

SUMMARY
The source, specificity, and plasticity of the forkhead box transcription factor 3 (Foxp3)+ regulatory T (Treg) and conventional T (Tconv) cell populations active at sites of autoimmune pathology are not well characterized. To evaluate this, we combined global repertoire analyses and functional assessments of isolated T cell receptors (TCR) from TCRα retrogenic mice with autoimmune encephalomyelitis. Treg and Tconv cell TCR repertoires were distinct, and autoantigen-specific Treg and Tconv cells were enriched in diseased tissue. Autoantigen sensitivity and fine specificity of these cells intersected, implying that differences in responsiveness were not responsible for lineage specification. Notably, autoreactive Treg and Tconv cells could be fully distinguished by an acidic versus aliphatic variation at a single TCR CDR3 residue. Our results imply that ontogenically distinct Treg and Tconv cell repertoires with convergent specificities for autoantigen respond during autoimmunity and argue against more than limited plasticity between Treg and Tconv cells during autoimmune inflammation.
INTRODUCTION
The T cell-specific forkhead box transcription factor 3 (Foxp3) endows T lymphocytes with regulatory activity (Fontenot and Rudensky, 2005; Pacholczyk and Kern, 2008; Horwitz et al., 2008; Tang and Bluestone, 2008). Foxp3+ regulatory T (Treg) cells are critical for immune homeostasis, and Foxp3 deficiency leads to overwhelming autoimmunity and early mortality. Treg cells predominantly form as a separate lineage in the thymus. Indeed, the TCR repertoires of Treg cells and conventional Foxp3− T (Tconv) cells are largely distinct, though ~10%–20% of TCR sequences are shared between the populations (Hsieh et al., 2006; Pacholczyk and Kern, 2008; Pacholczyk et al., 2006). Peripheral interconversion between Treg and Tconv cells is also observed (Liang et al., 2005; Lathrop et al., 2008; Coombes et al., 2007). Adaptive upregulation of Foxp3 has been hypothesized to be an important mechanism limiting inflammation-induced immunopathology.
During autoimmunity, Treg cells are often found in substantial numbers within affected organs. Defects or deficiencies in Treg cells have been identified in autoimmune patients, implicating them in pathogenesis (Viglietta et al., 2004; Zhang et al., 2009; Sugiyama et al., 2005). Adoptively transferred Treg cells can ameliorate or abrogate even ongoing organ-specific autoimmunity (Selvaraj and Geiger, 2008; Mekala et al., 2005; Tang and Bluestone, 2006; Kohm et al., 2002; Zhang et al., 2004). Although Treg cells may recognize foreign antigens, evidence indicates that they also bear an overrepresentation of self-specific TCR, and this self-specificity may be important for Treg cell restraint of immunopathologic responses (Hsieh et al., 2004, 2006; Andersson et al., 2007; Romagnoli et al., 2002; Jordan et al., 2001). Indeed, current evidence links specificity with Treg cell activity. Autoantigen-specific TCR transgenic (Tg) Treg cells are more potent than non-Tg Treg cells in downregulating models of experimental allergic encephalomyelitis, gastritis, and diabetes (Hori et al., 2002; Tang et al., 2004; O’Connor and Anderton, 2008; Huter et al., 2008). Importantly, myelin autoantigen-specific Treg cells have been directly identified by tetramer staining in the central nervous system (CNS) of mice with experimental allergic encephalomyelitis (EAE) and have been inferred in other studies (Korn et al., 2007; Yu et al., 2005; Reddy et al., 2004, 2005). In contrast, studies of a model of autoimmune uveitis failed to identify tissue specificity among involved Treg cells (Grajewski et al., 2006) and one analysis of Treg cell TCR specificity failed to find evidence for enhanced self-specificity (Pacholczyk et al., 2007).
TCR repertoire analyses have shown utility in surveying overlap between Treg and Tconv cell populations (Lathrop et al., 2008; Hsieh et al., 2004, 2006; Pacholczyk et al., 2006, 2007). For example, in a diabetes model, only limited Treg and Tconv cell overlap was seen in islets, suggesting that these cell types were not interconverting at the site of inflammation (Wong et al., 2007a). This type of analysis is powerful, though limited in that inferences are made by population shifts in the absence of specific knowledge about antigen reactivity. Likewise, studies of antigen reactivity among Treg and Tconv cells have typically been performed outside of the context of repertoire analyses that illuminate relationships between T cell populations.
To clarify the specificity and responsiveness of Treg cells, their relationship to effector T (Teff) cells during autoimmunity, and ultimately their source—Foxp3 upregulation in Teff cells or recruitment of a distinct regulatory population—we combined both repertoire analysis and studies of a large cohort of individual TCR. We developed a retroviral transgenic (retrogenic) mouse model of EAE in which the TCRα chain locus is fixed by the enforced expression of a TCRα from a myelin oligodendrocyte glycoprotein (MOG)-specific T cell. Vβ8.2 (TRBV13-2) is expressed by almost half of MOG-specific T cells in MOG-EAE (Mendel et al., 1995), and we focused on this disease-associated repertoire. Comparison of Vβ8.2+ TCRβ sequences indicated that the Treg and Tconv cell TCR repertoires have limited overlap within the periphery and CNS in mice with EAE. Further, shared TCR were predominantly expressed by a single cell type, Treg or Tconv cells. This argues against a primary influence for large-scale interconversion between Treg and Tconv cells in shaping CNS-infiltrating Treg and Teff cell responses. Isolation and expression in T lymphocytes of a subset of TCR sequences demonstrated marked enrichment of MOG-specific TCR in Tconv cells and, to a lesser extent, Treg cells infiltrating the CNS. Treg and Tconv cell TCR sensitivity and fine specificity for MOG autoantigen were similar, suggesting that these did not substantially influence T cell lineage assignment among responding cells. Importantly, conserved CDR3β features were associated with MOG specificity among the TCR analyzed. Independent features reliably distinguished MOG-specific Treg and Tconv cell TCR. These results demonstrate that the MOG-specific Treg and Teff cell responses during EAE arise from populations with different CDR3 imprints, identify only a more limited potential for interconversion between these populations, and suggest that cognate autoantigen sensitivity and presumably affinity is not a primary driving force in the specification of MOG-specific Treg cells responding in EAE. Implicitly, ontogenically distinct Treg and Tconv cell populations with convergent specificity for autoantigen are involved in the autoimmune response.
RESULTS
Retrogenic Mice Expressing a Single TCR Vα Chain
We generated TCRα retrogenic mice (Holst et al., 2006) incorporating the α chain of the MOG35–55-specific 1MOG244.2 TCR (Alli et al., 2008). TCRα cDNA was retrovirally transduced into Tcra−/−, GFP-Foxp3 gene-targeted hematopoietic progenitor cells (HPC), and these were adoptively transferred into sublethally irradiated Tcra−/− mice. Because of the endogenous Ca deficiency, all TCR incorporate the retrovirally encoded α chain; germline TCRβ freely rearranges.
At approximately 8 weeks post-HPC transfer, the retrogenic mice showed good lymphoid engraftment. Thymocyte subsets showed no significant differences (p > 0.05) compared with C57BL/6 mice, though trended toward increased DN cells (7.0% ± 6.8% 1MOG244.2α versus 3.0% ± 1.3% C57BL/6) and decreased CD4+ SP cells (1.7% ± 1.8% versus 7.0% ± 3.8%) (Figure S1A available online). A mean of 83 ± 6 × 106 total splenic lymphocytes was seen, primarily B cells. Engrafted T lymphocytes were decreased compared to C57BL/6 mice, and comprised 9.7% ± 2.9% of lymphocytes in the lymph nodes (LN) and 13.8% ± 4.9% in the spleen (Figures S1B and S1C; C57BL/6 data available at http://www.jax.org). The CD4/CD8 ratio was elevated, 6.2 ± 1.3 in the lymph nodes (LN) and 10.0 ± 2.7 in the spleen compared with ~1.6 in C57BL/6 mice (Figure S1D), indicating skewing of 1MOG244.2 TCRα T cells toward the CD4+ T cell lineage. CD4+ T cells expressed the GFP-Foxp3 transgene in proportions comparable to that observed in wild-type mouse strains, 12.8% ± 2.2% in LN and 7.5% ± 2.4% in spleen. The 1MOG244.2α retrogenic mice developed EAE with high penetrance after MOG35–55 immunization, with >90% of mice developing typical disease symptoms (Figure S2). Therefore mice expressing the 1MOG244.2α transgene develop a repertoire that incorporates normal proportions of CD4+ Treg and Tconv cells and are susceptible to EAE.
Increased TCR Vβ8.2 in CNS-Infiltrating 1MOG244.2α T Lymphocytes
As a low-resolution screen for EAE-associated alterations in the 1MOG244.2α TCR repertoire, we analyzed Vβ chain use by staining splenic or CNS CD4+ GFP-Foxp3− or GFP-Foxp3+ cells with Vβ-specific antibodies (Abs). Splenic T cell populations, either Treg or Tconv, did not differ in unimmunized mice and mice with EAE (Figures 1A and 1B). In contrast, a significant increase in Vβ8.1 or 8.2+ TCR use in CNS-infiltrating Tconv cells was seen compared with splenic Tconv cells (Figure 1A; p = 0.0135, ANOVA test; p = 0.018 after Hochberg’s adjustment for multiple tests). This is consistent with the documented over-representation of Vβ8.2 among MOG35–55-Ab-specific Teff cells (Mendel Kerlero de Rosbo and Ben-Nun, 1996; Mendel et al., 1995). A trend toward increased Vβ8.1 or 8.2 TCR use was also seen among CNS Treg cells (Figure 1B), though not statistically significant. Therefore, Vβ8.1- or 8.2-expressing T cells are concentrated in the CNS during EAE, implying increased MOG reactivity in the infiltrating T cell population.
Figure 1. Vβ Use by Retrogenic Treg and Tconv Cells.

CD4+ T lymphocytes from retrogenic mice, preimmune or 22–28 days after EAE induction, were flow cytometrically analyzed with a panel of TCR Vβ-specific antibodies. Comparisons were made between Tconv (A) and Treg (B) cells from unimmunized and immunized splenocytes and CNS T cells from mice with EAE. A significant increase in Vβ8.1 or 8.2 splenocytes was seen among Tconv cells (p = 0.018), and a decrease in Vβ8.3 TCR among Treg cells (p = 0.048) when comparing pre- or immune splenic and CNS-infiltrating populations. Data show mean + 1 SEM. *p < 0.05 among group by Hochberg-adjusted 1-way ANOVA.
TCR Jβ and CDR3 Use in MOG-EAE
To examine the Treg and Tconv cell TCR repertoires at higher resolution, we sacrificed 1MOG244.2α mice prior to immunization or 23–25 days after EAE induction. Spleen and, for mice with EAE, CNS cells were collected and pooled from 5–6 identically treated mice per group. Pooling was necessary because of low postisolation T cell yields from the CNS of retrogenic mice. The cells were sorted for CD4 and the presence or absence of GFP-Foxp3, and cDNA were prepared. TCR cDNA was amplified with Vβ8.2-Cβ primers to determine the distribution of Jβ among Vβ8.2+ TCR. Individual amplicons were cloned, sequenced, and analyzed for TCR gene use (Table 1). In total, 1829 sequences were assessed with the Vβ8.2-Cβ primer set. Proportions of Jβ used by either the Foxp3+ or Foxp3− populations showed no significant differences between the CNS and periphery in mice with EAE (p > 0.05, not shown). Comparison of pre- and postimmune splenocytes also failed to identify changes in Jβ use among Foxp3+ T cells and showed a significant difference in Foxp3− cells only for a single Jβ, Jβ2.4 (6.2% ± 0.5% pre versus 14.1% ± 0.5% post; p = 0.012). This indicates preserved diversity in the Vβ8.2+ population with EAE induction and when comparing the CNS-infiltrating and splenic T cell populations of diseased mice.
Table 1.
Isolation of TCR Vβ Sequences from Retrogenic Mice
| Isolated Sequences |
||||||
|---|---|---|---|---|---|---|
| Experiment | Disease Scores | T Cell Type | Vβ-Cβ | Vβ-Jβ | V-J φmax Upper 95% CI | |
| 1 (Preimmune) | NA | spleen Foxp3− | total | 137 | 155 | 2.25% |
| unique | 131 | 55 | ||||
| spleen Foxp3+ | total | 152 | 183 | 1.91% | ||
| unique | 150 | 64 | ||||
| 2 (Preimmune) | NA | spleen Foxp3− | total | 102 | 161 | 2.17% |
| unique | 97 | 54 | ||||
| spleen Foxp3+ | total | 128 | 123 | 2.85% | ||
| unique | 124 | 62 | ||||
| 3 (EAE, Day 23) | 1,2,3,3,4 | spleen Foxp3− | total | 159 | 167 | 2.09% |
| unique | 97 | 52 | ||||
| spleen Foxp3+ | total | 131 | 141 | 2.48% | ||
| unique | 57 | 46 | ||||
| CNS Foxp3− | total | 207 | 256 | 1.36% | ||
| unique | 90 | 42 | ||||
| CNS Foxp3+ | total | 147 | 226 | 1.54% | ||
| unique | 88 | 59 | ||||
| 4 (EAE, Day 25) | 1,1,1,2,2,2 | spleen Foxp3− | total | 160 | 208 | 1.68% |
| unique | 71 | 48 | ||||
| spleen Foxp3+ | total | 144 | 114 | 3.08% | ||
| unique | 82 | 49 | ||||
| CNS Foxp3− | total | 207 | 211 | 1.65% | ||
| unique | 82 | 45 | ||||
| CNS Foxp3+ | total | 155 | 207 | 1.68% | ||
| unique | 60 | 41 | ||||
For EAE studies, the day after immunization that analyses were performed and disease scores of individual mice studied are shown. Total numbers of sequences isolated and numbers of unique amino acid sequences identified are listed for Vβ8.2-Cβ and Vβ8.2-Jβ2.7 analyses. The φmax confidence interval (CI) refers to the upper frequency of a CDR3 sequence in the Vβ8.2-Jβ2.7 analysis for which there is 95% certainty of positive detection. NA, not applicable.
We next surveyed the more circumscribed Vβ8.2-Jβ2.7 repertoire. Jβ2.7 was selected because of its use in the original Vβ8.2+Jβ2.7+ TCRβ chain partner of the 1MOG244.2α TCR (Alli et al., 2008). Thus we knew that despite the theoretical potential for formation of >106 unique TCR sequences with this Vβ-Jβ constraint, Vβ8.2+Jβp2.7+ TCRβ had the potential to pair with the 1MOG244.2α to form MOG-specific TCR. In total, 2152 sequences were analyzed (Table 1). An estimated 95% confidence interval for the upper limit of the maximum frequency (φmax) of unobserved TCR CDR3 sequences for individual cell types and samples in different experiments ranged from 1.4% to 3.1% (Table 1). Therefore the collected sequences were adequate to assess common sequences in each cellular cohort. Among the Vβ8.2-Jβ2.7 sequences obtained, 452 unique amino acid sequences were identified. Interestingly, 54 (~12%) of these were shared among the 4 independent experiments or public sequences. Public sequences were overrepresented among more commonly identified sequences, with 74% of public sequences present within the 100 most frequent sequences. An increased probability of identifying sequences in multiple independent samples when their sequence frequency is high is expected and implies that the observed proportion of public sequences underestimates total representation because of non-identification of low-frequency public sequences. 16/54 public sequences were found in TCR exclusively used by Treg cells, 14/54 in Tconv cell TCR, and 24/54 in sequences identified both in Treg and Tconv cells. Abundance coverage estimator analysis (ACE) of pooled sequences, an indicator of total sequence diversity, indicated a limited diversity, with values of 271 and 337 for preimmune and 191 and 171 for postimmune Vβ8.2+, Jβ2.7+ Tconv and Treg splenocytes, respectively. Interestingly, the original β chain partner for the 1MOG244.2α TCR was among the public sequences identified in the CNS Foxp3− populations of both EAE induction experiments, though not in preimmune analyses. This suggests that receptors relevant to EAE in C57BL/6 mice, which served as the source of the original 1MOG244.2 hybridoma, play a role in the 1MOG244.2α retrogenic mice.
Among public TCR, 20 CDR3 sequences were identified as common to both preimmune T cells and T cells from mice with EAE. TCR sequences appearing predominantly in Foxp3+ or Foxp3− populations preimmune showed the same orientation, Foxp3+ or Foxp3−, in mice with EAE for 19 of the 20 receptors (not shown). This implies that TCR sequence rather than disease status is the primary force governing T cell Foxp3 expression.
Limited Overlap of Treg and Tconv Cell TCR Repertoires
Comparison of the unique CDR3 amino acid sequences for each cellular cohort revealed several notable features. Repertoire overlap was greater among a single class of cells (Treg or Tconv cells) across different organs (spleen and CNS) than among different classes of cells within an organ (Figure 2A). In experiment 3 (Table 1), 32.7% of unique splenic and CNS Treg cell TCR sequences and 23.9% of unique splenic and CNS Tconv cell TCR sequences overlapped. In contrast, only 11.0% of unique CNS and 10.1% of unique splenic sequences were shared between Treg and Tconv cells. In experiment 4, the values were 20.0%, 27.4%, 14.7%, and 6.6%, respectively. Further, among the minority of unique sequences that were shared by Treg and Tconv cells, the isolates of a particular sequence were typically skewed toward a single class of cells, either Treg or Tconv cells. Indeed, the Treg/Tconv cell identification ratio of shared TCR should form a Gaussian distribution, potentially around a ratio of ~1, if there was free and equal inter-conversion between Treg and Tconv cells. Yet 73% of shared CDR3 isolates were skewed by a ratio ≥2:1 toward a single cell class (Figure S3A; mean, median for majority Foxp3+ TCR: 6.1 ± 7.3, 2.5; Foxp3− TCR: 9.1 ±11.4, 5.1). This was influenced by the presence of CDR3 for which few sequences were isolated, and for which substantial skewing would therefore not be possible. Indeed, for shared sequences independently identified at least 10 times, 84% had a ratio ≥2:1 (Figure S3B; mean, median for Foxp3+: 9.3 ± 8.4, 9.1; Foxp3−: 13.4 ± 13.8, 10.0). Skewing of CDR3 toward a single cell class, Treg or Tconv cells, was also readily apparent in graphical mapping of the distribution among Foxp3+ or Foxp3− cells of the most common sequences isolated in individual experiments (Figures S4 and S5) and in an ecologic measure of population diversity, the Morisita Horn index, which demonstrated strong dissimilarity in Treg and Tconv cell CDR3 sequences in either spleen or CNS and greater similarity when an individual cell type was compared between spleen and CNS (Figure 2B).
Figure 2. Population Differences among TCR Subsets in Mice with EAE.

(A) Unique TCR sequences from the identified experiments (Table 1) were compared between population groups, and the frequency of sequences shared by Treg and Tconv cells within an organ (left) or of sequences from a specific cell type, Treg and Tconv cells, shared across organs (right) plotted. (B) The Morisita-Horn index, a measure of ecologic diversity among populations, is plotted for the same groups as (A). A value of 1.0 indicates population identity. Whereas (A) plots sharing of uniquely identified sequences, (B) measures differences in both sequence diversity and identification frequency between the indicated populations.
Comparative analyses were further performed on three structural features of the identified CDR3, length, charge, and hydrophilicity as potential correlates of specificity (Figure 3). The six cohorts of unique CDR3 sequences, acquired from postimmune CNS, postimmune spleen, and preimmune spleen Foxp3+ and Foxp3− cells, were compared via the Kruskal-Wallis test and significance was observed only for charge (p < 0.0001). With the Wilcoxon rank-sum test to compare any two cohorts within this group, the charge of the CNS Foxp3− CDR3 showed significant differences with each of the five other cohorts (p ≤ 0.02 for all comparisons after Hochberg’s adjustment for multiple tests). A significant difference was seen in only one other comparison group, CNS and preimmune splenic Foxp3+ CDR3 (p = 0.019). Therefore, CNS-infiltrating T cells, particularly Foxp3− cells, possess at least one distinguishing CDR3 characteristic, increased negative charge. Of note, the MOG35–55 epitope this CDR3 engages bears a potentially complementary positive charge and includes three arginine residues, two of which are predicted to lie in the Ab binding groove with their guanidinium moieties directed toward the TCR (Ben-Nun et al., 2006).
Figure 3. Length, Charge, and Hydrophilicity of Isolated CDR3.

Medians ± 95% CI for length (A), charge (B), and hydrophilicity (C) among unique CDR3 sequences identified in the six indicated populations is plotted. Analyzed sequences were determined as the regions bounded by but not including the conserved C and F residues bordering each CDR3β. *p < 0.05 according to the Hochberg-adjusted Wilcoxon rank sum test.
In summary, these results demonstrate distinct Treg and Tconv cell TCR repertoires in EAE in the CNS and periphery. Some overlap (~10%) was observed between CDR3 from the populations, though this was less than the overlap among a single class of cells, Treg or Tconv cells, compared in the spleen and CNS. Among CDR3 shared by Treg and Tconv cells, individual sequences tended to be dominantly represented within a single cell type. These results and the identification of distinct charge characteristics specifically among CNS-infiltrating Tconv cells implies that Treg and Tconv cells in EAE are distinct populations and do not substantially interconvert.
MOG Specificity among Treg and Tconv Cell TCR
To better delineate the properties of the responding TCR present within the CNS and periphery during EAE, we recreated full-length αβ TCR cDNAsfrom 40 identified CDR3 sequences (Table 2). The CDR3 were selected from sequences identified at high frequency (all were among the 50 most commonly identified sequences) to provide an approximately equal distribution of predominantly Foxp3+ and Foxp3− CDR3 of splenic or CNS origin. As above (Figures S3– S5), TCR with sequences shared by both Treg and Tconv cells were heavily skewed toward a single cell class (Table 2, far right column). A high representation of public TCR was seen among the selected CDR3 (21/40 sequences), reflecting the increased frequency of these sequences among common TCR. Reconstituted TCR were subcloned into a tricistronic retroviral vector essentially as we have previously described (Alli et al., 2008), with TCR α and β chains linked by the Thosea asigna virus 2A sequence to facilitate their stoichiometric expression and an IRES-linked cyan fluorescent protein (CFP). TCR-encoding retrovirus for these and the original 1MOG244.2αβ TCR were transduced into the CD4+ TCRαβ-deficient 4G4.CD4 T cell line. The cells were flow cytometrically sorted for similar TCR expression levels and stimulated with MOG35–55 peptide, and IL-2 production was measured as an indicator of MOG responsiveness.
Table 2.
Features of CDR3β Used to Reconstitute Full-Length TCR cDNA
| Clone # | Site | MOG-Specific | Foxp3+ | Sequence | Length | Public | Foxp3+ TCR | Foxp3− TCR | Foxp3+/Foxp3− Ratio |
|---|---|---|---|---|---|---|---|---|---|
| 4 | Spl | N | N | ASGDGGLGGRGEQY | 14 | Y | 0 | 18 | |
| 7 | Spl | N | N | ASGEKAEYEQY | 11 | Y | 1 | 15 | 0.067 |
| 10 | Spl | N | N | ASGTGGYSYEQY | 12 | N | 2 | 82 | 0.024 |
| 17 | Spl | N | N | ASGRQSSYEQY | 11 | N | 0 | 15 | |
| 30 | Spl | N | N | ASGGGSYEQY | 10 | N | 0 | 17 | |
| 31 | Spl | N | N | ASGAPDWGVPEQY | 13 | N | 3 | 33 | 0.090 |
| 32 | Spl | N | N | ASGEKTEYEQY | 11 | N | 2 | 30 | 0.067 |
| 36 | Spl | N | N | ASGDDWGGAYEQY | 13 | Y | 6 | 14 | 0.43 |
| 37 | Spl | N | N | ASGDGLASSYEQY | 13 | Y | 2 | 20 | 0.1 |
| 47 | Spl | N | N | ASGPVEQY | 8 | Y | 1 | 13 | 0.077 |
| 5 | CNS | N | Y | ASGDWDNYEQY | 11 | Y | 35 | 10 | 3.5 |
| 8 | CNS | N | Y | ASGGYEQY | 8 | Y | 33 | 2 | 16.5 |
| 15 | CNS | N | Y | ASGRDRGLEQY | 11 | N | 18 | 0 | |
| 20 | CNS | N | Y | ASGDWDVYEQY | 11 | N | 14 | 0 | |
| 34 | CNS | N | Y | ASGDGTGGVEQY | 12 | Y | 26 | 1 | 26 |
| 39 | CNS | N | Y | ASGDARLGGREQY | 13 | Y | 20 | 0 | |
| 3 | Spl | N | Y | ASGDGEQY | 8 | Y | 86 | 8 | 10.8 |
| 14 | Spl | N | Y | ASGGEVYEQY | 10 | N | 16 | 0 | |
| 24 | Spl | N | Y | ASAGWGLYEQY | 11 | N | 7 | 0 | |
| 26 | Spl | N | Y | ASGEGGFSYEQY | 12 | N | 20 | 0 | |
| 28 | Spl | N | Y | ASGDDWAYEQY | 11 | N | 13 | 0 | |
| 41 | Spl | N | Y | ASGRGTGGYEQY | 12 | Y | 19 | 0 | |
| 46 | Spl | N | Y | ASGDAWGVYEQY | 12 | Y | 15 | 0 | |
| 2 | CNS | Y | N | 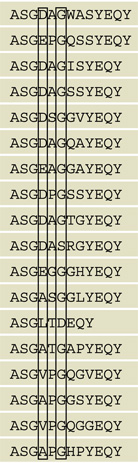 |
13 | N | 5 | 183 | 0.027 |
| 6 | CNS | Y | N | 13 | Y | 2 | 12 | 0.17 | |
| 11 | CNS | Y | N | 12 | Y | 7 | 32 | 0.22 | |
| 12 | CNS | Y | N | 12 | Y | 1 | 8 | 0.13 | |
| 19 | CNS | Y | N | 12 | Y | 3 | 15 | 0.2 | |
| 21 | CNS | Y | N | 12 | Y | 1 | 45 | 0.022 | |
| 29 | CNS | Y | N | 12 | Y | 3 | 14 | 0.21 | |
| 45 | CNS | Y | N | 12 | N | 0 | 16 | ||
| 244.2* | CNS | Y | N | 12 | Y | 0 | 7 | ||
| 44 | Spl | Y | N | 12 | N | 0 | 16 | ||
| 50 | Spl | Y | N | 12 | Y | 0 | 12 | ||
| 1 | CNS | Y | Y | 12 | N | 13 | 1 | 13 | |
| 9 | CNS | Y | Y | 9 | N | 25 | 0 | ||
| 16 | CNS | Y | Y | 12 | N | 16 | 0 | ||
| 18 | CNS | Y | Y | 12 | N | 13 | 0 | ||
| 52 | CNS | Y | Y | 12 | Y | 11 | 0 | ||
| 110 | CNS | Y | Y | 12 | N | 11 | 0 | ||
| 35 | Spl | Y | Y | 12 | Y | 22 | 0 |
Clone #, site of primary isolation; MOG specificity, association with Foxp3 expression; CDR3 length, isolation from multiple experiments (public) and frequency of identification from Foxp3+ and Foxp3− cDNA is indicated.
Original 1MOG244.2αβ TCR.
MOG specificity varied among the TCR cohorts (Figure 4). 2/12 (16.7%) of splenic Foxp3− TCR from immunized mice responded to MOG, whereas 1/8 (12.5%) of splenic Foxp3+ TCR responded. In contrast to this more modest representation of MOG-specific cells in the periphery, 8/8 (100%) of CNS Foxp3− and 6/12 (50%) of CNS Foxp3+ TCR-transduced cells produced IL-2 in response to MOG. These results therefore demonstrate substantial enrichment of MOG specificity within CNS-infiltrating Tconv cells of mice with EAE. Further, despite the evidence from our repertoire analyses for limited interconversion between Tconv and Treg cells and the use of a TCRα in the retrogenics derived from a Foxp3− T cell, a substantial proportion of the Treg cell response was directed against MOG.
Figure 4. MOG Specificity among Isolated TCR.

Forty CDR3β, distributed among Foxp3+ and Foxp3− isolates from the CNS or spleen, were used to construct retrovirus incorporating full-length αβ TCR. Retrovirally transduced 4G4.CD4 T cells were flow cytometrically sorted for similar surface expression of TCR and stimulated with Tcra−/− splenic APC with or without 100 µg/ml MOG35–55 peptide or with plate-bound CD3 antibody. IL-2 production was measured in cell-free supernatant at 24 hr by ELISA. Data indicate mean +1 SEM and are representative of three or more assays for each TCR.
Segregation of MOG-Specific Treg and Tconv Cell TCR Based on CDR3β Characteristics
Sequence information for all MOG-specific and nonspecific TCR was compared (Table 2). This provided several insights. First, MOG-specific CDR3β, Foxp3+ or Foxp3−, bore a consistent length of 12 (11.9 ± 0.8; 15/18 [83%] sequences length of 12). The non-MOG-specific TCR showed only slightly decreased average length (11.2 ± 1.6), but many fewer 12 amino acid sequences (5/23; 23%) and more diversity in this measure. MOG-specific TCR further more consistently had a G at the 6th CDR3 position (16/18; 89%) compared with non-MOG-specific TCR (4/23; 17%), implying that the absence of a side chain in that position is important for antigen recognition. More interestingly, among the MOG-specific TCR, CDR3 sequence distinguished TCR derived from primarily Foxp3+ and Foxp3− T cells. Acidic D or E residues were present at position 4 in 11/11 (100%) of Foxp3− but 0/7 (0%) of Foxp3+ MOG-specific CDR3. D and E would be anticipated to have a high prevalence at this location; a D is formed with Vβ8.2-Dβ recombination in the absence of junctional mutations or with a C→T replacement at the first D-region nucleotide. An E is formed with a C→A or G replacement at the same nucleotide. Indeed, 5/10 (50%) of Foxp3− and 8/13 (62%) of Foxp3+ nonspecific CDR3 bore a D or E at position 4, making its complete absence more surprising in the Foxp3+ MOG-specific population. In contrast, 100% of the MOG-specific Foxp3+ TCR bore an aliphatic A (4/7), V (2/7), or L (1/7) residue at that location, residues seen in only 1 of the 34 other CDR3 examined. Therefore, TCR expressed by MOG-specific Treg and Tconv cells comprise distinct families. Implicitly, these cells undergo a distinct selection process in 1MOG244.2α mice that is either not based on MOG recognition or is based on differential recognition of MOG.
Sensitivity and Fine Specificity of MOG-Specific TCR for Autoantigen
Current models of Treg cell lineage specification suggest that Treg cells possess a proclivity for self-reactivity and that thymocytes with higher affinity for self-antigen are diverted into the Foxp3+ lineage (Pacholczyk and Kern, 2008). MOG is expressed in the thymus, though in small amounts (Delarasse et al., 2003). If MOG is responsible for the selection of MOG-reactive Treg cells, then these cells would be anticipated to bear an increased affinity for it. Because the 4G4.CD4 TCR transductants were essentially identical except for the sequence of their TCR CDR3β, their relative sensitivity to Ag should primarily reflect their affinity. We therefore tested the sensitivity of all MOG-specific TCR to dilutions of MOG35–55 peptide (Figure 5). Interestingly, although Treg cell TCR trended toward increased sensitivity, substantial overlap was observed between the MOG-specific Treg and Tconv cell TCR. In each case, receptors were identified with high, moderate, and low sensitivity for MOG. Moreover, specific pairs of Foxp3+ and Foxp3− CDR3 with conserved amino acid sequences showed similar sensitivities for antigen (Table 2 and Figure 5). Clones 19 (Foxp3−) and 1 (Foxp3+), which besides the variation at position 4 only have a conserved V→L substitution at position 8, both responded vigorously to MOG stimulation. Clones 45 (Foxp3−) and 52 (Foxp3+), which differ only at position 4 and by a S→G at position 7, also responded similarly and more moderately. Therefore, although Treg cells tend toward increased sensitivity for MOG, this does not seem to be encoded into the position 4 variation and a MOG sensitivity threshold does not appear fundamental to response by Treg versus Tconv cell TCR.
Figure 5. Sensitivity of MOG35–55-Specific TCR for Autoantigen.

Cells bearing the indicated Foxp3− (A) or Foxp3+ (B) T cell-derived TCR were analyzed as in Figure 3 but stimulated with varied doses of MOG35–55 peptide. All cell lines were simultaneously analyzed for comparability. Data show mean ± SEM and are representative of three independent experiments with at least two independent transductions of each TCR.
These results suggest that other factors affected by the position 4 variation, for instance differential fine specificity for MOG or sensitivity to an alternative cross-reactive selecting Ag(s), are responsible for Treg cell lineage assignment. To test the former possibility, we examined the fine specificity of recognition of five of the MOG-specific Foxp3− TCR, including the index 1MOG244.2 TCR, and four of the Foxp3+ TCR. Prior studies with MOG35–55-specific TCR have identified critical TCR interacting residues, particularly R41, F44, R46, and V47 (Petersen et al., 2004). The different TCR demonstrated variability, through common patterns of reactivity when tested with alanine-substituted MOG35–55 peptides (Figure S6). An R41A substitution did not substantially influence recognition for 1MOG244.2 or most of the other TCR, whereas response was diminished to F44A, R46A, and V47A substitutions. However, with these and alanine substitutions at all residues from V37 to L50, no pattern was identified that distinguished the fine specificity of the Foxp3+ and Foxp3− T cell-derived TCR. Therefore, differential recognition of cognate MOG antigen does not appear to be associated with the binary differentiation of T cells with acidic or aliphatic residues at CDR3β position 4 alternatively into Tconv or Treg cells. Considering the absence of evidence for affinity or fine specificity-based segregation of Tconv and Treg cell TCR, our data would suggest that a MOG sensitivity threshold is not itself responsible for MOG-specific Treg cell recruitment, and presumably also lineage assignment.
Fidelity in TCR Lineage Specification
The 1MOG244.2α retrogenic mice were produced with Tcra−/− HPC and recipient mice. We were interested whether the MOG-specific Treg and Tconv cell TCR that we identified in 1MOG244.2α mice (Table 2) would similarly guide differentiation in a distinct system where the thymus is from a wild-type and not Tcra−/− mouse and most developing T cells are unmanipulated. To test this, we transduced MOG-specific TCRαβ pairs into CD45.1−CD45.2+ Rag1−/− HPC, which cannot express other TCR. These were diluted with an excess of congenic CD45.1+ CD45.2− T cell-depleted bone marrow cells and transplanted into CD45.1+CD45.2− mice. Seventeen mice demonstrated detectable CD45.2+CD4+ T cell engraftment (range 0.001%– 8.5% of CD4+TCR+ cells), including mice expressing three TCR derived from Treg cells (#1,9, 18; Table 2) and four from Tconv cells (#2, 6, 21, 45) (Figure S7). Overall, the Treg cell-derived TCR showed >20-fold increased engraftment into the Foxp3+ lineage when compared with the Tconv cell TCR (mean of Foxp3+ cells: 21.4% ± 10.6%; of Foxp3− cells: 0.95% ± 1.1%). Therefore, cells expressing TCR derived from Tconv cell TCR in 1MOG244.2α mice differentiate into Tconv and not Treg cells in this system. Cells expressing TCR derived from Treg cell TCR in 1MOG244.2α mice show increased Treg cell bias. Although not all of the retrogenic T cells expressing the Treg cell-derived TCR developed into Foxp3+ cells, this is not surprising because niche saturation for Treg cell development may occur even at very small numbers of Ag-specific T cells (Bautista et al., 2009). Despite heavy dilution with wild-type cells in these mixed chimeric mice, cells expressing the assayed TCR should nevertheless be increased in frequency compared with 1MOG244.2α mice. Therefore, Treg and Tconv cell lineage assignment in the 1MOG244.2 TCRα retrogenic mice is paralleled in a distinct and arguably more natural system, one in which the thymic stroma and the large majority of T cells are wild-type.
DISCUSSION
Treg and Tconv cells in naive mice bear distinct though overlapping repertoires (Hsieh et al., 2004, 2006; Pacholczyk et al., 2006; Pacholczyk and Kern, 2008; Wong et al., 2007b). Studies of adoptively transferred purified CD4+ T cells have indicated variable interconversion rates between Foxp3− and Foxp3+ populations (Lathrop et al., 2008; Fontenot et al., 2005; Liang et al., 2005). One recent analysis estimated a 4%–7% rate of Foxp3 upregulation after naive T cell transfer and identified a key role for TCR sequence and hence specificity in this (Lathrop et al., 2008). Mechanistic studies have provided a framework explaining plasticity between Treg and Tconv cells, with select signaling pathways regulating Foxp3 expression (Coombes et al., 2007; Quintana et al., 2008; Mucida et al., 2007; Selvaraj and Geiger, 2007). The extent to which interconversion influences active immune responses, and more specifically cells participating in autoimmunity, is uncertain. Some results suggest that adaptive upregulation of Foxp3 plays an important role in modulating autoimmunity (You et al., 2007; Manicassamy et al., 2009). Other studies failed to identify a role for Foxp3 induction (Korn et al., 2007; Wong et al., 2007a). During EAE, T cells activated in the periphery migrate to the CNS where they re-engage antigen on local dendritic cells and expand (Bailey et al., 2007). The early CNS response has a low frequency of Foxp3+ T cells, suggesting that substantial numbers of Treg cells are not stimulated to enter the CNS at this time. Yet Treg cells accumulate later in the disease and are associated with resolution (McGeachy et al., 2005). Therefore, either migration or expansion of this population is delayed or CNS-infiltrating Treg cells are forming through Teff cell conversion.
At a population level, our data fail to identify a substantial role for interconversion as a means of locally generating Treg cells in the CNS. The frequencies of shared CDR3 among unique TCR sequences in preimmune splenocytes (expt 1: 3.5%; expt 2: 6.4%) was only modestly lower than that in postimmune splenocytes (expt 3: 10.1%, expt 4: 6.6%). The difference between postimmune splenocytes and CNS-infiltrating cells (expt 3: 11%, expt 4: 14.7%) was likewise small. These trends may be consistent with limited interconversion between Treg and Tconv cell lineages; however, this and the heavy skewing of shared TCR toward a single cell type argue against extensive plasticity in Foxp3 expression during EAE.
Analysis of select receptors provided added insight into the composition of the autoreactive Treg and Teff cell responses. MOG-specific Teff cells were highly enriched in the CNS of 1MOG244.2α mice with EAE (100% of TCR). This contrasts with a low frequency (<10%) of MOG-specific CNS-infiltrating cells identified in a study with MOG-Ab tetramer, potentially reflecting our focus on a subpopulation of TCR (Vβ8.2+Jβ2.7+) that may tend toward MOG specificity. However, we were also unable to stain some MOG-specific T cells with an essentially identical tetramer, including the 1MOG244.2TCR studied here, suggesting that this tetramer may underestimate the actual frequency of MOG-specific cells (R. Alli and T.L.G., unpublished observations).
More interestingly, we identified a concentration of MOG-reactive TCR among the CNS-infiltrating Foxp3+ population (50% versus 12.5% among Foxp3+ splenocytes). This functionally confirms the presence of cognate autoantigen-reactive Treg cells in the CNS of mice with EAE at high frequency. Unlike Teff cells, which were uniformly MOG specific, 50% of Vβ8.2+Jβ2.7+ Treg cell TCR tested did not recognize MOG. These cells may be bystanders or effectors specific for alternative antigens. Indeed, Treg cells need not be specific for cognate autoantigen, and response toward tissue-specific or even tissue-unrestricted self antigens released during inflammation may potentially stimulate regulatory functions.
Among the MOG-specific T cells, a length of 12 amino acids and a G at position 6 in the Vβ8.2+Jβ2.7+ CDR3β was highly correlated with MOG specificity. Importantly, MOG-specific Teff cells during EAE were fully distinguished by a D or E at position 4, and Treg cells by an aliphatic residue, most commonly an A. Implicitly, Treg and Teff cells during autoimmunity arise from distinct population reservoirs, identifiable by the variation at residue 4. Additional structural criteria overlayed on this must be met for MOG specificity. It would seem likely that these distinct populations arise from differences in the origination and selection of the T cells.
Expression of high-affinity cognate antigen on thymic stroma promotes enhanced Treg cell differentiation for some TCR (Picca et al., 2006). MOG is expressed at low levels in the thymus (Delarasse et al., 2003). If MOG sensitivity guides Treg cell development, these cells should be more sensitive to MOG than Teff cells. Overall, Treg cell TCR showed higher MOG sensitivity than did Teff cell TCR. However, the substantial overlap in Treg and Teff cell TCR sensitivity implies that this is not fundamental to their selection. Further, differential fine specificity for MOG was not identified in Treg and Teff cell TCR. The simplest explanation for the similar MOG response yet different and conserved CDR3 features of Treg and Teff cell TCR is that lineage selection for the corresponding cell types is not MOG dependent, but rather that alternative Ag(s) promote selection.
Recognition of the acidic versus aliphatic variation in Tconv and Treg cell CDR3 was possible only by examining a narrow slice of the repertoire, one with a fixed TCRα and limited to Vβ8.2 and Jβ2.7. This focused repertoire analysis required the production and study of a manipulated mouse strain, the 1MOG244.2α retrogenic mice. The consistent identification of specific public TCR within Treg or Tconv cell repertoires in these mice indicates consistency in T cell differentiation patterns within them. However, the 1MOG244.2α mice do use a Tcra−/− stem cell recipient, whose thymi developed in the absence of normal T cell maturation. Further, premature expression of TCRα in transgenic or retrogenic systems, such as 1MOG244.2α mice, can influence pTα signaling and decrease the efficiency of the DN to DP transition, potentially altering the balance of different thymocyte subsets (Huang and Kanagawa, 2004; Borowski et al., 2004; Aifantis et al., 2006). We do not find evidence for these alterations influencing Treg cell lineage assignment. Grossly, proportions of Treg cells are normal in 1MOG244.2α mice, and overlap among Treg and Tconv cell TCR is similar to that observed in other systems (Hsieh et al., 2006; Pacholczyk et al., 2006). More importantly, precursor cells retrogenic for specific Treg or Tconv cell TCR, when heavily diluted with wild-type bone marrow and transplanted into wild-type mice, show the same lineage predilection as in 1MOG244.2α mice. This suggests that TCR-driven Treg and Tconv cell lineage specification in 1MOG244.2α mice is indicative of the potentials of the same TCR in normal thymi as well.
In summary, we demonstrate through studies of repertoire and individual TCR only limited overlap between Treg and Tconv cells responding during MOG-EAE. MOG-specific Treg cells form a substantial though not exclusive component of the CNS-infiltrating Treg cell repertoire. Examination of MOG-specific Treg and Teff cell TCR with a fixed TCRα and restricted TCRβ usage fails to find evidence for sensitivity or fine specificity for MOG influencing Treg versus Tconv cell lineage assignment, but does reveal lineage segregation based on conserved CDR3 features. Our results favor a model in which Treg cell response during EAE is developmentally encoded in the TCR repertoire and argue against anything more than limited plasticity between Treg and Tconv cells during the autoinflammatory response.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6 and Tcra−/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME). GFP-FoxP3 gene-targeted mice were provided by A. Rudensky (U. Washington). Mice were bred under specific-pathogen-free, including all detectable strains of Helicobacter, conditions. Experiments were performed in accordance with institutional animal care and use procedures.
Peptides, Antibodies, and Flow Cytometry
MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) and alanine substitutions of it were synthesized by the St. Jude Hartwell Center for Biotechnology and HPLC purified prior to use. Monoclonal antibodies specific for CD4 (clone L3T4), CD8 (clone 53-6.7), CD25 (clone 7D4), TCRβ (clone H57–597), TCR Vβ2 (clone B20.6), TCR Vβ3 (clone KJ25), TCR Vβ4 (clone KT4), TCR Vβ5.1, 5.2 (clone MR9-4), TCR Vβ6 (clone RR4–7), TCR Vβ7 (clone TR310), TCR Vβ8.1 and 8.2 (clone MR5-2), TCR Vβ8.3 (clone 1B3.3), TCR Vβ10b (clone B21.5), TCR Vβ11 (clone RR3–15), TCR Vβ12 (clone MR11-1), and TCR Vβ14 (clone 14-2) were obtained from BD Biosciences. Flow cytometry was performed on a FACS Calibur (BD Biosciences), and flow cytometric sorting was on a MoFlo (DakoCytomation) or iCyt reflection (iCyt, Champaign, Ill) cell sorter.
Retroviral TCR Construct Synthesis
To generate the 1MOG244.2 TCRα construct, the 1MOG244.2 TCRα (Alli et al., 2008) was PCR amplified and inserted into EcoRI/XhoI cloning sites in the MSCV-I-CFP vector (Persons et al., 1997). To generate other TCR, we amplified and TA cloned CDR3β cDNA isolated during repertoire analyses with primers incorporating an endogenous Vβ8.2 BamHI site and inserting a XhoI restriction site within the Cβ (5′ primer: GGTGCTGGATCCACTGAGAAAGGA GATATCCC; 3′ primer: GCCTCGAGAACCGTGAGCCTGGTGC). The introduced XhoI site did not alter amino acid sequence. The cDNA were then subcloned into the previously described 1MOG244.2αβ MSCV-I-CFP vector (Alli et al., 2008) modified with the Quick change site directed mutagenesis kit (Stratagene) to incorporate the XhoI. Construct integrity was verified by DNA sequencing.
Retrovirus Production and Cellular Transduction
Retrovirus transduction of hematopoietic progenitor cells (HPC) or murine surface TCR-deficient 4G4.CD4 T hybridoma cells was as previously described (Alli et al., 2008).
Generation of Retrogenic Mice
Bone marrow cells were harvested from the femurs of Tcra−/− mice 48 hr after the administration of 0.15 mg 5-fluorouracil g−1 body weight, cultured in complete Click’s medium (Invitrogen) with 20% FCS, IL-3 (20 ng ml−1), IL-6 (50 ng ml−1), and stem cell factor (50 ng m−1) for 48 hr and then cocultured for an additional 48 hr with irradiated (1200 rad) GP+E86 retrovirus producer cells. Transduced progenitor cells were harvested, washed with PBS, transduction confirmed by flow cytometry for CFP, and injected into sublethally irradiated (450 rad) Tcra−/− recipients at a ratio of 1.5 recipient mice per bone marrow donor.
EAE Induction and Clinical Evaluation
EAE was induced by subcutaneous immunization of mice 8–10 weeks post-stem cell transfer with 100 µg of MOG35–55 peptide in complete Freund’s adjuvant containing 0.4 mg Mycobacterium tuberculosis H37RA (Difco). 200 ng pertussis toxin (List Biological Laboratories) was administered i.v. on days 0 and 2. Clinical scoring was: 1, limp tail; 2, hind limb paresis or partial paralysis; 3, total hind limb paralysis; 4, hind limb paralysis and body or front limb paresis or paralysis; 5, moribund.
Cell Isolation
At the indicated time points, 5–6 mice per cohort were sacrificed, spleens were removed, the circulation was perfused with PBS, and brain and spinal cord removed. T cells were isolated from CNS tissue by density centrifugation as described (Selvaraj and Geiger, 2008). Isolated cells were flow cytometrically analyzed with the indicated antibodies or stained with CD4-specific Ab and immediately sorted into Tconv cell (CD4+GFP−) and Treg cell (CD4+GFP+) populations.
RNA Isolation and cDNA Transcription
Total RNA was extracted with the RNeasy Protect MiniKit (QIAGEN). cDNA was generated with the Omniscript RT kit (QIAGEN).
CDR3 Sequencing and Analysis
cDNA was PCR amplified with either Cβ- or Jβ2.7-specific primers (Cβ, 5′-CAA GGAGACCTTGGGTGGAGTCACATT; Jβ2.7, 5′-TAAAACCGTGAGCCTGGTG CCGG) and a Vβ8.2 primer (Vβ8.2, 5′-ATGTCTAACACTGCCTTCCCTGACCC). Five independent PCR reactions were performed with each sample per primer pair. The ~400 bp PCR products were gel purified and TA cloned into the pCR 2.1 vector (Invitrogen). cDNA-containing plasmid was isolated and −50 clones/PCR reaction sequenced. Vβ, Jβ, and CDR3 were identified with IMGT-V-Quest software (Immunogenetics Information System; http://www.imgt.cines.fr). CDR3 charge and hydrophilicity analyses were performed with the Innovagen peptide calculator (http://www.innovagen.se).
Cytokine Analysis
TCR-transduced 4G4.CD4 cells (1 × 105) were flow cytometrically sorted for equivalent TCR expression levels and cultured in duplicate in the presence of 3 × 105 APCs and the indicated stimulus. 100 µl of culture supernatant was collected at 24 hr and analyzed for IL-2 by sandwich ELISA (BD PharMingen) or Bio-Plex (Bio-Rad).
Statistics
One-factor ANOVA (GLM procedure, SAS, v9.1.3, SAS Institute Inc., Cary, NC) was applied to compare three or more groups, and two-sample t test (TTEST procedure, SAS) was applied to compare any two groups, if the data followed a normal distribution. If the data were not normally distributed, the Kruskal-Wallis test (NPAR1WAY procedure, SAS) was used to test differences among three or more groups, and the Wilcoxon rank-sum test (NPAR1WAY procedure, SAS) was used to compare the distribution between any two groups. A nonparametric method (Kaigh and Cheng, 1991) was used to estimate medians along with their 95% confidence intervals. A p value < 0.05 was considered significant in all analyses. p values from parametric or nonparametric tests were adjusted for multiple comparisons via the Hochberg method (MULTTEST procedure, SAS). Means are displayed ±1 SD. Morisita-Horn indices and abundance coverage estimator (ACE) analyses were calculated with EstimateS software (Hsieh et al., 2006; Wong et al., 2007a, 2007b). The upper limit of 95% confidence interval (CI) of the maximum frequency (φmax) of an unobserved TCR CDR3 transcript in a sample was calculated via the estimation of binomial parameter (StatXact-8 8.0.0, Cytel Studio) and the upper limit of 95% CI of the Blyth-Still-Casella estimation reported (Pacholczyk et al., 2007).
Supplementary Material
ACKNOWLEDGMENTS
We thank R. Cross, Y. He, and the Immunology Core Flow Cytometry facility for assistance with flow cytometric sorting and J.S. Geiger for assistance with CDR3 sequence analysis. This work was supported by the National Institutes of Health Grant R01 AI056153 (to T.L.G.) and by the American Lebanese Syrian Associated Charities (ALSAC)—St. Jude Children’s Research Hospital (to all authors).
Footnotes
SUPPLEMENTAL DATA
Supplemental Data include seven figures and can be found with this article online at http://www.cell.com/immunity/supplemental/S1074-7613(09)00501-9.
REFERENCES
- Aifantis I, Mandal M, Sawai K, Ferrando A, Vilimas T. Regulation of T-cell progenitor survival and cell-cycle entry by the pre-T-cell receptor. Immunol. Rev. 2006;209:159–169. doi: 10.1111/j.0105-2896.2006.00343.x. [DOI] [PubMed] [Google Scholar]
- Alli R, Nguyen P, Geiger TL. Retrogenic modeling of experimental allergic encephalomyelitis associates T cell frequency but not TCR functional affinity with pathogenicity. J. Immunol. 2008;181:136–145. doi: 10.4049/jimmunol.181.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson J, Stefanova I, Stephens GL, Shevach EM. CD4+CD25+ regulatory T cells are activated in vivo by recognition of self. Int. Immunol. 2007;19:557–566. doi: 10.1093/intimm/dxm021. [DOI] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)−17 cells in relapsing EAE. Nat. Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- Bautista JL, Lio CW, Lathrop SK, Forbush K, Liang Y, Luo J, Rudensky AY, Hsieh CS. Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat. Immunol. 2009;10:610–617. doi: 10.1038/ni.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Nun A, Kerlero de Rosbo N, Kaushansky N, Eisenstein M, Cohen L, Kaye JF, Mendel I. Anatomy of T cell autoimmunity to myelin oligodendrocyte glycoprotein (MOG): Prime role of MOG44F in selection and control of MOG-reactive T cells in H-2b mice. Eur. J. Immunol. 2006;36:478–493. doi: 10.1002/eji.200535363. [DOI] [PubMed] [Google Scholar]
- Borowski C, Li X, Aifantis I, Gounari F, von Boehmer H. Pre-TCRalpha and TCRalpha are not interchangeable partners of TCRbeta during T lymphocyte development. J. Exp. Med. 2004;199:607–615. doi: 10.1084/jem.20031973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes JL, Siddiqui KR, Arancibia-Caárcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delarasse C, Daubas P, Mars LT, Vizler C, Litzenburger T, Iglesias A, Bauer J, Della Gaspera B, Schubart A, Decker L, et al. Myelin/ oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J. Clin. Invest. 2003;112:544–553. doi: 10.1172/JCI15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: Regulatory T cell development and the forkhead family transcription factor Foxp3. Nat. Immunol. 2005;6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Grajewski RS, Silver PB, Agarwal RK, Su SB, Chan CC, Liou GI, Caspi RR. Endogenous IRBP can be dispensable for generation of natural CD4+CD25+ regulatory T cells that protect from IRBP-induced retinal autoimmunity. J. Exp. Med. 2006;203:851–856. doi: 10.1084/jem.20050429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA. Generation of T-cell receptor retrogenic mice. Nat. Protoc. 2006;1:406–417. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- Hori S, Haury M, Coutinho A, Demengeot J. Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proc. Natl. Acad. Sci. USA. 2002;99:8213–8218. doi: 10.1073/pnas.122224799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz DA, Zheng SG, Gray JD. Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol. 2008;29:429–435. doi: 10.1016/j.it.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat. Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- Huang CY, Kanagawa O. Impact of early expression of TCR alpha chain on thymocyte development. Eur. J. Immunol. 2004;34:1532–1541. doi: 10.1002/eji.200424870. [DOI] [PubMed] [Google Scholar]
- Huter EN, Stummvoll GH, DiPaolo RJ, Glass DD, Shevach EM. Cutting edge: Antigen-specific TGF beta-induced regulatory T cells suppress Th17-mediated autoimmune disease. J. Immunol. 2008;181:8209–8213. doi: 10.4049/jimmunol.181.12.8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat. Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- Kaigh WC, Cheng C. Subsampling quantile estimators standard errors with applications. Commun. Stat. Theory Methods. 1991;20:977–995. [Google Scholar]
- Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Bäck-ström BT, Sobel RA, Wucherpfennig KW, Strom TB, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 2007;13:423–431. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop SK, Santacruz NA, Pham D, Luo J, Hsieh CS. Antigen-specific peripheral shaping of the natural regulatory T cell population. J. Exp. Med. 2008;205:3105–3117. doi: 10.1084/jem.20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Alard P, Zhao Y, Parnell S, Clark SL, Kosiewicz MM. Conversion of CD4+ CD25- cells into CD4+ CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J. Exp. Med. 2005;201:127–137. doi: 10.1084/jem.20041201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manicassamy S, Ravindran R, Deng J, Oluoch H, Denning TL, Kasturi SP, Rosenthal KM, Evavold BD, Pulendran B. Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat. Med. 2009;15:401–409. doi: 10.1038/nm.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J. Immunol. 2005;175:3025–3032. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- Mekala DJ, Alli RS, Geiger TL. IL-10-dependent infectious tolerance after the treatment of experimental allergic encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Proc. Natl. Acad. Sci. USA. 2005;102:11817–11822. doi: 10.1073/pnas.0505445102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel I, Kerlero de Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: Fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur. J. Immunol. 1995;25:1951–1959. doi: 10.1002/eji.1830250723. [DOI] [PubMed] [Google Scholar]
- Mendel Kerlero de Rosbo N, Ben-Nun A. Delineation of the minimal encephalitogenic epitope within the immunodominant region of myelin oligodendrocyte glycoprotein: Diverse V beta gene usage by T cells recognizing the core epitope encephalitogenic for T cell receptor V beta b and T cell receptor V beta a H-2b mice. Eur. J. Immunol. 1996;26:2470–2479. doi: 10.1002/eji.1830261030. [DOI] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- O’Connor RA, Anderton SM. Multi-faceted control of autoaggression:Foxp3+ regulatory T cells in murine models of organ-specific autoimmune disease. Cell. Immunol. 2008;251:8–18. doi: 10.1016/j.cellimm.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Pacholczyk R, Kern J. The T-cell receptor repertoire of regulatory T cells. Immunology. 2008;125:450–458. doi: 10.1111/j.1365-2567.2008.02992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006;25:249–259. doi: 10.1016/j.immuni.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Pacholczyk R, Kern J, Singh N, Iwashima M, Kraj P, Ignatowicz L. Nonself-antigens are the cognate specificities of Foxp3+ regulatory T cells. Immunity. 2007;27:493–504. doi: 10.1016/j.immuni.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persons DA, Allay JA, Allay ER, Smeyne RJ, Ashmun RA, Sorrentino BP, Nienhuis AW. Retroviral-mediated transfer of the green fluorescent protein gene into murine hematopoietic cells facilitates scoring and selection of transduced progenitors in vitro and identification of genetically modified cells in vivo. Blood. 1997;90:1777–1786. [PubMed] [Google Scholar]
- Petersen TR, Bettelli E, Sidney J, Sette A, Kuchroo V, Bäckström BT. Characterization of MHC- and TCR-binding residues of the myelin oligodendrocyte glycoprotein 38–51 peptide. Eur. J. Immunol. 2004;34:165–173. doi: 10.1002/eji.200324669. [DOI] [PubMed] [Google Scholar]
- Picca CC, Larkin J, 3rd, Boesteanu A, Lerman MA, Rankin AL, Caton AJ. Role of TCR specificity in CD4+ CD25+ regulatory T-cell selection. Immunol. Rev. 2006;212:74–85. doi: 10.1111/j.0105-2896.2006.00416.x. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, Sobel RA, Wucherpfennig KW, Kuchroo VK. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 2004;101:15434–15439. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J, Waldner H, Zhang X, Illes Z, Wucherpfennig KW, Sobel RA, Kuchroo VK. Cutting edge: CD4+CD25+ regulatory T cells contribute to gender differences in susceptibility to experimental autoimmune encephalomyelitis. J. Immunol. 2005;175:5591–5595. doi: 10.4049/jimmunol.175.9.5591. [DOI] [PubMed] [Google Scholar]
- Romagnoli P, Hudrisier D, van Meerwijk JP. Preferential recognition of self antigens despite normal thymic deletion of CD4(+)CD25(+) regulatory T cells. J. Immunol. 2002;168:1644–1648. doi: 10.4049/jimmunol.168.4.1644. [DOI] [PubMed] [Google Scholar]
- Selvaraj RK, Geiger TL. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J. Immunol. 2007;178:7667–7677. doi: 10.4049/jimmunol.178.12.7667. [DOI] [PubMed] [Google Scholar]
- Selvaraj RK, Geiger TL. Mitigation of experimental allergic encephalomyelitis by TGF-beta induced Foxp3+ regulatory T lymphocytes through the induction of anergy and infectious tolerance. J. Immunol. 2008;180:2830–2838. doi: 10.4049/jimmunol.180.5.2830. [DOI] [PubMed] [Google Scholar]
- Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, McCormick TS, Cooper KD. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J. Immunol. 2005;174:164–173. doi: 10.4049/jimmunol.174.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA. Regulatory T-cell physiology and application to treat autoimmunity. Immunol. Rev. 2006;212:217–237. doi: 10.1111/j.0105-2896.2006.00421.x. [DOI] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: A jack of all trades, master of regulation. Nat. Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viglietta V, Baecher-Allan C, Weiner HL, Hafler DA. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med. 2004;199:971–979. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Mathis D, Benoist C. TCR-based lineage tracing: No evidence for conversion of conventional into regulatory T cells in response to a natural self-antigen in pancreatic islets. J. Exp. Med. 2007a;204:2039–2045. doi: 10.1084/jem.20070822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C. Adaptation of TCR repertoires to self-peptides in regulatory and non-regulatory CD4+ T cells. J. Immunol. 2007b;178:7032–7041. doi: 10.4049/jimmunol.178.11.7032. [DOI] [PubMed] [Google Scholar]
- You S, Leforban B, Garcia C, Bach JF, Bluestone JA, Chatenoud L. Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc. Natl. Acad. Sci. USA. 2007;104:6335–6340. doi: 10.1073/pnas.0701171104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, Jain R, Waldner H, Hardaway JC, Collins M, et al. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J. Immunol. 2005;174:6772–6780. doi: 10.4049/jimmunol.174.11.6772. [DOI] [PubMed] [Google Scholar]
- Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int. Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang HB, Chi LJ, Wang WZ. The role of FoxP3+CD4+CD25hi Tregs in the pathogenesis of myasthenia gravis. Immunol. Lett. 2009;122:52–57. doi: 10.1016/j.imlet.2008.11.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.