cell divisions before they appear in the circulation.13,38 With increasing self-renewal, the cells appear to be ‘trapped’ in hematopoiesis longer than normal cells. Consequently, our model predicts that with imatinib therapy, circulating hematopoietic cells should have an increase in average telomere length, compared to their length before initiation of therapy, a phenomenon that has been experimentally proven.39
In the absence of tyrosine kinase inhibitor therapy, CML progenitors have a fitness advantage compared to normal cells (inset of Figure 3A), and most of the time: (i) there are many more CML progenitor cells (~105) than LSC (~1); (ii) the former replicate faster (~once every 8–10 weeks) than the latter (~once per year); (iii) genomic instability imparted by BCR-ABL increases the mutation rate of CML progenitor cells, which raises the risk of resistance and transformation;40 (iv) CML progenitors contribute longer to hematopoiesis than their normal counterparts due to enhanced self-renewal;20 and (v) CML progenitors undergo a higher number of replications during their lifetime.13,27,41 Consequently, without therapy, the effective population of CML progenitors is large and at risk of additional mutations that may lead to resistance and/or blast transformation. In view of this, it is perhaps not surprising that blast crisis arises from cells belonging to the colony-forming unit – granulocyte-macrophage pool rather than the LSC.3 Moreover, any therapy that effectively suppresses the CML progenitor pool should significantly reduce the probability of treatment failure and blast transformation. In the present framework of CML dynamics, the progressive decline in CML progenitors under imatinib explains why the risk of treatment failure decreases with time from 5.5% after 1 year of therapy to 0.4% after 4 years.21 Similarly, the risk of progression to accelerated phase or blast transformation decreases from 2.1% after 1 year to essentially 0% after 4 years for patients who achieve a complete cytogenetic response (~3 log reduction in the BCR-ABL/BCR ratio, Figure 5).21 Finally, studies of newly diagnosed patients treated with imatinib show that the risk of transformation is reduced compared to that of patients on standard therapy with interferon-α and cytosine arabinoside with an improvement in survival.21 Overall, these findings strongly suggest that imatinib is capable of doing what is necessary – to suppress the uncontrolled growth of CML progenitor cells. In this respect, the second generation tyrosine kinase inhibitors — dasatinib and nilotinib — are expected to give even better results although they tend to be more toxic.
In the present context, it is also clear why therapies involving non-specific inhibitors of cell proliferation (cytosine arabinoside and hydroxyurea), do not alter the natural history of the disease including progression to blast crisis. Unlike imatinib, these agents do not reverse the fitness advantage of the CML progenitor cells and do not discriminate between normal and CML cells; hence, the clone will grow, persistently dominate hematopoiesis and maintain the risk of transformation due to its large size. The only exception has been interferon-α, which also reduces the fitness of CML progenitor cells compared to their normal counterparts.32 Interestingly, some long-term survivors have been reported who have been treated with interferon-α alone, and some appear to be operationally cured.42 Recently, it was shown that interferon-α increases the replication rate of hematopoietic stem cells;43 perhaps by doing so, interferon-α increases the probability of stochastic extinction of the LSC clone, thus providing an explanation of the operational cures that have been observed.
The same arguments above can be used to explain why the incidence of resistance to tyrosine kinase inhibitors will decrease with more efficient progenitor cell elimination. Clearly, the development of resistance will be more likely the larger the population of BCR-ABL-expressing progenitors. By reducing the population of cells at risk, imatinib remains an effective drug. It appears that the incidence of imatinib resistance is lower in patients who received the agent immediately after diagnosis than in patients who received prior therapies before treatment with imatinib.44 Thus, early diagnosis of CML paired with immediate treatment with a tyrosine kinase inhibitor may be the key to successful therapy. Our results further suggest that higher doses of the tyrosine kinase inhibitor will be more effective in treating CML. Importantly, the tyrosine kinase inhibitor need not be taken for the rest of the patient’s life (Figure 5) since it can cure CML.11
A prior model of CML also proposed that imatinib therapy can cure the disease under some circumstances.12 This model was based on the premise of a selective functional effect of imatinib on LSC, a feature distinctly different from our model in which LSC are not affected by tyrosine kinase inhibitor therapy. Rather, it is the combined effect of the intrinsic stochastic dynamics within the hematopoietic and leukemic stem cell pools together with the impact of tyrosine kinase inhibitor therapy on progenitor cells that can lead to cure of the disease. More specifically, in our model, LSC that drive CML undergo stochastic extinction independently of the drug.
Tyrosine kinase inhibitors, despite not hitting LSC directly, are providential since they effectively hit the most dangerous element of CML dynamics – the progenitor cells. They confer a relative fitness disadvantage to treated mutant cells compared to normal cells, enabling the latter to benefit from the hierarchical architecture to regain a dominant contribution to hematopoiesis. In doing so, tyrosine kinase inhibitors not only effectively contribute to improving the patients’ condition, but they should also contribute to effectively curing CML in most patients, given the high probability of extinction of the LSC giving rise to the disease.
Footnotes
Funding: JMP is supported by FCT-Portugal. AT is supported by the Emmy-Noether program of the German Research Foundation. DD is supported by an Early Career Development Award from the Mayo Clinic.
Authorship and Disclosures
TL: concept and design, collection of data, data analysis, manuscript writing, final approval; JMP: concept and design, data analysis, manuscript writing, final approval; AT: concept and design, data analysis, manuscript writing, final approval; DD: concept and design, data analysis manuscript writing, final approval.
The authors reported no potential conflicts of interest.
References
- 1.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247(4944):824–30. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 2.Goldman JM, Melo JV. Chronic myeloid leukemia – advances in biology and new approaches to treatment. N Engl J Med. 2003;349(15):1451–64. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 3.Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657–67. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 4.Martin PJ, Najfeld V, Hansen JA, Penfold GK, Jacobson RJ, Fialkow PJ. Involvement of the B-lymphoid system in chronic myelogenous leukaemia. Nature. 1980;287 (5777):49–50. doi: 10.1038/287049a0. [DOI] [PubMed] [Google Scholar]
- 5.Zhao RC, Jiang Y, Verfaillie CM. A model of human p210(bcr/ABL)-mediated chronic myelogenous leukemia by transduction of primary normal human CD34(+) cells with a BCR/ABL-containing retroviral vector. Blood. 2001;97(8):2406–12. doi: 10.1182/blood.v97.8.2406. [DOI] [PubMed] [Google Scholar]
- 6.Michor F, Iwasa Y, Nowak MA. The age incidence of chronic myeloid leukemia can be explained by a one-mutation model. Proc Natl Acad Sci USA. 2006;103(40):14931–4. doi: 10.1073/pnas.0607006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quintas-Cardama A, Cortes J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–30. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–25. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 9.Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435(7046):1267–70. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 10.Kavalerchik E, Goff D, Jamieson CH. Chronic myeloid leukemia stem cells. J Clin Oncol. 2008;26(17):2911–5. doi: 10.1200/JCO.2008.17.5745. [DOI] [PubMed] [Google Scholar]
- 11.Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O, et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood. 2007;109(1):58–60. doi: 10.1182/blood-2006-03-011239. [DOI] [PubMed] [Google Scholar]
- 12.Roeder I, Horn M, Glauche I, Hochhaus A, Mueller MC, Loeffler M. Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implications. Nat Med. 2006;12(10):1181–4. doi: 10.1038/nm1487. [DOI] [PubMed] [Google Scholar]
- 13.Dingli D, Traulsen A, Pacheco JM. Chronic myeloid leukemia: origin, development, response to therapy, and relapse. Clinical Leukemia. 2008;2:133–9. [Google Scholar]
- 14.Gordon MY, Blackett NM. Routes to repopulation – a unification of the stochastic model and separation of stem-cell subpopulations. Leukemia. 1994;8(6):1068–73. [PubMed] [Google Scholar]
- 15.Dingli D, Pacheco JM. Allometric scaling of the active hematopoietic stem cell pool across mammals. PLoS ONE. 2006;1:e2. doi: 10.1371/journal.pone.0000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemischka IR, Raulet DH, Mulligan RC. Developmental potential and dynamic behavior of hematopoietic stem cells. Cell. 1986;45(6):917–27. doi: 10.1016/0092-8674(86)90566-0. [DOI] [PubMed] [Google Scholar]
- 17.Goldman JM. Chronic myeloid leukemia stem cells: now on the run. J Clin Oncol. 2009;27(2):313–4. doi: 10.1200/JCO.2008.19.2260. [DOI] [PubMed] [Google Scholar]
- 18.Dingli D, Traulsen A, Pacheco JM. Stochastic dynamics of hematopoietic tumor stem cells. Cell Cycle (Georgetown, Tex) 2007;6(4):461–6. doi: 10.4161/cc.6.4.3853. [DOI] [PubMed] [Google Scholar]
- 19.Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell. 2004;6(6):587–96. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 20.Marley SB, Gordon MY. Chronic myeloid leukaemia: stem cell derived but progenitor cell driven. Clin Sci (Lond) 2005;109(1):13–25. doi: 10.1042/CS20040336. [DOI] [PubMed] [Google Scholar]
- 21.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 22.Dingli D, Traulsen A, Pacheco JM. Compartmental architecture and dynamics of hematopoiesis. PLoS ONE. 2007;2(4):e345. doi: 10.1371/journal.pone.0000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buescher ES, Alling DW, Gallin JI. Use of an X-linked human neutrophil marker to estimate timing of lyonization and size of the dividing stem cell pool. J Clin Invest. 1985;76(4):1581–4. doi: 10.1172/JCI112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnston MD, Edwards CM, Bodmer WF, Maini PK, Chapman SJ. Examples of mathematical modeling: tales from the crypt. Cell Cycle (Georgetown, Tex) 2007;6(17):2106–12. doi: 10.4161/cc.6.17.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnston MD, Edwards CM, Bodmer WF, Maini PK, Chapman SJ. Mathematical modeling of cell population dynamics in the colonic crypt and in colorectal cancer. Proc Natl Acad Sci USA. 2007;104(10):4008–13. doi: 10.1073/pnas.0611179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marley SB, Deininger MW, Davidson RJ, Goldman JM, Gordon MY. The tyrosine kinase inhibitor STI571, like interferon-alpha, preferentially reduces the capacity for amplification of granulocyte-macrophage progenitors from patients with chronic myeloid leukemia. Exp Hematol. 2000;28(5):551–7. doi: 10.1016/s0301-472x(00)00142-9. [DOI] [PubMed] [Google Scholar]
- 27.Holyoake TL, Jiang X, Drummond MW, Eaves AC, Eaves CJ. Elucidating critical mechanisms of deregulated stem cell turnover in the chronic phase of chronic myeloid leukemia. Leukemia. 2002;16(4):549–58. doi: 10.1038/sj.leu.2402444. [DOI] [PubMed] [Google Scholar]
- 28.Moran PAP. The Statistical Processes of Evolutionary Theory. Oxford, UK: Clarendon; 1962. [Google Scholar]
- 29.Dingli D, Pacheco JM, Traulsen A. Multiple mutant clones in blood rarely coexist. Phys Rev. 2008;77(2 Pt 1):021915. doi: 10.1103/PhysRevE.77.021915. [DOI] [PubMed] [Google Scholar]
- 30.Dingli D, Luzzatto L, Pacheco JM. Neutral evolution in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci USA. 2008;105(47):18496–500. doi: 10.1073/pnas.0802749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ichimaru M, Ishimaru T, Mikami M, Yamada Y, Ohkita T. Incidence of leukemia in a fixed cohort of atomic bomb survivors and controls, Hiroshima and Nagasaki October 1950–December 1978: Technical Report RERF TR 13-81. Radiation Effects Research Foundation; Hiroshima: 1981. 1981. [Google Scholar]
- 32.Marley SB, Davidson RJ, Goldman JM, Gordon MY. Effects of combinations of therapeutic agents on the proliferation of progenitor cells in chronic myeloid leukaemia. Br J Haematol. 2002;116(1):162–5. doi: 10.1046/j.1365-2141.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 33.Marley SB, Lewis JL, Gordon MY. Progenitor cells divide symmetrically to generate new colony-forming cells and clonal heterogeneity. Br J Haematol. 2003;121(4):643–8. doi: 10.1046/j.1365-2141.2003.04338.x. [DOI] [PubMed] [Google Scholar]
- 34.Primo D, Sanchez ML, Espinosa AB, Tabernero MD, Rasillo A, Sayagues JM, et al. Lineage involvement in chronic myeloid leukaemia: comparison between MBCR/ABL and mBCR/ABL cases. Br J Haematol. 2006;132(6):736–9. doi: 10.1111/j.1365-2141.2005.05944.x. [DOI] [PubMed] [Google Scholar]
- 35.Verma D, Kantarjian H, Jain N, Cortes J. Sustained complete molecular response after imatinib discontinuation in a patient with chronic myeloid leukemia not previously exposed to interferon alpha. Leuk Lymphoma. 2008;49(7):1399–402. doi: 10.1080/10428190802043903. [DOI] [PubMed] [Google Scholar]
- 36.Branford S, Seymour JF, Grigg A, Arthur C, Rudzki Z, Lynch K, et al. BCR-ABL messenger RNA levels continue to decline in patients with chronic phase chronic myeloid leukemia treated with imatinib for more than 5 years and approximately half of all first-line treated patients have stable undetectable BCR-ABL using strict sensitivity criteria. Clin Cancer Res. 2007;13(23):7080–5. doi: 10.1158/1078-0432.CCR-07-0844. [DOI] [PubMed] [Google Scholar]
- 37.Lopes JV, Pacheco JM, Dingli D. Acquired hematopoietic stem-cell disorders and mammalian size. Blood. 2007;110(12):4120–2. doi: 10.1182/blood-2007-05-089805. [DOI] [PubMed] [Google Scholar]
- 38.Brummendorf TH, Rufer N, Holyoake TL, Maciejewski J, Barnett MJ, Eaves CJ, et al. Telomere length dynamics in normal individuals and in patients with hematopoietic stem cell-associated disorders. Ann NY Acad Sci. 2001;938:293–304. doi: 10.1111/j.1749-6632.2001.tb03598.x. [DOI] [PubMed] [Google Scholar]
- 39.Brummendorf TH, Ersoz I, Hartmann U, Balabanov S, Wolke H, Paschka P, et al. Normalization of previously shortened telomere length under treatment with imatinib argues against a preexisting telomere length deficit in normal hematopoietic stem cells from patients with chronic myeloid leukemia. Ann NY Acad Sci. 2003;996:26–38. doi: 10.1111/j.1749-6632.2003.tb03229.x. [DOI] [PubMed] [Google Scholar]
- 40.Slupianek A, Nowicki MO, Koptyra M, Skorski T. BCR/ABL modifies the kinetics and fidelity of DNA double-strand breaks repair in hematopoietic cells. DNA Repair (Amst) 2006;5(2):243–50. doi: 10.1016/j.dnarep.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brummendorf TH, Holyoake TL, Rufer N, Barnett MJ, Schulzer M, Eaves CJ, et al. Prognostic implications of differences in telomere length between normal and malignant cells from patients with chronic myeloid leukemia measured by flow cytometry. Blood. 2000;95(6):1883–90. [PubMed] [Google Scholar]
- 42.Kujawski LA, Talpaz M. The role of interferon-alpha in the treatment of chronic myeloid leukemia. Cytokine Growth Factor Rev. 2007;18(5–6):459–71. doi: 10.1016/j.cytogfr.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 43.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458 (7240):904–8. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 44.Jabbour E, Kantarjian H, Jones D, Talpaz M, Bekele N, O’Brien S, et al. Frequency and clinical significance of BCR-ABL mutations in patients with chronic myeloid leukemia treated with imatinib mesylate. Leukemia. 2006;20(10):1767–73. doi: 10.1038/sj.leu.2404318. [DOI] [PubMed] [Google Scholar]