

Abstract
Background
The role of the Wilms tumor 1 gene (WT1) in acute leukemias has been underscored by mutations found in acute myeloid leukemia identifying patients with inferior survival. Furthermore, aberrant expression of WT1 in acute myeloid leukemia was associated with an increased risk of relapse. No larger studies have performed a combined approach including WT1 mutation and expression analyses in acute T-lymphoblastic leukemia.
Design and Methods
We analyzed the WT1 mutations and the expression status in a total of 252 consecutive adult patients with newly diagnosed T-lymphoblastic leukemia, who were registered on the GMALL 06/99 and 07/03 protocols and had sufficient material available. The GMALL protocols included intensive chemotherapy as well as stem cell transplantation according to a risk-based model with indication for stem cell transplantation in first complete remission for early and mature T-lymphoblastic leukemia patients; patients with thymic T-lymphoblastic leukemia were allocated to a standard risk group and treated with intensive chemotherapy.
Results
Twenty of the 238 patients analyzed had WT1 mutations (WT1mut) in exon 7. WT1mut cases were characterized by immature features such as an early immunophenotype and higher WT1 expression. In thymic T-lymphoblastic leukemia, WT1mut patients had an inferior relapse-free survival compared to WT1 wild-type patients. T-lymphoblastic leukemia patients with aberrant WT1 expression (high or negative) showed a higher relapse rate and an inferior outcome compared to patients with intermediate WT1 expression. In the standard risk group of thymic T-lymphoblastic leukemia, aberrant WT1 expression was predictive for an inferior relapse-free survival as compared to patients with intermediate expression. In multivariate analysis, WT1 expression was of independent prognostic significance for relapse-free survival.
Conclusions
WT1 mutations were associated with an inferior relapse-free survival in standard risk thymic T-lymphoblastic leukemia patients. Moreover, altered expression associated with inferior outcome also suggests a role of WT1 in T-lymphoblastic leukemia and the potential use of molecularly-based treatment stratification to improve outcome.
Keywords: adult acute T-lymphoblastic leukemia, WT1, gene expression, mutations
Introduction
The Wilms tumor 1 gene, WT1, encodes a transcription factor involved in normal and malignant hematopoiesis.1 WT1 was first recognized as a tumor suppressor when congenital malformation syndromes with predisposition to childhood kidney cancer were linked to WT1 germline mutations.2 In acute leukemia, WT1 mutations have been reported in 10% of patients with acute myeloid leukemia (AML). However, mutations were also observed in selected cases of acute T-lymphoblastic leukemia (TALL), as well as undifferentiated/biphenotypic leukemia.3 The mutations reported included frame shift mutations in exon 7, resulting in a truncated protein lacking the major DNA binding portion of the WT1 protein.4 Most importantly, in intermediate risk AML with normal cytogenetics, WT1 mutations identified patients with an inferior outcome.5,6
Apart from WT1 mutations, overexpression of WT1 was found in AML patients and to a lower extent in acute lymphoblastic leukemia (ALL).7 There are conflicting studies regarding the prognostic impact of WT1 expression levels in newly diagnosed AML. Some studies have associated high WT1 expression levels in pre-treatment leukemic samples with an inferior outcome, whereas other studies could not demonstrate the prognostic significance of WT1 expression levels in AML patients.8–10 Only a few studies have so far evaluated the prognostic relevance of WT1 expression in ALL and have suggested an association between high expression and an inferior outcome.11,12
In normal hematopoiesis, expression of WT1 is mainly restricted to the progenitor compartment, whereas mature hematopoietic cells lack WT1 expression. Enforced expression of WT1 in CD34 positive hematopoietic precursors has been shown to induce growth arrest.13,14 The ability of WT1 to repress or activate gene transcription is dependent on WT1 levels, isoforms, cell type, and interaction of WT1 with other proteins.15 Moreover, the complexity of its role in hematopoiesis may be explained by its differentiation dependent function, as it was shown that WT1 maintains primitive stem cells in a quiescence state, while it promotes differentiation of more mature progenitors.16
So far, studies have mainly focused on AML and underscored the role of WT1 by exploring its aberrant expression and mutational status. Interestingly WT1 mutations were predominantly observed in immature AML subtypes, in biphenotypic acute leukemias, and overexpression of WT1 was correlated with a more undifferentiated phenotype. In a recent study by Tosello et al. mutations in the WT1 gene were found in a subset of adult as well as pediatric T-ALL patients.17 Together these findings suggest that altered WT1 function might also be implicated in other leukemic subgroups and reflect transformation of a primitive hematopoietic cell of origin with retained multi-lineage potential.3
We had previously shown that AML and T-ALL share molecular markers with prognostic significance that are indicative of an immature leukemic subtype.18 Thus, we have examined the prognostic implications of WT1 mutations and expression levels in a large cohort of adult T-ALL within the German Multicenter ALL (GMALL) study group. As multiple genetic hits cooperate in different cellular pathways and are required for leukemic transformation,19,20 WT1 alterations were analyzed in the context of other genetic alterations.
Design and Methods
Patients and treatment
We studied a total of 252 consecutive adult patients with newly diagnosed T-ALL who were registered on the GMALL 06/99 and 07/03 protocols and had sufficient material available.21,22 The GMALL protocols included intensive chemotherapy as well as stem cell transplantation according to a risk-based model with indication for stem cell transplantation in first complete remission for early and mature T-ALL patients; patients with thymic T-ALL were allocated to a standard risk group and treated with intensive chemotherapy. All patients gave written informed consent to participate in the study according to the Declaration of Helsinki. The study was approved by the ethics board of the Goethe University Frankfurt/Main, Germany.
Molecular characterization
Pre-treatment bone marrow (BM) samples were centrally collected, enriched for the blast fraction by density-gradient centrifugation, and stored in liquid nitrogen. Immunophenotyping of fresh samples was centrally performed by flow cytometry in the GMALL reference laboratory at the Charité, Berlin, Germany. Immunophenotyping was carried out as previously described.23 CD1a positive, cortical (III) stage T-ALL was referred as thymic T-ALL in the GMALL study group.
From pre-treatment bone marrow samples (n=252) sufficient genomic DNA (n=238) and RNA (n=223) were isolated using the Trizol reagent (Invitrogen, Karlsruhe, Germany). Specimens of the 238 patients were studied for WT1 mutations in exons 7 and 9 by DNA sequencing of amplified PCR products.5 Mutations were confirmed by cloning the specific PCR products and sequencing up to 14 independent clones. Mutations in the NOTCH1 and the FBXW7 genes were determined by direct sequencing of PCR-amplified products.24,25 FLT3 mutations [internal tandem duplications (ITD) and mutations in the tyrosine kinase domain (TKD835)] were analyzed using the FLT3 mutation assay (InVivoScribe Technologies, San Diego, USA). The mRNA expression of HOX11, HOX11L2, BAALC, and ERG were determined by real-time RT-PCR.18 Expression analysis of WT1 was performed in 223 samples by a comparative real-time RT-PCR assay using primers WT1F CAGGCCAGGATGTTTCCTAA and WT1R AATGAGTGGTTGGGGAACTG with a WT1-probe 5′FAM-CCGCTATTCGCAATCAGGGTTACA-TAMRA. Multiplex PCR was performed with beta-glucuronidase (GUS) as a housekeeping gene in duplicates.26 PCR conditions were as follows: initial denaturation with 95°C for 10 min, annealing at 60°C and extension at 72°C. GUS and WT1 were coamplified using 2 μL cDNA, 1x master mix (IQ Mix, BioRad, Munich, Germany). All reactions were carried out using the Rotor Gene Real-time PCR 3000 Machine (Corbett Research, Qiagen, Germany). The comparative cycle threshold (CT) method was used to determine the relative expression levels of WT1, and the cycle number difference (ΔCT=GUS-WT1) was calculated using the mean of ΔCT from the two replicates, that is μ(ΔCT), and expressed as 2(ΔμCT). In all samples, amplification of GUS reached the threshold within 30 cycles. For samples without detectable WT1 amplification within 60 cycles, WT1 expression values were set at 0. A calibrator (cDNA from the cell line KG1a) included in each run was used for standardization between runs. Positive and negative controls were included in all assays.
Statistical analyses
Comparisons of baseline clinical variables across groups were made using the χ2 Fisher’s exact test for categorical data; the non-parametric Mann-Whitney U test was applied for quantitative variables. A P value ≤0.05 (two-sided) was considered to indicate a significant difference. Clinical follow-up data were available from 215 T-ALL patients with a median follow-up time of 20.5 months (range: 0.5 to 81.2 months). Complete remission was assessed after completion of induction chemotherapy. Overall survival and relapse-free survival were calculated using the Kaplan-Meier method and the log-rank test was used to compare differences between survival curves. Overall survival was measured from the protocol on-study date until the date of death of any cause. Relapse-free survival was measured from time of complete remission date until the date of relapse.18 For outcome analyses patients were censored at the time of stem cell transplantation.
Three WT1 expression groups were defined as follows: after categorizing WT1 expression levels into quintiles, a logistic regression analysis with relapse as the dependent and WT1 grouping as the independent variable was performed. In this model, T-ALL patients with the highest quintile (WT1 high, n=45; median expression: 0.04, range: 0.01–0.4) and patients with no detectable WT1 expression (WT1 negative, n=97) differentiated substantially with respect to relapse (i. e. the differences in regression coefficients had the magnitude of about two standard errors) compared to the remaining patients with intermediate WT1 expression levels (WT1 intermediate, n=81; median expression: 0.0001, range: 2×10−9 to 10−2). The follow-up time and censoring was equivalent between the three expression groups.
In order to identify independent prognostic factors and effect modifiers (i. e. interactions between different factors), Cox’s proportional hazards models were constructed. The following covariates were included into the full model: BAALC expression (low versus high), ERG expression (low versus high), white blood count (WBC as continuous), age (< 35 versus > 35 years), immunophenotype (thymic versus early/mature). The impact of effect modifiers was verified by the inclusion of interaction terms into the multiple Cox’s regression analyses. Stepwise forward and backward selections were performed. All calculations were performed using the SPSS software, version 17 (SPSS Inc., Chicago, IL, USA).
Results
WT1 mutations in adult T-ALL
WT1 mutations (WT1mut) were detectable in 20 (8%) of the 238 T-ALL patients. Mutations in exon 7 (WT1mut7) were identified in all 20, with 2 patients having coexisting mutations in exon 9 (WT1mut9). WT1mut7 were frameshift or nonsense mutations predicted to result in a truncated WT1 protein, whereas WT1mut9 were missense mutations leading to single amino-acid substitutions (Online Supplementary Table S1). WT1 wild-type amplicons were present in the majority of samples; thus it is likely that mutations were heterozygous, though the presence of residual normal cells or subpopulations of leukemic cell clones without WT1 mutations cannot be excluded. We focused on WT1 exons 7 and 9 as these regions have previously been recognized as mutational hot spots in AML. However, we cannot exclude that mutations in other regions also exsist.4,17
Association of WT1 mutations with clinical and molecular characteristics
There was no significant difference between WT1mut and WT1 wild-type (WT1wt) patients with respect to clinical parameters at diagnosis such as WBC, age, sex, mediastinal mass, CNS involvement (Table 1; data not shown). WT1mut cases were characterized by immature features such as an early immunophenotype (45% of WT1mut showed an early T-ALL as compared to only 25% of WT1wt; P=0.07). WT1mut cases also showed higher CD34 levels as determined by flow cytometry (mean CD34 expression: WT1mut: 37% vs. WT1wt: 20%; P=0.02), and more frequently co-expressed the myeloid marker CD13 (P=0.03; Table 1). Moreover, WT1mut cases showed a higher frequency of aberrant HOX11L2 expression (47% of WT1mut vs. 7% of WT1wt expressed HOX11L2; P<0.001) and WT1mut cases had significantly higher WT1 mRNA expression levels as compared to WT1wt cases (P<0.001). Expression of both mutated and wild-type WT1 transcripts were verified by PCR using mutation specific primers in 2 representative cases (data not shown). There was no significant correlation between the WT1 mutation status and expression of the previously characterized molecular risk markers ERG, BAALC, and HOX11.
Table 1.
Clinical and molecular characteristics of T-ALL patients with respect to the WT1 mutation status.
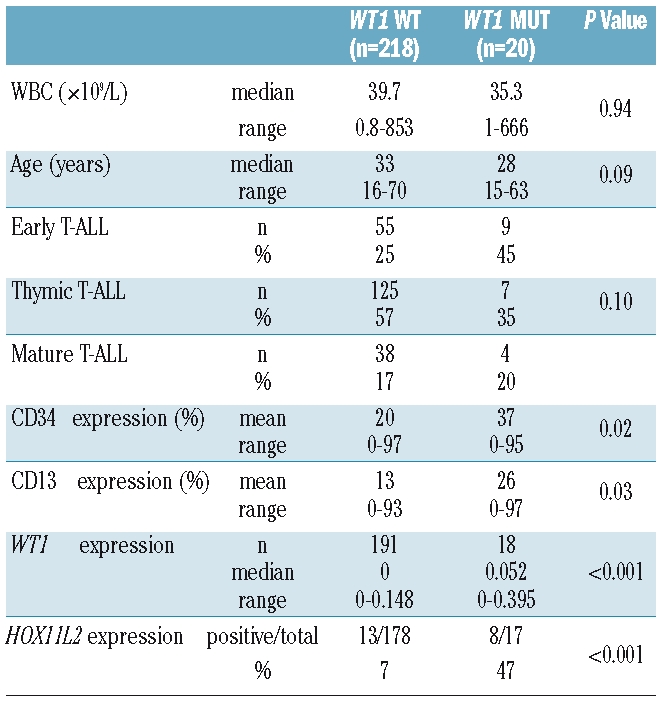
The presence of coexisting gene mutations was further investigated. Similar to the mutation frequencies observed in WT1wt patients, WT1mut cases showed gene mutations in NOTCH1 (10/20, 50% of cases) and FBXW7 (2/20, 10% of cases). In addition, cases were also analyzed for FLT3 mutations, given a high frequency of FLT3 ITD mutations observed in WT1mut AML.4,6 Though FLT3 mutations are a rare event in T-ALL (found in up to only 3%),27,28 in WT1mut cases, we identified one T-ALL patient with co-existence of an FLT3 ITD (1/20) and 3 patients with FLT3 TKD mutations (3/20). Thus 20% of WT1mut T-ALL harbored simultaneous FLT3 mutations (Online Supplementary Figure S1). In contrast, only one WT1wt patient showed a FLT3 ITD (1/108 WT1wt cases) and 2 WT1wt patients had an FLT3 TKD mutation (2/101 WT1wt cases).
WT1 mutations and outcome
No differences were observed in the complete remission rates between mutated and wild-type WT1 patients. We found no significant difference in the relapse-free survival between the WT1mut and WT1wt patients in the overall cohort (Figure 1A). However, within the standard risk group of thymic T-ALL, the small subgroup of WT1mut showed an inferior relapse-free survival as compared to WT1wt thymic patients (P=0.03; Figure 1B).
Figure 1.
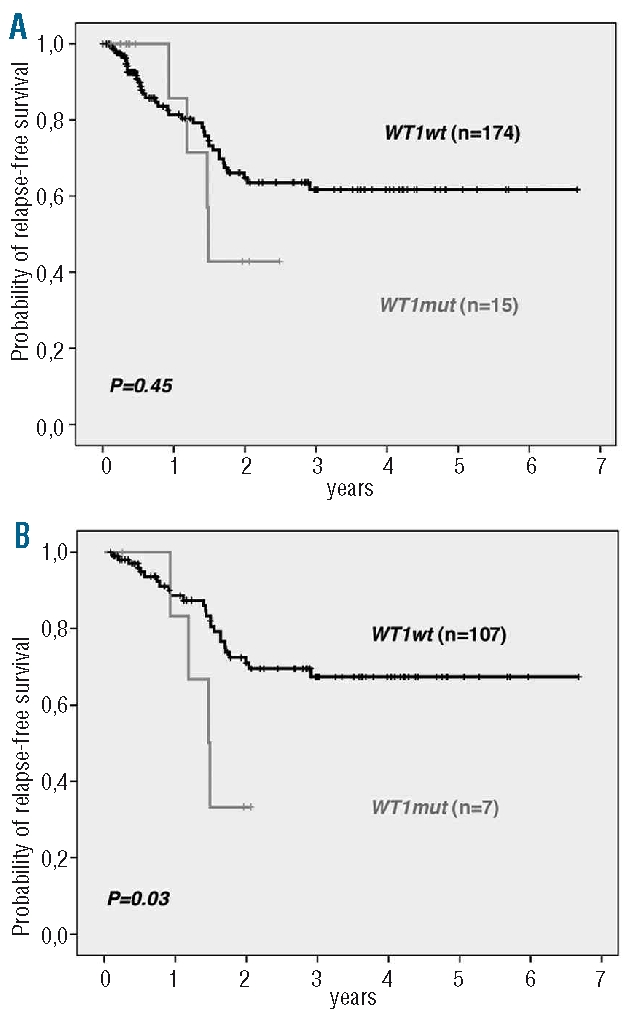
Relapse-free survival of adult T-ALL patients with respect to the WT1 mutation status. (A) Overall T-ALL cohort. (B) Thymic TALL subgroup.
WT1 mRNA expression in adult T-ALL
Expression levels of WT1 were analyzed in 223 patients. Three expression groups including WT1 negative (n=97), WT1 intermediate (n=81), and WT1 high (n=45) were defined as outlined above (see Statistical analyses). These groups did not differ with respect to clinical parameters at diagnosis (including age, WBC, CNS involvement, mediastinal mass; data not shown). Patients with high WT1 expression were characterized by immature features such as an early immunophenotype, high BAALC expression levels, and aberrant expression of CD13 and HOX11L2. WT1 negative and WT1 intermediate cases had predominantly a thymic phenotype (Table 2). A strong correlation was observed between the WT1 expression and mutation status: WT1 mutations were predominantly found in the WT1 high group (13/41; 29% were WT1mut) compared to only 1/94 (1%) of WT1 negative and 4/74 (5%) of WT1 intermediate cases harboring WT1 mutations (P<0.001). No differences were seen in the frequencies of NOTCH1 or FBXW7 gene mutations across the WT1 expression groups (data not shown).
Table 2.
Molecular characteristics and outcome with respect to WT1 expression.

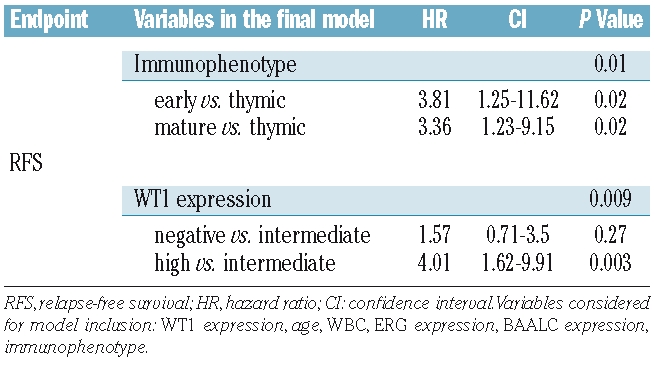
Complete remission rates were similar between patients of the different WT1 expression groups. However, T-ALL patients with negative or high WT1 expression relapsed more frequently (35% and 67%, respectively) compared to patients with intermediate WT1 expression (21%; overall P=0.002; Table 2). In the overall cohort, T-ALL patients with high expression showed an inferior outcome with only 25.8% of patients remaining relapse-free at four years as compared to 57.0% of WT1 negative and 77.2% of WT1 intermediate patients (overall P=0.001; Table 2, Figure 2A). Moreover, in the multivariate analysis (Table 3), WT1 expression was of independent prognostic significance (P=0.009). For T-ALL patients with high WT1 expression a Hazard Ratio of 4.0 and for patients lacking WT1 expression a Hazard Ratio of 1.6 were observed.
Figure 2.
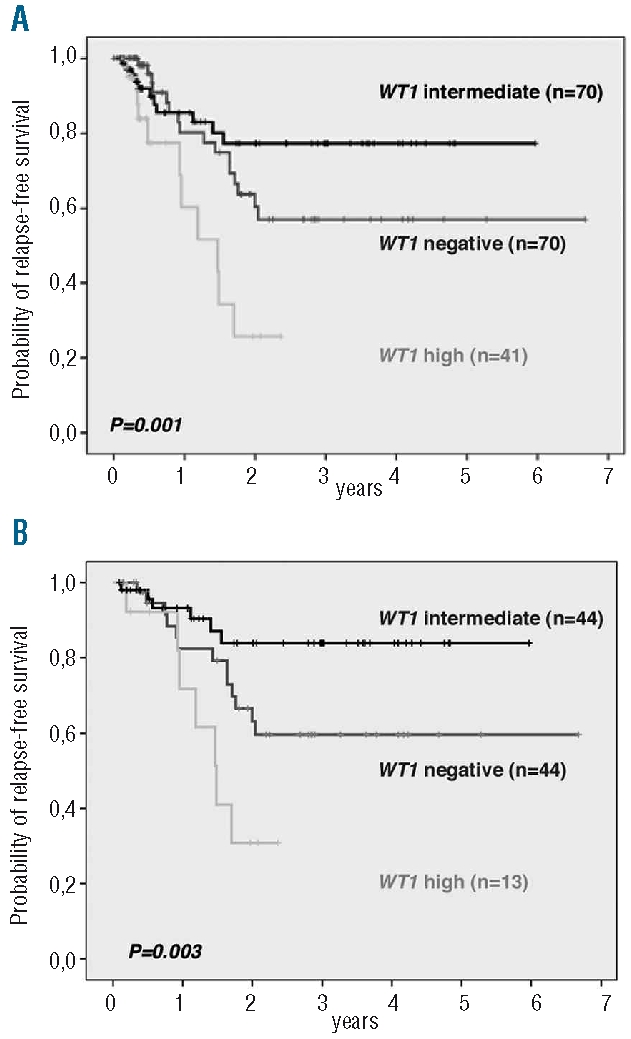
Relapse-free survival of adult T-ALL patients with respect to WT1 expression. (A) Overall T-ALL cohort. (B) Thymic T-ALL subgroup. The overall P values are given. Comparisons between the separate WT1 expression groups were as follows: 2A: WT1 intermediate versus WT1 negative: P=0.26; WT1 intermediate versus WT1 high: P=0.003; WT1 negative versus WT1 high: P=0.002. 2B: WT1 intermediate versus WT1 negative: P=0.055; WT1 intermediate versus WT1 high: P=0.054; WT1 negative versus WT1 high: P=0.001.
Table 3.
Multivariate analysis for relapse-free-survival.
Prognostic relevance of WT1 mRNA expression in adult T-ALL subgroups
We next investigated the prognostic impact of WT1 expression in the standard risk group of thymic T-ALL. An unfavorable outcome was observed for patients with WT1 high (relapse-free at four years: 30.8%) or negative expression (relapse-free at four years: 59.6%) as compared to thymic T-ALL with intermediate WT1 expression (relapse-free at four years: 83.9%; overall P=0.003; Table 2; Figure 2B).
Moreover, when patients with negative or high WT1 expression were combined to a WT1 high-risk group and compared to the favorable group of patients with intermediate WT1 expression (WT1 low-risk group), thymic TALL patients within the WT1 high-risk group showed an inferior overall survival (P=0.037; Figure 3A).
Figure 3.
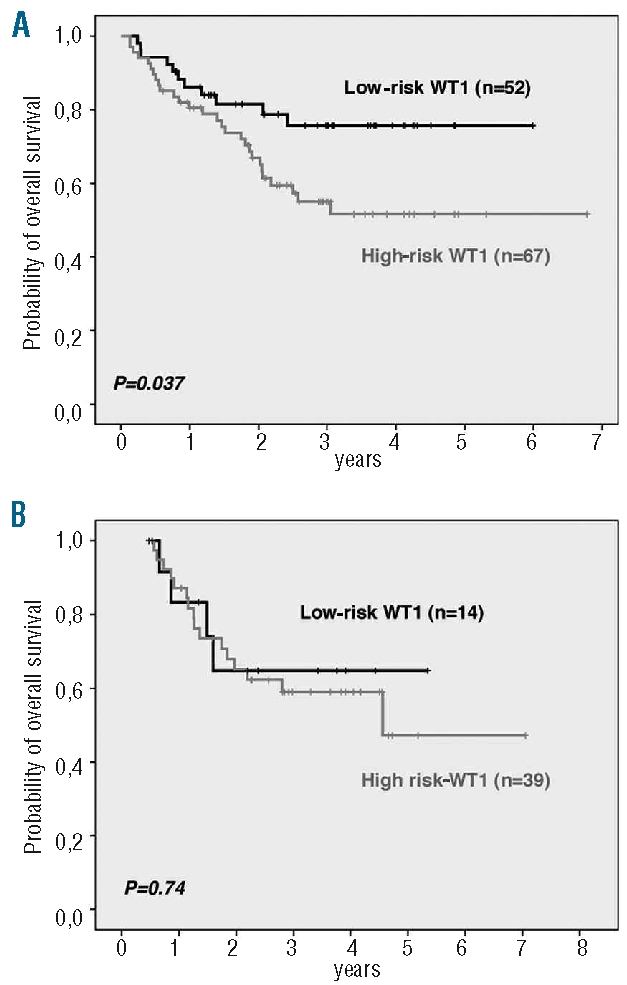
Overall-survival with respect to WT1 expression in T-ALL subgroups. (A) WT1 expression risk groups in thymic T-ALL. (B) WT1 expression risk groups in early/mature T-ALL (including only patients undergoing allogeneic stem cell transplantation as assigned by the protocol).
In the GMALL protocols, early and mature T-ALLs are defined high risk and allocated to allogeneic stem cell transplantation in first complete remission. For these patients, WT1 expression was no longer of prognostic relevance as a similar outcome for WT1 low-risk and WT1 high-risk patients was observed (P=0.74; Figure 3B).
Discussion
We present a study investigating genotype and expression alterations of WT1 in a large cohort of adult T-ALL. Overall, WT1 mutations were found in 8% of T-ALL patients at initial diagnosis and associated with a more immature T-ALL subtype; WT1 mutated patients showed a higher relapse rate. In addition, altered WT1 expression was of independent prognostic significance with negative or high WT1 expression levels predicting inferior outcome.
WT1 mutations have previously been reported in about 10% of AML patients with normal cytogenetics and were of adverse prognostic significance predicting inferior overall survival, relapse-free survival, as well as response to induction therapy.3–6 Interestingly, WT1 mutations were associated with high expression of the progenitor markers ERG and BAALC, which have been shown to share prognostic significance in AML and T-ALL.5,18 We, therefore, reasoned that WT1 mutations might also be of prognostic relevance in T-ALL. Frequency and localization were very similar to the reports in AML. WT1 mutations in exon 7 are expected to result in a truncated WT1 protein that acts in a dominant negative manner by impaired DNA binding and abolished protein interaction abilities.2 Thus, WT1 may function as a tumor suppressor not only contributing to myeloid but may also be implicated in T-cell leukemogenesis as the repression of downstream targets is likely impaired in mutated WT1 leukemic cells. Moreover, the coexistence of gene mutations, 70% (14/20) of T-ALL patients with WT1 mutations revealed additional mutations in NOTCH1, FBXW7, or FLT3, suggests that loss of WT1 function may act in cooperation with other genetic hits likely affecting differentiation and proliferation.
Whereas WT1 mutations were not predictive for primary chemotherapy resistance in our T-ALL cohort, in contrast to the observation in AML,6 WT1 mutated patients showed a higher relapse rate compared to WT1wt T-ALL patients and within the standard risk group of thymic T-ALL, WT1mut patients had an adverse relapse-free survival. These outcome analyses suggesting an adverse effect of WT1 mutations in T-ALL remain limited due to the small number of WT1 mutated cases. Interestingly, in a recent study by Tosello et al. WT1 mutations were found in a similar frequency of adult T-ALL patients (11.7%); however no significant prognostic impact was observed in the overall T-ALL cohort.17
In addition to mutational events, alterations of WT1 expression (both under- and overexpression) have been described in various malignancies. In acute leukemia, WT1 expression levels have mainly been studied in AML with inconsistent results with respect to their prognostic impact at initial diagnosis.15,29 Studies investigating WT1 expression as marker of minimal residual disease have shown more consistency with rising WT1 expression predicting relapse.30,31 However, the prognostic impact of WT1 expression has not yet been determined in a larger cohort of adult T-ALL. T-ALL patients with high WT1 expression levels displayed a specific molecular signature as shown by an immature phenotype, aberrant CD13 expression, and positivity for BAALC and HOX11L2 (both markers associated with inferior outcome).18,31 As it was shown that enforced expression of WT1 in thymocytes blocked intrathymic differentiation, high expression of WT1 in TALL blasts may reflect the cellular origin of progenitors (e.g. early thymocyte progenitor) that physiologically express WT1 at high levels.1,32,33 Importantly, WT1 expression remained of prognostic significance independently of the other factors including the immunophenotype.34 Similar to data in pediatric ALL by Boublikova et al., we also observed that in addition to patients with high WT1 expression, patients with no WT1 expression showed an inferior outcome compared to patients with intermediate expression levels.11 In particular, in the standard risk group of thymic T-ALL intermediate WT1 expression was associated with a relapse-free survival of over 80% at four years, whereas WT1 negative and WT1 high expressers did significantly worse (59.6% and 30.8%, respectively).
The identification of WT1 mutations acting in a dominant negative fashion as well as the ability of WT1 to induce growth inhibition and suppress tumorgenicity in mice has supported its role as tumor suppressor.14,15 We propose that in addition to its mutational inactivation, lack of WT1 expression may act in a similar manner in TALL. So far combined expression and mutation analyses of WT1 have not yet been performed, but gene expression analyses identified a subgroup of AML patients with very low WT1 expression and indicated that WT1 is specifically deregulated in subgroups of leukemia.35 In fact, the WT1 negative expression group rarely showed WT1 mutations (only one T-ALL patient lacking WT1 expression harbored a WT1 mutation) suggesting that lack of mRNA might be sufficient for a pathophysiological impact and would not require additional mutational inactivation. Thus, lack of physiological WT1 function resulting in altered regulation of downstream targets might be implicated in leukemogenesis and may confer chemotherapy resistance similar to mutational inactivation of WT1.
On the other hand, an oncogenic role has also been suggested by WT1 overexpression where enforced expression of WT1 in murine hematopoietic progenitors resulted in expansion of undifferentiated cells and the addition of a second genetic hit blocking differentiation-induced leukemic transformation.36 In leukemia, high levels of WT1 expression may promote proliferation and protect cells from apoptosis as it was shown that WT1 overexpression is associated with chemotherapy resistance in vitro due to repression of the proapoptotic gene BAK and induction of BCL2.37,38 In T-ALL, these findings can be recapitulated as leukemic blasts with high level WT1 expression displaying an immature phenotype and patients with WT1 high expression showing a higher relapse rate and an inferior survival. The observation that WT1 mutated cases show frequent overexpression of WT1 (expression of the WT1 wild type as well as mutated allele) might be a result of its negative auto-regulation, which is impaired by the mutations affecting the DNA binding domain.39
In summary, these data suggest that the transcriptional control of WT1 resembles Janus-like characteristics with abilities in repression as well as activation of different downstream pathways. Thus altered WT1 function, either by mutational inactivation or lack of mRNA expression, as well as aberrant overexpression might contribute in different ways. Though the precise alterations of WT1 downstream pathways in these scenarios will have to be explored in T-ALL, the prognostic implications already ask for validation. Until further studies confirm these findings, immunophenotyping remains at this point the most established factor used for risk adapted treatment stratification.
Acknowledgments
we thank Liliana Mochmann for critical reading of the manuscript.
Footnotes
Funding: supported by a grant from the Deutsche Krebshilfe (Max-Eder Nachwuchsförderung) to CD Baldus.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
SH conducted the study, performed the laboratory work and wrote the manuscript. NG and DH provided the clinical data and critically reviewed the manuscript. AS participated in the statistical analysis. CS, TB, SS, OB, UK, AB provided molecular data. ET and WKH co-ordinated the research. CDB was the principal investigator, wrote the manuscript, and takes primary responsibility for the paper.
The authors reported no potential conflicts of interest.
References
- 1.Ariyaratana S, Loeb DM. The role of the Wilms tumour gene (WT1) in normal and malignant haematopoiesis. Expert Rev Mol Med. 2007;9(14):1–17. doi: 10.1017/S1462399407000336. [DOI] [PubMed] [Google Scholar]
- 2.Little M, Wells C. A clinical overview of WT1 gene mutations. Hum Mutat. 1997;9(3):209–25. doi: 10.1002/(SICI)1098-1004(1997)9:3<209::AID-HUMU2>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 3.King-Underwood L, Pritchard-Jones K. Wilms’ tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood. 1998;91(8):2961–8. [PubMed] [Google Scholar]
- 4.Summers K, Stevens J, Kakkas I, Smith M, Smith LL, Macdougall F, et al. Wilms’ tumour 1 mutations are associated with FLT3-ITD and failure of standard induction chemotherapy in patients with normal karyotype AML. Leukemia. 2007;21(3):550–1. doi: 10.1038/sj.leu.2404514. [DOI] [PubMed] [Google Scholar]
- 5.Paschka P, Marcucci G, Ruppert AS, Whitman SP, Mrózek K, Maharry K, et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2008;26(28):4595–602. doi: 10.1200/JCO.2007.15.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Virappane P, Gale R, Hills R, Kakkas I, Summers K, Stevens J, et al. Mutation of the Wilms’ Tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: The United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol. 2008;26(33):5429–35. doi: 10.1200/JCO.2008.16.0333. [DOI] [PubMed] [Google Scholar]
- 7.Menssen HD, Renkl HJ, Rodeck U, Maurer J, Notter M, Schwartz S, et al. Presence of Wilms’ tumor gene (wt1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia. 1995;9(6):1060–7. [PubMed] [Google Scholar]
- 8.Schmid D, Heinze G, Linnerth B, Tisljar K, Kusec R, Geissler K, et al. Prognostic significance of WT1 gene expression at diagnosis in adult de novo acute myeloid leukemia. Leukemia. 1997;11(5):639–43. doi: 10.1038/sj.leu.2400620. [DOI] [PubMed] [Google Scholar]
- 9.Barragán E, Cervera J, Bolufer P, Ballester S, Martín G, Fernández P, et al. Prognostic implications of Wilms’ tumor gene (WT1) expression in patients with de novo acute myeloid leukemia. Haematologica. 2004;89(8):926–33. [PubMed] [Google Scholar]
- 10.Bergmann L, Miething C, Maurer U, Brieger J, Karakas T, Weidmann E, Hoelzer D. High levels of Wilms’ tumor gene (wt1) mRNA in acute myeloid leukemias are associated with a worse long-term outcome. Blood. 1997;90(3):1217–25. [PubMed] [Google Scholar]
- 11.Boublikova L, Kalinova M, Ryan J, Quinn F, O’Marcaigh A, Smith O, et al. Wilms’ tumor gene 1 (WT1) expression in childhood acute lymphoblastic leukemia: a wide range of WT1 expression levels, its impact on prognosis and minimal residual disease monitoring. Leukemia. 2006;20(2):254–63. doi: 10.1038/sj.leu.2404047. [DOI] [PubMed] [Google Scholar]
- 12.Chiusa L, Francia di Celle P, Campisi P, Ceretto C, Marmont F, Pich A. Prognostic value of quantitative analysis of WT1 gene transcripts in adult acute lymphoblastic leukemia. Haematologica. 2006;91(2):270–1. [PubMed] [Google Scholar]
- 13.Baird PN, Simmons PJ. Expression of the Wilms’ tumor gene (WT1) in normal hemopoiesis. Exp Hematol. 1997;25(4):312–20. [PubMed] [Google Scholar]
- 14.Svedberg H, Richter J, Gullberg U. Forced expression of the Wilms tumor 1 (WT1) gene inhibits proliferation of human hematopoietic CD34(+) progenitor cells. Leukemia. 2001;15(12):1914–22. doi: 10.1038/sj.leu.2402303. [DOI] [PubMed] [Google Scholar]
- 15.Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21(5):868–76. doi: 10.1038/sj.leu.2404624. [DOI] [PubMed] [Google Scholar]
- 16.Ellisen LW, Carlesso N, Cheng T, Scadden DT, Haber DA. The Wilms tumor suppressor WT1 directs stage-specific quiescence and differentiation of human hematopoietic progenitor cells. EMBO J. 2001;20(8):1897–909. doi: 10.1093/emboj/20.8.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tosello V, Mansour MR, Barnes K, Paganin M, Sulis ML, Jenkinson S, et al. WT1 mutations in T-ALL. Blood. 2009;114(5):1038–45. doi: 10.1182/blood-2008-12-192039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baldus CD, Martus P, Burmeister T, Schwartz S, Gökbuget N, Bloomfield CD, et al. Low ERG and BAALC expression identifies a new subgroup of adult acute T-lymphoblastic leukemia with a highly favorable outcome. J Clin Oncol. 2007;25(24):3739–45. doi: 10.1200/JCO.2007.11.5253. [DOI] [PubMed] [Google Scholar]
- 19.Gilliland DG, Jordan CT, Felix CA. The molecular basis of leukemia. Hematology Am Soc Hematol Educ Program. 2004:80–97. doi: 10.1182/asheducation-2004.1.80. [DOI] [PubMed] [Google Scholar]
- 20.Van Vlierberghe P, Homminga I, Zuurbier L, Gladdines-Buijs J, van Wering ER, Horstmann M, et al. Cooperative genetic defects in TLX3 rearranged pediatric TALL. Leukemia. 2008;22(4):762–70. doi: 10.1038/sj.leu.2405082. [DOI] [PubMed] [Google Scholar]
- 21.Brüggemann M, Raff T, Flohr T, Gökbuget N, Nakao M, Droese J, et al. German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia. Clinical significance of minimal residual disease quantification in adult patients with standard-risk acute lymphoblastic leukemia. Blood. 2006;107(3):1116–23. doi: 10.1182/blood-2005-07-2708. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz S, Rieder H, Schläger B, Burmeister T, Fischer L, Thiel E. Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(−)/CD24(−)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia. 2003;17(8):1589–95. doi: 10.1038/sj.leu.2402989. [DOI] [PubMed] [Google Scholar]
- 23.Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, van’t Veer MB. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL) Leukemia. 1995;9(10):1783–6. [PubMed] [Google Scholar]
- 24.Weng AP, Ferrando AA, Lee W, Morris JP, 4th, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 25.Nowak D, Mossner M, Baldus CD, Hopfer O, Thiel E, Hofmann WK. Mutation analysis of hCDC4 in AML cells identifies a new intronic polymorphism. Int J Med Sci. 2006;3(4):148–51. doi: 10.7150/ijms.3.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beillard E, Pallisgaard N, van der Velden VH, Bi W, Dee R, van der Schoot E, et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR) - a Europe against cancer program. Leukemia. 2003;17(12):2474–86. doi: 10.1038/sj.leu.2403136. [DOI] [PubMed] [Google Scholar]
- 27.Paietta E, Ferrando AA, Neuberg D, Bennett JM, Racevskis J, Lazarus H, Dewald G, Rowe JM, Wiernik PH, Tallman MS, Look AT. Activating FLT3 mutations in CD117/KIT(+) T-cell acute lymphoblastic leukemias. Blood. 2004;104(2):558–60. doi: 10.1182/blood-2004-01-0168. [DOI] [PubMed] [Google Scholar]
- 28.Van Vlierberghe P, Meijerink JP, Stam RW, van der Smissen W, van Wering ER, Beverloo HB, Pieters R. Activating FLT3 mutations in CD4+/CD8- pediatric T-cell acute lymphoblastic leukemias. Blood. 2005;106(13):4414–5. doi: 10.1182/blood-2005-06-2267. [DOI] [PubMed] [Google Scholar]
- 29.Lapillonne H, Renneville A, Auvrignon A, Flamant C, Blaise A, Perot C, et al. High WT1 expression after induction therapy predicts high risk of relapse and death in pediatric acute myeloid leukemia. J Clin Oncol. 2006;24(10):1507–15. doi: 10.1200/JCO.2005.03.5303. [DOI] [PubMed] [Google Scholar]
- 30.Weisser M, Kern W, Rauhut S, Schoch C, Hiddemann W, Haferlach T, Schnittger S. Prognostic impact of RT-PCR-based quantification of WT1 gene expression during MRD monitoring of acute myeloid leukemia. Leukemia. 2005;19(8):1416–23. doi: 10.1038/sj.leu.2403809. [DOI] [PubMed] [Google Scholar]
- 31.Baak U, Gökbuget N, Orawa H, Schwartz S, Hoelzer D, Thiel E, Burmeister T German Multicenter ALL Study Group. Thymic adult T-cell acute lymphoblastic leukemia stratified in standard- and high-risk group by aberrant HOX11L2 expression: experience of the German multicenter ALL study group. Leukemia. 2008;22(6):1154–60. doi: 10.1038/leu.2008.52. [DOI] [PubMed] [Google Scholar]
- 32.Hosen N, Sonoda Y, Oji Y, Kimura T, Minamiguchi H, Tamaki H, et al. Very low frequencies of human normal CD34+ haematopoietic progenitor cells express the Wilms’ tumour gene WT1 at levels similar to those in leukaemia cells. Br J Haematol. 2002;116(2):409–20. doi: 10.1046/j.1365-2141.2002.03261.x. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Oka Y, Tsuboi A, Yamagami T, Miyazaki T, Yusa S, et al. The lck promoter-driven expression of the Wilms tumor gene WT1 blocks intrathymic differentiation of T-lineage cells. Int J Hematol. 2003;77(5):463–70. doi: 10.1007/BF02986614. [DOI] [PubMed] [Google Scholar]
- 34.Thiel E, Kranz BR, Raghavachar A, Bartram CR, Löffler H, Messerer D, et al. Prethymic phenotype and genotype of pre-T (CD7+/ER−)-cell leukemia and its clinical significance within adult acute lymphoblastic leukemia. Blood. 1989;73(5):1247–58. [PubMed] [Google Scholar]
- 35.Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350(16):1617–28. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- 36.Nishida S, Hosen N, Shirakata T, Kanato K, Yanagihara M, Nakatsuka S, et al. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood. 2006;107(8):3303–12. doi: 10.1182/blood-2005-04-1656. [DOI] [PubMed] [Google Scholar]
- 37.Ito K, Oji Y, Tatsumi N, Shimizu S, Kanai Y, Nakazawa T, et al. Antiapoptotic function of 17AA(+)WT1 (Wilms’ tumor gene) isoforms on the intrinsic apoptosis pathway. Oncogene. 2006;25(30):4217–29. doi: 10.1038/sj.onc.1209455. [DOI] [PubMed] [Google Scholar]
- 38.Mayo MW, Wang CY, Drouin SS, Madrid LV, Marshall AF, Reed JC, Weissman BE, Baldwin AS. WT1 modulates apoptosis by transcriptionally upregulating the bcl-2 proto-oncogene. EMBO J. 1999;18(14):3990–4003. doi: 10.1093/emboj/18.14.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rupprecht HD, Drummond IA, Madden SL, Rauscher FJ, 3rd, Sukhatme VP. The Wilms’ tumor suppressor gene WT1 is negatively autoregulated. J Biol Chem. 1994;269(8):6198–206. [PubMed] [Google Scholar]