

Abstract
Platelet-type von Willebrand disease (PT-VWD) is a rare autosomal dominant bleeding disorder which is due to a mutation in the gene encoding for platelet glycoprotein Ibα (GPIbα) resulting in enhanced affinity for von Willebrand factor (VWF). PT-VWD is often mistakenly diagnosed as type 2B VWD for the similarities between these two conditions. We characterized a new case of PT-VWD and evaluated the usefulness of a flow cytometric assay in the differential diagnosis between PT-VWD (n=1) and type 2B VWD (n=4). The flow cytometric assay was able to highlight the increased affinity of VWF for GPIbα as much as did RIPA and to differentiate the two diseases through mixing tests. Genetic analysis revealed a heterozygous point mutation in codon 239 of the GPIbα gene leading to a methionine to valine substitution (M239V). Flow cytometry represents a useful tool for the diagnosis of PT-VWD.
Keywords: von Willebrand disease, PT-VWD, GPIbα
Introduction
Platelet-type von Willebrand disease (PT-VWD) is a rare autosomal dominant bleeding disorder due to a mutation in the gene encoding for platelet glycoprotein Ibα (GPIbα) confering to GPIbα an enhanced affinity for von Willebrand factor (VWF).1–5 As a consequence, platelets from patients with PT-VWD bind spontaneously high molecular weight (HMW) multimers of VWF and are then cleared from the circulation, resulting in thrombocytopenia and the loss of HMW VWF multimers.
Patients with PT-VWD present a mild thrombocytopenia with large platelets and often platelet aggregates in blood smears and have a prolonged bleeding time associated with mucocutaneous bleeding or hemorrhage after surgery. Ristocetin induced platelet aggregation (RIPA) is enhanced, plasma von Willebrand factor antigen (VWF:Ag) is normal or mildly reduced while von Willebrand factor activity is reduced, resulting in a low VWF:activity/VWF:Ag ratio. Given the clinical and laboratory similarities, patients with PT-VWD are often wrongly diagnosed as type 2B VWD6,7 and the real prevalence of PT-VWD is therefore probably underestimated.
The differential diagnosis between these two conditions relies on genetic analysis, which is technically demanding and not available to most laboratories. Alternatively, differential diagnosis requires several assays, including mixing tests and cryoprecipitate challenge, which are rather cumbersome. The last method in particular often gives ambiguous results, because false positives can be found in some individuals with type 2B VWD.8
Differential diagnosis is important because the two diseases must be treated differently: patients with type 2B VWD with normal exogenous VWF while patients with PT-VWD with platelet transfusions.5
We have recently described a flow cytometric assay to quantify VWF-binding induced by ristocetin to fresh autologous or to formalin-fixed donor platelets.9 The assay is useful in the diagnosis of VWD and for the monitoring of DDAVP treatment and in particular is able to detect enhanced affinity of VWF for GPIb, similarly to the RIPA, and therefore allows the diagnosis of type 2B VWD.
We report here a patient with a lifelong mucocutaneous bleeding disorder in which we made the differential diagnosis of PT-VWD versus type 2B VWD by applying the flow cytometric assay and confirmed this by genetic analysis.
Design and Methods
Case Report
A 30-year old female with a mild bleeding diathesis and a previous diagnosis of VWD was referred to our Center for further evaluation. She had had a history of mild thrombocytopenia and easy bruising, either spontaneous or following minor trauma, since birth. Bone marrow aspirate was normal with a mild increase of megakaryocytes. We also studied 5 other members of her family (mother, father, sister and 2 sons), with no history of bleeding, who resulted normal. As controls we simultaneously studied 11 healthy subjects who had not ingested any drugs within the previous 10 days and 4 patients previously characterized as type 2B VWD.9 All patients studied gave their informed, written consent to the studies performed; all studies were carried out in conformity with the declaration of Helsinki.
Laboratory studies
A standardized bleeding time test (BT) was performed, as previously described.10 RIPA was evaluated with citrated platelet-rich plasma (PRP) using an optical aggregometer (APACT-4, Helena Biosciences Europe, Sunderland, UK) by challenging platelets with 0.3 to 2.0 mg/ml ristocetin (Mascia Brunelli S.p.A., Milan, Italy); VWF:RCo was assessed with formalin-fixed platelets using a commercial kit (Helena Biosciences Europe, Sunderland, UK). VWF:Ag (Diagnostica Stago, Asnieres, France), and VWF:CB (Gradipore Ltd, French Forest NSW, Australia) were evaluated by ELISA. PFA-100® (Dade-Behring, Deerfield, IL, USA) was performed using collagen/ADP (C/ADP) and collagen/epinephrine (C/Epi) cartridges, as described.9
The binding of VWF induced by ristocetin to either fresh autologous or donor formalin-fixed platelets was evaluated by flow cytometry using a mouse anti-human VWF antibody, clone 4f9 (Immunotech, Marseille, France) and a FITC-conjugated goat against-mouse IgG (Beckman Coulter, Miami, FL, USA), as previously described.9 The expression of platelet membrane glycoproteins was investigated by whole blood flow cytometry using FITC-conjugated monoclonal antibodies anti GPIIb (P2), GPIIIa (SZ21), GPIbα (SZ2), GPIX (SZ1), GPIV (FA6-152), CD9 (ALB6) and CD31 (5.6E) and a PE conjugated platelet-specific monoclonal antibody, as described.11, 12
Samples were analyzed in an EPICS XL-MCL flow cytometer (Coulter Corporation, Miami, Florida, USA).
Aggregometric and flow cytometric mixing assays were performed by adding patient or control platelet-poor plasma (PPP) to patient or control platelet pellets, in the following combinations:13 patient platelets/patient plasma, patient platelets/control plasma, control platelets/control plasma, control platelets/patient plasma. Platelet aggregation induced by cryoprecipitate, in an amount equivalent to 60 U/dL of VWF, was studied as previously described.14
For the identification of the molecular defect, the patient’s genomic DNA was extracted and the entire coding region of GPIBA was amplified by PCR, in four overlapping fragments, using a series of oligonucleotide primer pairs. PCR products were purified and the complete sequence of the GPIBA coding region was obtained by direct sequencing.2
Results and Discussion
The patient was studied on three different occasions over an eight month period. Routine blood clotting assays were normal. Platelet count was mildly reduced (130–168×106/mL) with a mean platelet volume of 11.7 fL (normal 8.0–12.0 fL). Bleeding time was over 20 min and PFA-100® C/Epi and C/ADP CT over 300sec. Platelet aggregation in response to collagen, ADP, arachidonic acid and adrenaline was normal while the RIPA was markedly increased (0.3–0.4 mg/mL; normal values 1.1±0.15 mg/mL, range 0.9–1.35 mg/mL) (Figure 1B); the patient’s platelets showed a mild spontaneous aggregation (Figure 1A). All the main platelet glycoproteins, including GPIbα, were normal.
Figure 1.
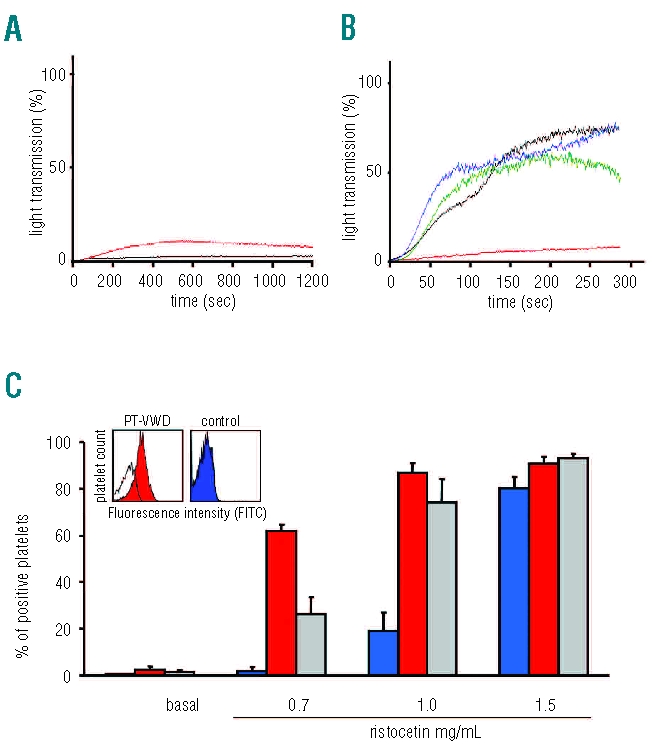
(A) Representative platelet aggregation tracings showing mild spontaneous aggregation in a PT-VWD patient (red); black tracing represents a healthy control. (B) Representative platelet aggregation tracings in response to ristocetin in PT-VWD (green: 0.3 mg/mL; blue: 0.9 mg/mL) and control (red: 0.3 mg/mL; black: 0.9 mg/mL). (C) Binding of VWF to control (blue), PT-VWD (red) and type 2B VWD (gray) platelets induced by ristocetin (0, 0.7, 1, 1.5 mg/mL) as assessed by flow cytometry. Data represent mean±SEM for 3 repeated measures obtained for 1 PT-VWD patient and mean±SEM obtained of 4 different type 2B VWD patients. The inset shows flow cytometric histograms representing VWF binding to patient’s or control’s platelets induced by ristocetin (0.7 mg/mL). The white histograms represent the sample in absence of ristocetin.
VWF:Ag was within the normal range (63–84 U/dL; normal 55.7–129.6 U/dL) while VWF:RCo varied from 27 to 60 U/dL (normal 40.9–124.8 U/dL) and the VWF:RCo/VWF:Ag ratio was reduced (0.43–0.75; normal 0.6–1.2). VWF:CB was 45.7–61.0 U/dL (normal 57.0–100.9) and the VWF:CB/VWF:Ag ratio was 0.7 (normal 0.7–1.1).
Ristocetin-induced VWF-binding to autologous fresh platelets, by flow cytometry, was markedly increased as well as in 4 patients with type 2B VWD (Figure 1C) while ristocetin-induced VWF-binding to donor formalin-fixed platelets was mildly reduced (36.6 U/dL; normal 51.5–129.8 U/dL).
Mixing tests performed by either light transmission aggregometry or flow cytometry showed that the patient’s plasma had no effect on normal platelets and that the patient’s platelets maintained enhanced VWF-binding when mixed with normal plasma (Figure 2), demonstrating the platelet origin of the defect. On the contrary, when the mixing tests were performed with samples from 4 patients with type 2B VWD, it emerged that the patient’s plasma induced enhanced VWF binding when added to normal platelets while patient’s platelets bound a normal amount of VWF when mixed with control plasma, demonstrating the plasmatic origin of the defect (Figure 2A).
Figure 2.
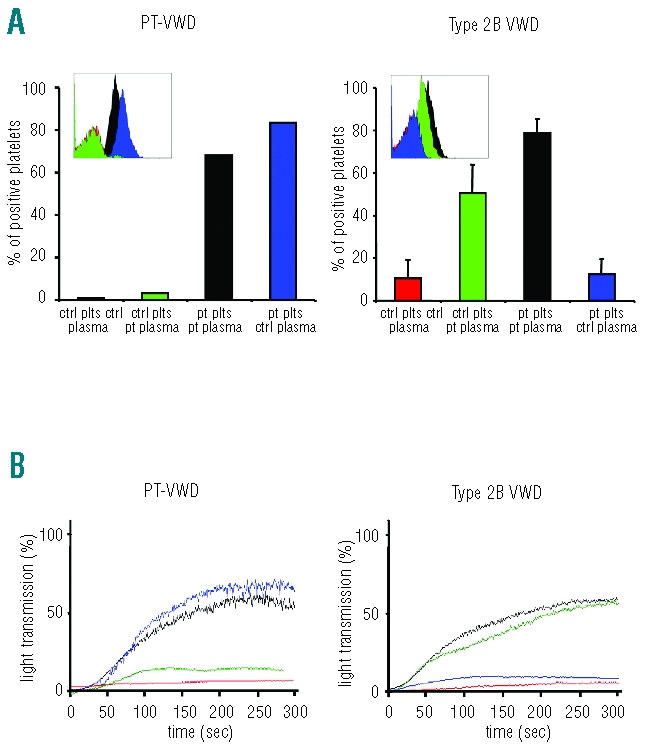
Aggregometric and flow cytometric mixing assays were performed by adding patient (PT-VWD or type 2B VWD) or control PPP to patient or control platelet pellets. VWF binding and RIPA were determined in the resulting mixed samples: control platelets/control plasma (red), control platelets/patient plasma (green), patient platelets/patient plasma (black) and patient platelets/control plasma (blue). Binding of VWF to platelets induced by ristocetin (1mg/mL) was detected by flow cytometry and expressed as % of positive platelets (A). The insets show flow cytometric histograms representing VWF binding to platelet GPIbα. Ristocetin induced platelet aggregation was evaluated by light transmission aggregometry (B). Data represent mean obtained for 1 PT-VWD patient and mean±SEM obtained from 4 different type 2B VWD patients.
Finally, the addition of cryoprecipitate to the patient’s platelets induced aggregation as well as VWF-binding, while cryoprecipitate was inactive when added to control platelets or platelets from type 2B VWD (Figure 3).
Figure 3.
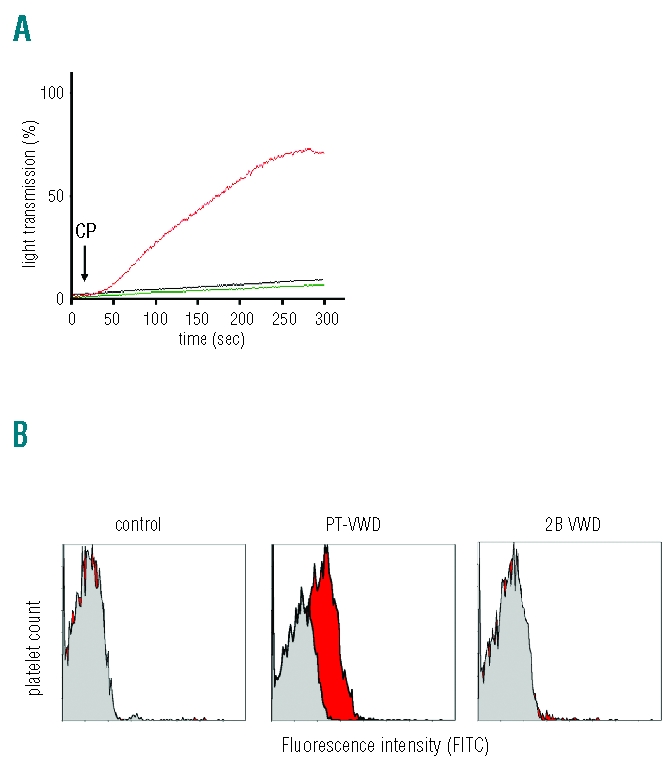
Representative platelet aggregation tracings in response to cryoprecipitate (CP), of a patient with PT-VWD (red), a patient with type 2B VWD (black) and a healthy subject (green) (A). Flow cytometric histograms representing VWF binding to platelet GPIbα in a healthy subject, in PT-VWD patient and in a type 2B VWD patient in response to cryoprecipitate (red) or to vehicle (gray) (B).
DNA sequencing revealed a heterozygous point mutation in codon 239 of GPIBA, an A to G exchange leading to a methionine-to-valine aminoacid substitution (M239V), a molecular defect previously described in 8 patients from 4 different families with PT-VWD.2,15,16
PT-VWD is a rare bleeding disorder, with 42 patients belonging to 16 unrelated families so far described; but its real prevalence is not known. A few published series suggest that the prevalence of PT-VWD may be about 10% that of type 2B VWD, but in fact it could represent a larger proportion because affected patients are often misdiagnosed as type 2B VWD. Based on our limited case series in patients with suspected type 2B VWD, up to 20% may actually be PT-VWD. In fact, out of 5 patients with enhanced affinity between VWF and platelets, one resulted to be a PT-VWD. Our findings confirm a likely underestimate of the prevalence of PT-VWD. However, for a reliable estimate, a large multicenter and prospective study has to be performed before making any conclusion on the prevalence of PT-VWD versus type 2B VWD, and the ongoing Canadian PT-VWD project is certainly a means to obtain this information.17,18
Enhanced platelet aggregation in response to low doses of ristocetin is the first laboratory evidence of an increased affinity of VWF for GPIbα orienting towards either type 2B VWD or PT-VWD. Further assays are then required to differentiate these two rare hemorrhagic disorders, including platelet aggregation mixing tests. Genetic analysis of the von Willebrand factor or GPIbα genes is the conclusive test, but is only available in a few laboratories. We have applied a recently described flow cytometric assay of the binding of VWF to platelets induced by ristocetin9 to the diagnosis of PT-VWD.
The assay performed with fresh autologous platelets clearly highlighted the increased affinity of VWF for GPIbα and, when applied to mixing tests, it was able to differentiate PT-VWD from type 2B VWD demonstrating the platelet origin of the defect. Moreover, the flow cytometric test was able to detect VWF-binding to the patient’s platelets induced by cryoprecipitate, a feature characterizing PT-VWD. The advantages of flow cytometry for the diagnosis of PT-VWD are its special ability to detect enhanced affinity of VWF for GPIbα. In fact, VWF binding induced by low doses of ristocetin (0.7 mg/mL) in type 2B VWD (n=4) was increased about 14.6 fold compared to that found in healthy controls (n=37), while in the same subjects ristocetin-induced light transmission was only increased 3.6 fold compared to healthy controls. Similarly, in PT-VWD the increase of VWF binding was 34.5 fold and that of light transmission 3.7 fold compared to healthy controls. Thus, flow cytometry can discriminate better between patients with enhanced affinity of VWF for GPIbα and healthy controls. Other advantages of flow cytometry are the small volume of sample required (about 150μL), the rapidity of the test, and the possibility of performing mixing tests and cryoprecipitate challenge with small blood volumes, including in pediatric samples. Moreover, flow cytometry is now a widely used technique available to most hematology laboratories and this may allow PT-VWD to be diagnosed also in laboratories not specifically involved in platelet function studies. In conclusion, flow cytometry may represent a simple and useful tool for the diagnosis of VWF and for the discrimination of different VWD types, although this should be validated in a larger study.
Acknowledgments
We thank Dr. S. Ballanti (Hematology Department, University of Perugia) for referring the patient to us.
Footnotes
Funding: this work was supported in part by a grant from Fondazione Cassa di Risparmio di Perugia (project n. 2007-0130-020), Fondazione Cassa di Risparmio di Perugia (project 2008.021.329), a grant from the Italian Ministry of University and Scientific Research (MIUR 2007, Prot. 20073KBBHC_003) and a grant from the Italian Ministry of Health (RFPS-2006-8-334062) to Prof. P. Gresele.
Authorships and Disclosures
PG performed the clinical assessment of the patient and planned the study. SG, AMM and LC performed the laboratory assays. PG and SG wrote the manuscript. PG critically revised manuscript content and gave final approval for its submission.
The authors reported no potential conflicts of interest.
References
- 1.Franchini M, Montagnana M, Lippi G. Clinical, laboratory and therapeutic aspects of platelet-type von Willebrand disease. Int J Lab Hematol. 2008;30(2):91–4. doi: 10.1111/j.1751-553X.2007.00978.x. [DOI] [PubMed] [Google Scholar]
- 2.Russell SD, Roth GJ. Pseudo-von Willebrand disease: a mutation in the platelet glycoprotein Ib alpha gene associated with a hyperactive surface receptor. Blood. 1993;81(7):1787–91. [PubMed] [Google Scholar]
- 3.Matsubara Y, Murata M, Sugita K, Ikeda Y. Identification of a novel point mutation in platelet glycoprotein Ibα, Gly to Ser at residue 233, in a Japanese family with platelet-type von Willebrand disease. J Thromb Haemost. 2003;1(10):2198–205. doi: 10.1046/j.1538-7836.2003.00369.x. [DOI] [PubMed] [Google Scholar]
- 4.Miller JL, Cunningham D, Lyle VA, Finch CN. Mutation in the gene encoding the α chain of platelet glycoprotein Ib in platelet-type von Willebrand disease. Proc Natl Acad Sci USA. 1991;88(11):4761–5. doi: 10.1073/pnas.88.11.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Othman M, Notley C, Lavender FL, White H, Byrne CD, Lillicrap D, O'Shaughnessy DF. Identification and functional characterization of a novel 27-bp deletion in the macroglycopeptide-coding region of the GPIBA gene resulting in platelet-type von Willebrand disease. Blood. 2005;105(11):4330–6. doi: 10.1182/blood-2002-09-2942. [DOI] [PubMed] [Google Scholar]
- 6.Federici AB. Diagnosis of inherited von Willebrand disease: a clinical perspective. Semin Thromb Hemost. 2006;32(6):555–65. doi: 10.1055/s-2006-949661. [DOI] [PubMed] [Google Scholar]
- 7.Sadler JE, Budde U, Eikenboom JCJ, Favaloro EJ, Hill FGH, Holmberg L, et al. The working party on von Willebrand disease classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand factor. J Thromb Haemost. 2006;4(10):2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 8.Favaloro EJ. 2B or not 2B? Differential identification of type 2B, versus pseudo-von Willebrand disease. Br J Haematol. 2006;135(1):141–2. doi: 10.1111/j.1365-2141.2006.06249.x. [DOI] [PubMed] [Google Scholar]
- 9.Giannini S, Mezzasoma AM, Leone M, Gresele P. Laboratory diagnosis and monitoring of desmopressin treatment of von Willebrand’s disease by flow cytometry. Haematologica. 2007;92(12):1647–54. doi: 10.3324/haematol.11313. [DOI] [PubMed] [Google Scholar]
- 10.Ciferri S, Emiliani C, Guglielmini G, Orlacchio A, Nenci GG, Gresele P. Platelets release their lysosomal content in vivo in humans upon activation. Thromb Haemost. 2000;83(1):157–64. [PubMed] [Google Scholar]
- 11.Gresele P, Falcinelli E, Giannini S, D'Adamo P, D'Eustacchio A, Corazzi T, et al. Dominant inheritance of a novel integrin {beta}3 mutation associated with a hereditary macrothrombocytopenia and platelet dysfunction in two Italian families. Haematologica. 2009;94(5):663–9. doi: 10.3324/haematol.2008.002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giannini S, Mezzasoma AM, Guglielmini G, Rossi R, Falcinelli E, Gresele P. A new case of acquired Glanzmann’s thrombasthenia: diagnostic value of flow cytometry. Cytometry B Clin Cytom. 2008;74(3):194–9. doi: 10.1002/cyto.b.20396. [DOI] [PubMed] [Google Scholar]
- 13.Favaloro EJ, Patterson D, Denholm A, Mead S, Gilbert A, Collins A, Estell J, et al. Differential identification of a rare form of platelet-type (pseudo-) von Willebrand disease (VWD) from Type 2B VWD using a simplified ristocetin-induced-platelet-agglutination mixing assay and confirmed by genetic analysis. Br J Haematol. 2007;139(4):623–6. doi: 10.1111/j.1365-2141.2007.06850.x. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi H, Handa M, Watanabe K, Ando Y, Nagayama R, Hattori A, et al. Further characterization of platelet-type von Willebrand’s disease in Japan. Blood. 1984;64(6):1254–62. [PubMed] [Google Scholar]
- 15.Takahashi H, Murata M, Moriki T, Anbo H, Furukawa T, Nikkuni K, et al. Substitution of Val for Met at residue 239 of platelet glycoprotein Ib alpha in Japanese patients with platelet-type von Willebrand disease. Blood. 1995;85(3):727–33. [PubMed] [Google Scholar]
- 16.Kunishima S, Heaton DC, Naoe T, Hickton C, Mizuno S, Saito H, Kamiya T. De novo mutation of the platelet glycoprotein Ib alpha gene in a patient with pseudo-von Willebrand disease. Blood Coagul Fibrinolysis. 1997;8(5):311–5. doi: 10.1097/00001721-199707000-00009. [DOI] [PubMed] [Google Scholar]
- 17.Favaloro EJ. Phenotypic identification of platelet-type von Willebrand disease and its discrimination from type 2B von Willebrand disease: a question of 2B or not 2B? A story of nonidentical twins? Or two sides of a multidenominational or multifaceted primary-hemostasis coin? Semin Thromb Hemost. 2008;34(1):113–27. doi: 10.1055/s-2008-1066019. [DOI] [PubMed] [Google Scholar]
- 18.Othman M, Favaloro EJ. Genetics of type 2B von Willebrand disease: “true 2B,” “tricky 2B,” or “not 2B.” What are the modifiers of the phenotype? Semin Thromb Hemost. 2008;34(6):520–31. doi: 10.1055/s-0028-1103363. [DOI] [PubMed] [Google Scholar]