

Abstract
In this paper we propose a novel approach to the design and implementation of knowledge-based decision support systems for translational research, specifically tailored to the analysis and interpretation of data from high-throughput experiments. Our approach is based on a general epistemological model of the scientific discovery process that provides a well-founded framework for integrating experimental data with preexisting knowledge and with automated inference tools.
In order to demonstrate the usefulness and power of the proposed framework, we present its application to Genome-Wide Association Studies, and we use it to reproduce a portion of the initial analysis performed on the well-known WTCCC dataset. Finally, we describe a computational system we are developing, aimed at assisting translational research. The system, based on the proposed model, will be able to automatically plan and perform knowledge discovery steps, to keep track of the inferences performed, and to explain the obtained results.
Keywords: Automated reasoning, Knowledge-based systems, High-throughput research, Genome-wide association studies
1. Introduction
The rapid evolution of high-throughput experimental methods in the past decade has led to a revolution in the way biomedical research is performed, opening the way to large-scale translational approaches. The exponential increase of the amount of data produced by each experiment, at all levels (from next-generation DNA sequencing to genotyping, to gene expression analysis, to proteomics, to high-level observations on genotype/phenotype correlations) and at a steadily decreasing cost, has opened up unprecedented new opportunities for studying biological systems on a large scale, taking a holistic perspective that promises to expand our understanding of biological processes and of their connections with clinically relevant findings. “In-silico” experiments, that are becoming part of the standard process of knowledge discovery, consist of a complex sequence of iterative data analysis steps, each of which produces intermediate data and results that need to be properly stored and maintained. However, the availability of such high volumes of data, combined with the need to access large amounts of heterogeneous information available on the World-Wide Web, poses major challenges in terms of data management and data analysis. The need for improved computational environments oriented to data and knowledge integration has been widely recognized. Resources like SRS [1] and NCBI's Entrez [2] have been empowered with sessions and query management capabilities; the adoption of Web Services technology has allowed the creation of complex, distributed data analysis tools (e.g., Soaplab [3], BioMOBY [4,5] and BioMart [6]); the application of workflow management technology to biomedical research has led to the implementation of IT platforms able to coordinate interdependent analysis steps [7] (e.g., stand-alone tools like Orange [8], the Taverna workbench [9,10], and client-server systems, like Pegasys [11] and BioWMS [12]).
In this paper we propose and discuss the application of KB-DSS to the field of translational bioinformatics. As recognized in a recent seminal paper by Aniba et al. [13], the availability of technological solutions is not enough, in itself, to cope with the data management and knowledge discovery challenges encountered in current biomedical research. Drawing on past experiences in other areas of biomedical informatics [14], we propose an architecture for the implementation of knowledge-based decision support systems (KB-DSS) specifically tailored to translational research. Such systems should be able to select and perform the data gathering, analysis, and interpretation “actions” that would be the most appropriate towards solving a given task, to automatically plan and perform knowledge discovery steps, keeping track of the inferences performed, and to explaining the obtained results. Moreover, they should be able to formally represent and manage multiple alternative hypotheses at the same time, and to use them for planning experiments, and to update them according to the experiment results.
A recent example of a complete running system for automated discovery in molecular biology is represented by the Robot Scientist project [15], which developed an autonomous system able to generate hypotheses explaining the available evidence, to plan experiments to test them, to run the experiments in a fully automated laboratory, and to interpret their results, starting new cycles if needed. The idea of an explicit, structured representation of hypotheses has been explored in a recent work by Roos et al [16], but without a well-defined reasoning framework to operate on them. Past examples include a wide variety of medical expert systems for diagnosis [17,18], therapy planning [19], patient monitoring and critical care [20].
Although the basic principles are similar, the use of KB-DSS for translational bioinformatics presents some significant differences compared to the above-described experiences. To start, while the traditional use of KB-DSS is aimed at diagnostic and therapeutic reasoning, in the translational bioinformatics field the goal is, instead, to support scientific discovery. Moreover, the classical architecture of a KB-DSS consists of an integrated knowledge base and a general inference mechanism able to reason on the available data and knowledge. In the context of translational bioinformatics, this model needs to evolve to take into account both the very large scale of the datasets being studied (while a traditional biomedical expert system normally handles up to a few hundred variables at most, high-throughput experimental techniques can sample millions of variables at once), and the availability of an extremely large corpus of background knowledge, in essentially unstructured form, in online repositories. As a consequence, we believe that in order for a KB-DSS to be successful in this context, it should be based on a conceptual framework designed to support the reasoning processes specific to translational research. In this scenario, the goal is not to perform complete inferential and experimental cycles, but to provide researchers with more efficient tools to better structure and organize the research process, and to more efficiently perform its repetitive aspects. The conceptual model should therefore include meta-models of reasoning in scientific discovery, specialized to molecular medicine, and a powerful and general information management architecture [13].
We address these requirements by proposing an automated reasoning model that accurately describes the current practice of scientific discovery in molecular medicine. The model can be used to guide the development of KB-DSS for translational research, specifically tailored to the analysis and interpretation of data from high-throughput experiments. Our approach is based on a general epistemological model of scientific discovery process that provides a well-founded framework for integrating experimental data with preexisting knowledge and with automated inference tools. The model, called Select and Test Model (ST-Model) [21,22], was initially developed in the field of Artificial Intelligence in Medicine to support the design and implementation of expert systems. We will show that the ST-Model can be instantiated to guide the development of KB-DSS for high-throughput biomedical research. We will also describe a computational system we are developing, which allows investigators to explicitly formulate and represent hypotheses grounded in existing biomedical knowledge, to validate them against the available experimental data, and to refine them in a structured, iterative process.
As a proof of concept we will focus, in particular, on Genome-Wide Association Studies (GWAS), which aim at discovering relationships between one or more variables at the molecular level and a phenotype. Case-control association studies attempt to find statistically significant differences in the distribution of a set of markers between a group of individuals showing a trait of interest (the cases) and a group of individuals who do not exhibit the trait (the controls). GWAS rely on large-scale genotyping techniques to analyze a very large set of genetic markers, in order to achieve a sufficiently good coverage of the entire genome, a strategy that is appropriate when there is little or no a priori information about the location of the genetic cause of the phenotype being studied. Because of their increasing importance in the field of molecular medicine, of the constant advances in the technology they are based on, and of the analytical challenges they pose, GWAS are an ideal example to demonstrate the application of our proposed approach.
This paper is structured as follows: Section 2 describes the ST-Model in detail; Section 3 presents the application of the ST-Model to GWAS, Section 4 is devoted to an overview of the design and implementation of the computational system we are developing, and Section 5 describes a case study in which the ST-Model is applied to a well-known GWAS. The paper ends with some conclusions summarizing the methodology described in the article and discussing its applicability to translational research.
2. The ST-Model
Cognitive science research shows that experts engaged in a problem-solving task typically perform a fixed sequence of inferential steps that may be repeated cyclically. In our context, the task consists in generating and evaluating new explanatory hypotheses, starting from a definition of the research problem and a set of available data. Following the well-known Generate-and-Test paradigm [23], those steps are: i) a hypotheses selection phase, in which the initially available information is used to generate a set of candidate hypotheses, and ii) a hypotheses testing phase, in which hypotheses selected in the previous step are used to predict expected consequences, that are then matched with available or other (possibly new) information in order to confirm or disprove them.
As reported in [21], this can be described as a process of abduction, interpreted as an inference to the best explanation. Formally, abduction is a method of logical inference introduced by Peirce [24], corresponding to the logical fallacy known as “affirming the consequent”: if it is known that α implies β, and β is observed to be true, then it can be assumed that α is true. Since β may be true because of other causes, this inference may be wrong. This kind of inference is defeasible and thus non-monotonic (since its conclusions may be disproved by additional evidence), and is at the basis of scientific discovery, theory revision, and both selective and creative reasoning [21].
The ST (Select and Test) Model is a general framework for automated reasoning that formalizes the process of inference to the best explanation as an iterative sequence of elementary inferential steps. Each step in the model is implemented by a specific inference type, as shown in the schema in Figure 1. The first step of the process is an abstraction, through which a set of high-level features are extracted from the initial data and information. This is followed by an abduction step, in which the abstracted features are used to construct one or more hypotheses, each of which is a potential explanation for the observed data. Indeed, part of the power of the framework comes from its ability to handle multiple competing hypotheses at the same time. Hypotheses are then ranked to define the order in which they will be examined in the following steps, according to preference criteria which can be application-dependent, or defined on the basis of prior knowledge. The purpose of ranking is to ensure that the “best” hypotheses are examined first, a heuristic strategy aimed at accelerating convergence to the optimal solution. Next, a deduction step examines the best-ranked hypotheses and derives a set of consequences that are expected to be true from each one. The deductive step will, in general, make use of background domain knowledge. Predictions are then matched against the available data, in the induction phase: hypotheses whose consequences match the available data are retained, while those that contradict the available data are discarded. The process can then be repeated cyclically: existing hypotheses can be refined, or new ones generated, on the basis of additional data, the resulting set of hypotheses is re-ranked, and their expected consequences are compared against the available experimental data. The process terminates when no hypotheses are left, or when a sufficiently small number of hypotheses is reached.
Figure 1.

The ST-Model.
Although the ST-Model is a general epistemological model of scientific reasoning, it can be directly translated into a set of concrete computational steps. We will illustrate this through a simple example using propositional rules (representing implications of the form “IF the antecedent is true, THEN the consequent is true”). Let us imagine a knowledge base containing rules expressing the relationship between transcription factors and the genes they regulate (see Table 1). Each rule expresses a know fact of the form: “If transcription factor T is expressed, then gene G is upregulated.” Let us also imagine we have performed a gene expression microarray experiment, and that analysis of its results allows claiming that genes G1 and G3 are up-regulated and that gene G2 is unchanged, while no information is available on T1 and T2 (this is the outcome of the abstraction step, in which “raw” numerical values are converted into domain-specific assertions about the behavior of one or more biological objects). Starting from the observation that gene G1 is up-regulated, the abduction step uses rules 1 and 3 to generate the two alternative hypotheses “T1” and “T2”, since both of them cause gene G1 to be up-regulated. The ranking step now orders the hypotheses, for example on the basis of a certainty factor associated with each rule. Let us imagine that according to the ranking function used here, hypothesis “T1” should be tested before “T2”. The deductive step now uses rule 2 to derive the fact that gene G2 should be up-regulated as an expected consequence of hypothesis “T1”. The next step consists in verifying whether this predicted consequence is actually confirmed by the available data. Since in this example gene G2 is not up-regulated, the eliminative induction step will rule out hypothesis “T1”. Hypothesis “T2” is tested next, and rule 4 produces the expected consequence that gene G3 should be up-regulated. Since this is verified against the experimental evidence, hypothesis “T2” is selected as the best explanation for the available data.
Table 1.
Knowledge base of propositional rules.
| 1. T1 (transcription factor T1 is expressed) →G1 (gene G1 is upregulated) |
| 2. T1 (transcription factor T1 is expressed) →G2 (gene G2 is upregulated) |
| 3. T2 (transcription factor T2 is expressed) →G1 (gene G1 is upregulated) |
| 4. T2 (transcription factor T2 is expressed) →G3 (gene G3 is upregulated) |
In the following sections we show how the ST-Model can be used as the basis for the implementation of KB-DSS in translational bioinformatics, providing a sound way to organize complex workflows and experiments (both in silico and in vitro), and grounding the analysis process on a clear epistemological framework. We will substantiate our claim by applying the ST-Model to the problem of defining a KB-DSS for supporting GWAS.
3. The ST-Model for Genome-Wide Association studies
The ultimate goal of GWAS is to unravel the molecular mechanisms underlying complex phenotypic traits, by searching for statistically significant differences in the distribution of a set of genetic markers between a set of cases and one of controls. Since GWAS normally assume little or no a priori knowledge about the genetic cause of the trait under study, they need to essay a very large set of genetic markers, in order to sample the entire genome with a sufficiently high granularity.
Although the technology to perform GWAS has in recent years seen dramatic improvements, thanks to the development of genotyping microarrays and deep sequencing, these studies still suffer from limitations that reduce their ability to tackle complex diseases [25]. In GWAS, as in all high-throughput molecular medicine contexts, the number of genetic markers tested is much higher than what can be analyzed manually. While the ability to sample hundreds of thousands of variables in parallel provides great benefits in terms of throughput and experimental costs, it also makes it harder to ensure that the results obtained are statistically significant. The number of available subjects is often too small to guarantee a statistical power sufficient to detect small causal effects, which are likely to be present in the case of complex genetic disorders. Variables are treated as being independent of each other, while it is well known that genetic factors are often correlated with each other (for example, due to genetic linkage), and that the majority of phenotypes are caused by the interplay of multiple genetic factors. Finally, these studies provide limited explanation capabilities: even when the analysis phase is able to identify one or more genetic factors that are significantly associated with the phenotype, it does not necessarily provide an indication of the mechanism through which they affect the phenotype, something that instead has to be worked out a posteriori by the investigator.
A KB-DSS for GWAS would help properly design and perform each inferential step, including formulating new hypotheses, comparing them with the existing evidence, and planning confirmatory experiments. The ST-Model described in the previous section provides a foundation for automated reasoning and a meta-architecture for computational environments, since it represents a general epistemological model. In the following, we describe the instantiation of the ST-Model for case-control GWAS.
A classical GWAS can be represented in the following way:
- Abstraction: the first step consists of selecting the clinical measurements that are needed to properly define the phenotype, and an initial set of individuals sharing the defined phenotype. Phenotype definition, data pre-processing, variable summarization, SNP selection, subpopulation handling, and correction for stratification, may all be viewed as part of the abstraction step, which allows moving from a generic definition of the study to a “computable” problem [26].
- Abduction: hypotheses are generated by testing the SNPs in the available dataset for association with the phenotype. The analysis tool used to perform the statistical association test produces a set of candidate SNPs as result; each SNP in this set therefore represents an independent hypothesis, of the form “the alleles of SNP x are significantly associated with the phenotype”. This step involves a creative abduction: at the start of the process, all SNPs can potentially be associated with the phenotype, just by virtue of being part of the genotyping dataset, but none of these associations is supported by evidence. Only after statistical analysis those SNPs that are potential “good statistical explanations” for the phenotype are selected, and the hypothesis space is populated. This creative step differs from the selective abduction performed in diagnostic reasoning, where a set of already established hypotheses is present in the knowledge-base, and the data are only used to select the ones that may be a good explanation of the data.
- Ranking: candidate SNPs are ordered according to their biological or statistical significance. For example, the ranking function could be based on the p-values measuring the statistical significance of the association, or on the location of each SNP in the genome.
- Deduction: once the hypothesis space is populated, the validity of each hypothesis (i.e., of the association of each individual SNP with the phenotype) is assessed, relying on the biological knowledge available on that SNP. In this phase, additional information about each top-ranked candidate SNP is derived, with the goal of establishing a biologically-founded relationship between SNPs and the phenotype that may explain the observed statistical association. A common strategy is to identify genes located close to the SNPs, and to study the metabolic pathways or GeneOntology classes they belong to, under the assumption that SNPs act as markers for candidate genes. In the translational bioinformatics context, this step can rely on the extremely extensive collections of biomedical information that are available in online repositories, in order to identify possible consequences of the hypothesis under consideration. Although the volume and depth of such information is constantly increasing, it is in general formalized and represented in different, possibly incompatible ways in different sources. Moreover, these information repositories are dynamic: their contents may change often as a consequence of research advances, and the results of the deductive step are therefore non-monotonic. For this reason, it will be increasingly important to develop data integration tools, able to provide a uniform, consistent, and dynamically updated view of a collection of related data elements, possibly coming from disparate sources.
- Induction: If an over-representation of consequences matching the phenotype is found, this provides evidence that the hypotheses under consideration are correct; otherwise, the hypotheses just tested are discarded. Eliminative induction allows reduction of the hypothesis space by evaluating if any of the results of the abduction step are ruled out by the currently available knowledge.
After the first run of the model, a list of candidate SNPs is retained. These SNPs can be then tested in confirmatory studies, or can be validated through meta-analysis, by running a new deduction / eliminative induction cycle. Alternatively, the researcher may want to refine the phenotype definition and repeat the entire analysis on a different set of subjects.
Analysis at the SNP level is usually followed by analysis at the gene level, whereby SNPs are treated as markers for the genes they belong to (and that are assumed to be the real causal factors for the phenotype). This suggests that the reasoning process involved in modern biological research proceeds not just through cyclic tasks, but also by changing the space in which hypotheses are formulated.
Let us consider again the problem of finding a relationship between the genotype and a disease phenotype. In general terms, this problem is intractable since the overall hypotheses space is extraordinarily large (every possible combination of all genetic factors) and for the most part unobservable. In practice, the hypothesis space is reduced to the set of markers (e.g., SNPs) that current technologies can sample; this allows abduction to be performed as a “creative” step, searching through a finite space of potential hypotheses, in which experiments are feasible and the problem is, at least in theory, solvable. Alternatively, the abstraction step can create new hypotheses involving genes rather than SNPs. Again, this can be modeled through the ST-Model: the problem is restated, becoming “are the genes containing the associated SNPs related to the phenotype?”, and the reasoning process then proceeds at this higher level of abstraction. Gene expression analysis or knock-out experiments can be used in the inductive phase to confirm or disprove the new hypotheses, now expressed in terms of genes instead of SNPs. In the same way, individual genes can be abstracted again into pathways or functional classes, leading to another run of the ST-Model at an even higher level of abstraction (Figure 3). This is indeed the process through which complex experiments fill the gap between the “punctual” analytical approach and the “global” solution of the overall problem being studied. This reasoning path reflects the requirement, necessary for the proper interpretation of GWAS, to relate a statistically significant association with a causal, biological explanation.
Figure 3.

A GWAS represented by multiple instances at the ST-Model at different levels of abstraction.
While the large-scale model depicted in Figure 3 is general enough to represent the overall reasoning process and to guide the implementation of an actual computational system, this step is in general quite complex, because of the need to integrate a variety of different analytical methods specific for different domains and the ability to manage different hypotheses spaces at the same time, and is therefore outside the scope of this work. In this paper we concentrate on the implementation of the portion of the model that handles the selection of candidate SNPs, as described in Figure 2. The next section will describe the current state of this work, and Section 5 will present its application to a case study.
Figure 2.

The ST-Model for abductive inference in GWAS studies.
4. A computational infrastructure for Genome-Wide Association Studies based on the ST-Model
We are developing a distributed, modular computational environment, based on the ST-Model, to support the above described style of research. The purpose of our system is to facilitate the analysis and interpretation of experimental results by automating the most common data management and integration tasks, as well as the required reasoning steps. In this section we briefly describe the system components and their role in the overall development of the discovery process.
The main components of the system in its current version are:
The Phenotype Miner, a module for phenotypic data management and inspection. We previously proposed the application of data warehouse concepts to facilitate the investigation of biomedical data by researchers lacking technical expertise and database skills. The Phenotype Miner provides a simple and effective tool to organize, represent and explore phenotypic data along multiple dimensions, and to easily create sets of subjects based on one or more phenotypes of interest [27,28].
The GWAS assistant, a module that helps perform the GWAS quality-control phase [29]. The GWAS assistant implements formal methods, based on Multi-Criteria Decision Making theory, for setting appropriate genotyping rate thresholds for GWAS.
Genephony, a knowledge management tool for genomic datasets, designed to support large-scale, exploratory research at the genome-wide level by assisting researchers in manipulating and exploring large datasets of genomic information. Genephony offers researchers a set of integrated and automated tools to easily create new datasets containing both experimental data and background knowledge from public resources, to annotate them and to export them in a variety of common formats [30]. The main functionalities provided by the platform are: the ability to easily define and handle very large, integrated datasets of genomic information; a simple, consistent and easy-to-use interface; and high interoperability with other commonly used software tools, achieved through the use of standard data exchange formats and communication protocols.
A high-level module to support the analysis and interpretation of results. This module, still under development, will provide hypothesis generation and management capabilities, will coordinate the interactions between the system components, and will facilitate access to biomedical data and knowledge repositories.
The components, which were initially developed as independent, stand-alone systems, are now being integrated into a comprehensive decision support system. While at the practical level integration is accomplished using Web Services protocols such as SOAP [28], at the conceptual level the coordination and interplay between these modules is guided by the ST-Model framework, as described in the following paragraphs:
Abstraction
A first, required step in the computational investigation of the genetic bases of diseases is an accurate definition of the phenotype under study. This is necessary to guide both the selection of the subjects to be studied and the choice of the experimental strategy to be followed (e.g., whether to perform a genome-wide scan or a more targeted analysis). The first component of the system, the Phenotype Miner, fulfills these requirements by providing a data collection infrastructure, a data warehouse system for phenotypic data, and a tool to formally define the phenotypes of interest. Phenotypes are defined by specifying a set of variables and the ranges of values they may take. Our system provides a tool to automatically select a set of subjects whose clinical data satisfy the definition of the phenotype of interest, without requiring the user to write database queries (that are instead automatically generated by the system). Phenotypes are formalized as a set of conditions in the form of attribute/value pairs, combined using logical operators (AND, OR) to define more and more complex phenotypes. In particular, the AND operator allows the specialization of a defined phenotype, while the OR operator is used to merge different phenotypes into a single more comprehensive one. A graphical wizard facilitates the creation of the rules that define a phenotype. Once these rules are defined, they are stored in the phenotype definition tables, and the SQL statement to select the subgroup of individuals satisfying them is automatically generated. Once the genotypes for the selected subjects are retrieved, the GWAS assistant can be used to perform the necessary quality control and pre-processing steps, including setting the genotyping rate and correcting for stratification.
Abduction and ranking
Performing a “classical” SNP-based association study on all SNPs in the dataset (using an appropriate analysis program such as PLINK [31]), the high-level module identifies those that show the strongest statistical association with the phenotype, and uses them to populate the hypothesis space. Candidate SNPs can then be ranked according to different criteria; for example their p-value, or their functional and biological properties, determined through the use of a large-scale annotation tool such as Genephony.
Deduction
After selecting the set of genomic markers that are thought to be related to the phenotype and generating a corresponding set of hypotheses, the system exploits the data integration and manipulation features offered by Genephony to derive their expected consequences. Using the annotation functions provided by Genephony, it is easy to determine, for example, the set of genes that contain the SNPs found in the previous step. A hypothesis based on SNPs can thus be transformed into a hypothesis based on genes, on the assumption that SNPs may be used as genetic markers for genes. The user may now work towards verifying the gene-level hypotheses, or proceed to generate new hypotheses at a different abstraction level, for example by retrieving the pathways that the genes belong to, and analyzing all SNPs belonging to the genes in the pathways.
Induction and hypothesis space maintenance
The high-level module will provide a “controller” interface by which the user can generate, test and select hypotheses according to the conceptual framework described by the ST-Model. The controller communicates with the other components of the system through appropriate Web Services interfaces, and uses their functionalities to implement the inferential steps that constitute the model. The controller will also provide a function to rule out hypothesis that, after the deduction step, appear to be, irrelevant or trivial. Since the eliminative induction step is strongly dependent on the users' preferences, we plan to leave this feature under their direct control.
The current version of the integrated module interface consists of three main components (snapshots are shown in Figure 4): i) a section for dataset uploading, ii) a hypotheses workspace, dynamically populated by the sets of hypotheses generated at each step of the analysis, and iii) a central interactive section, used both to display the contents of each hypothesis and to provide commands to operate on it. Commands are customized on the basis of the hypotheses contents, so that the possible next steps of the analysis are automatically generated by the system, leaving to the user the choice of which specific reasoning path to explore.
Figure 4.

A schematic representation of an analysis tasks that may be performed using the system. PM indicates the Phenotype Miner, while arrows labeled GP represent remote calls to Genephony.
4. The ST-Model in Genome-Wide Association Studies: a case study
In order to show that the ST-Model can be useful to model molecular medicine research in general, and GWAS in particular, here we apply it to the well-known Wellcome Trust Case-Control Consortium (WTCCC) study, a large scale association study that has collected and genotyped samples on 14,000 subjects, affected by one of seven common diseases, and on 3,000 controls [32]. In this section, we will detail each phase of this study using the conceptual framework provided by the ST-Model.
Abstraction
As previously described, the initial phase of a GWAS analysis consists in abstracting the raw data into a set of “usable” problem variables. This requires a precise definition of the phenotype, and a thorough data collection and validation process. In the WTCCC study, quality control analysis was performed on the overall data set (17,000 subjects and 3,000 controls), leading to the exclusion of approximately 8% of the available SNPs and of around 800 patients. The control groups, population structures and substructures, and effects of geographical variation were also analyzed to avoid confounders.
Abduction
The hypothesis space was generated by running a set of statistical association tests. Both standard statistical analysis (trend and genotype test) and Bayesian approaches (Bayes factor calculation) were applied. The result is a collection of SNPs that are determined to be significantly associated with membership in the case or control groups.
Ranking
It is interesting to note that different ranking strategies were applied to the hypotheses. SNPs were initially ranked on the basis of their p-values (or of their Bayes Factor), also taking the statistical power of the association tests into consideration. In a second stage, a higher ranking was assigned to SNPs belonging to clusters of correlated, statistically significant SNPs.
Deduction/Induction
To start, other published studies were searched to confirm or rule-out the hypotheses so far generated. In particular, the first search was aimed at determining whether the top-ranked SNPs were already known to be associated with the diseases under study. The following steps are described as follows in the WTCCC report: “…assessments on the basis of positional candidacy carry considerable weight, and, as we show, these already allow us, for selected diseases, to highlight pathways and mechanisms of particular interest. Naturally, extensive re-sequencing and fine-mapping work, followed by functional studies will be required before such inferences can be translated into robust statements about the molecular and physiological mechanisms involved” [32]. In other words, in this case the reasoning process proceeds by identifying the potential functional implications of each candidate SNP, on the basis of background knowledge retrieved from the available repositories. The induction phase then uses evidence from the literature and the data available in knowledge-bases to affect the ranking of the hypotheses or even, when sufficient knowledge is available, to rule some of them out entirely.
To better exemplify the application of the ST-Model to the WTCCC study, we focus here on the results obtained for Type 2 Diabetes Mellitus (T2D). We will concentrate in particular on the ranking, deduction, and induction steps, which require knowledge-based analysis (see Figure 5).
Figure 5.
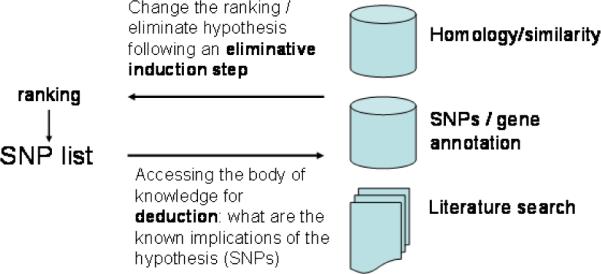
The ranking/deduction/induction steps of GWAS as implemented in the WTCCC study.
The first task in the analysis consisted in checking whether the selected SNPs were related to three gene variants known to be diabetes-related: PPARG (Peroxisomal Proliferative Activated Receptor Gamma; P12A102), KCNJ11 (the inwardly-rectifying Kir6.2 component of the pancreatic beta-cell KATP channel) and TCF7L2 (transcription factor 7-like 2). In this case, a cluster of SNPs related to TCF7L2 gave the strongest association signal for T2D. The hypothesis set was also found to contain SNPs in close proximity with a SNP that had previously been shown to be highly associated with diabetes, but that was not present on the microarray platform used in the study. This shows how hypothesis ranking can be affected by the available domain knowledge.
The analysis then proceeded by considering the remaining highly associated SNPs, following a line of reasoning which involves searching for clusters of associated SNPs, linking them with genes, and analyzing their functional properties, role in pathways, shared protein domains, etc. As reported in the previous section, this perfectly corresponds to iterating the deduction and induction phases by invoking full cycles of the ST-Model in which the hypotheses are formulated at different abstraction levels (genes, proteins, pathways, etc). In order to provide a further example of this, we carried out an additional analysis step following the ST-Model framework. Given the discovery, resulting from the previous stage, that SNPs in the TCF7L2 gene are significantly associated with the presence of Type 2 Diabetes, we wanted to generate new hypotheses at the metabolic pathways level. TCF7L2 is known to be a critical component of the Wnt signaling pathway, that has recently been linked to Type 2 Diabetes [33,34], and therefore represents a plausible candidate for a new hypothesis. Once again, we have performed an abstraction (generalizing from a single gene to one of the pathways that contain it) followed by an abduction (formulating the hypothesis that this pathway explains the available phenotype). We then performed the deduction step, in which we derive expected consequences from our hypothesis and check them against the available data, to confirm or disprove the hypothesis. To this end, we generated a set of 659 representative SNPs, using the annotation tools described in the previous section: after generating the set of all SNPs belonging to the genes in the Wnt signaling pathway, we extracted those for which genotype data is available in the WTCCC study, and we further selected a “prioritized” subset, giving preference to non-synonymous coding SNPs and to SNPs in promoter regions, and ensuring an equal number of SNPs from each gene. Finally, we removed SNP “rs4506565” from this set, since this is the SNP that was found to be significantly associated with T2D in the WTCCC study, and therefore represents a hypothesis that was already tested. We then assigned a score to each individual in the T2D cohort, calculated on the basis of his/her genotypes for all the SNPs in the set, and we performed the same operation on the two control groups (NBS and 58C). By applying the Wilcoxon test (p < 0.01) we found a significant difference between the scores obtained on the T2D cohort and the scores obtained in either one of the control groups. In other words, we were able to prove the hypothesis that there is a relationship between Type 2 Diabetes and the Wnt signaling pathway using the available genotype data, rediscovering a known finding through the proposed automated reasoning framework. A similar result was found for a similar-sized group of individuals in the T1D (Type 1 Diabetes) cohort, using the same set of SNPs, something that could indicate that the involvement of the Wnt pathway is common to both types of diabetes. Table 2 summarizes the results of this experiment.
Table 2.
Results of the comparison between two diabetes datasets (T2D and T1D) and two control datasets (NBS and 58C).
| Datasets | p-value | |
|---|---|---|
| T2D | NBS | 0.00116 |
| T2D | 58C | 0.00778 |
| T1D | NBS | 0.00050 |
| T1D | 58C | 0.00418 |
| T2D | T1D | 0.41520 |
| NBS | 58C | 0.25004 |
Each dataset contains genotype data for 659 SNPs in 1,400 subjects. Each subject received a score based on his/her genotypes, and the numbers in the third column indicate the significance of the difference of the average score for subjects in the two compared groups. Numbers in bold represent differences that are significant at the 0.01 level.
Note that the purpose of this experiment was to test whether the system would be able to reproduce an already known result, starting from a hypothesis (deduction step). We have not yet validated the overall induction / deduction mechanism, but we assume that its errors (false positives, false negatives) will be determined by the properties and performance of the algorithm applied in each separate step (for example, relying on association study p-values in the induction phase). To “globally” assess the performance of our system we would need to perform a study involving real users (data analysts / biologists / physicians), something that is outside the scope of this paper.
6. Discussion and conclusions
In this paper we have shown how the ST-Model, an epistemological model of the knowledge discovery process, can be used to formally describe the reasoning processes performed by researchers in the context of high throughput molecular research in general, and of GWAS in particular. Our main claims are that the ST-Model is able to describe the reasoning processes underlying current practice in large-scale molecular medicine studies, and that this model is amenable to be turned into a general computational architecture for decision support in translational bioinformatics.
The first claim is supported by the observation that creative abduction, as reported by Magnani [21] and Peirce [24], is the fundamental step in the inference to the best explanation. In this approach, the hypothesis space is dynamically created, starting from a phenotype of interest, and progressively refined on the basis of the available knowledge, through a series of deduction/induction cycles, possibly at different abstraction levels, and/or through additional experiments.
The second claim directly addresses the usefulness of the ST-Model for translational bioinformatics. The availability of large-scale datasets generated by high-throughput methods and of easily accessible repositories of background knowledge makes it now possible to combine the advantages of hypothesis-free research with those of hypothesis-driven research. We believe that a clear, formal definition of the conceptual steps that compose the discovery process can greatly benefit the design of computational systems supporting high-throughput research, thus moving beyond the typical “pipeline” model in which successive analysis steps are concatenated in a fixed, uni-directional sequence. A system based on the ST-Model explicitly distinguishes the hypothesis definition, hypothesis ranking, and hypothesis validation phases, and organizes them in a cyclical exploratory process. Moreover, access to the literature, to databases and to knowledge bases can be made “intentional”, i.e. the activity can be recorded as part of the intention of the user to find a particular type of evidence which may confirm or rule out a hypothesis.
We therefore believe that the ST-Model can be used as a well-founded framework to design Knowledge-Based Decision Support Systems,able to perform complete reasoning cycles, going through the abstraction, abduction, ranking, deduction, and induction steps as necessary, and keeping track of the inferences performed and of the intermediate hypotheses generated. Our experience shows that it is feasible and practical to build computational systems, such as the one we are developing, based on the ST-Model. However, it is important to remark that, although the ST-Model is an effective approach for descriptive and computational purposes, it is not necessarily the best possible choice for all different applications. Its purpose is to provide a general schema that fits the most common patterns of scientific discovery, but we recognize that there will always be cases that are not appropriately captured by this model.
Turning the steps of the conceptual model into actual software tools requires a significant design and implementation effort, and in this respect our work is still in its preliminary stage. Although the epistemological model we described lends itself well to the development of “plug-and-play” software architectures, in which each different component implements a specific reasoning task independently of the rest of the system, our goal at this stage is to provide researchers with a tool to effectively support their discovery process, rather than a fully-automated system. We envision that this experience will allow us to use the ST-Model as a design principle to build a new generation of KB-DSS for translational research in medicine, able to effectively integrate existing knowledge and experimental data in an architecture for automated discovery.
Acknowledgments
The authors wish to thank Alireza Nazarian for his help in analyzing the WTCCC data, and Alberto Malovini for his help in implementing the GWAS analysis pipeline. This work was partially supported by NIH grant R01 HL87681-01, “Genome-Wide Association Studies in Sickle Cell Anemia and in Centenarians” (A.R.), by the Fondazione Cariplo grant “Bioinformatics for Tissue Engineering: Creation of an International Research Group” (A.N.), and by FIRB project “ITALBIONET – Rete Italiana di Bioinformatica” (R.B.).
This study makes use of data generated by the Wellcome Trust Case Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Etzold T, Ulyanov A, Argos P. SRS: information retrieval system for molecular biology data banks. Methods in Enzymology. 1996;266:114–128. doi: 10.1016/s0076-6879(96)66010-8. [DOI] [PubMed] [Google Scholar]
- [2].Schuler GD, Epstein JA, Ohkawa H, Kans JA. Entrez: molecular biology database and retrieval system. Methods in Enzymology. 1996;266:141–162. doi: 10.1016/s0076-6879(96)66012-1. [DOI] [PubMed] [Google Scholar]
- [3].Senger M, Rice P, Oinn T. In: Soaplab: a unified sesame toor to analysis tools. Cox Simon J., editor. University of Southampton; Southampton, UK: 2003. pp. 509–13. [Google Scholar]
- [4].Wilkinson MD, Links M. BioMOBY: an open source biological web services proposal. Briefings in Bioinformatics. 2002 Dec.3:331–341. doi: 10.1093/bib/3.4.331. [DOI] [PubMed] [Google Scholar]
- [5].Wilkinson M, Schoof H, Ernst R, Haase D. BioMOBY successfully integrates distributed heterogeneous bioinformatics Web Services. The PlaNet exemplar case. Plant Physiology. 2005 May;138:5–17. doi: 10.1104/pp.104.059170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A. BioMart--biological queries made easy. BMC Genomics. 2009;10:22. doi: 10.1186/1471-2164-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Romano P. Automation of in-silico data analysis processes through workflow management systems. Briefings in Bioinformatics. 2008 Jan.9:57–68. doi: 10.1093/bib/bbm056. [DOI] [PubMed] [Google Scholar]
- [8].Demsar J, Zupan B, Leban G, Curk T. Proceedings of PKDD. Pisa, Italy: 2004. Orange: from experimental machine learning to interactive data mining; pp. 537–539. [Google Scholar]
- [9].Oinn T, Addis M, Ferris J, Marvin D, Senger M, Greenwood M, Carver T, Glover K, Pocock MR, Wipat A, Li P. Taverna: a tool for the composition and enactment of bioinformatics workflows. Bioinformatics. 2004 Nov.20:3045–3054. doi: 10.1093/bioinformatics/bth361. [DOI] [PubMed] [Google Scholar]
- [10].Kawas E, Senger M, Wilkinson MD. BioMoby extensions to the Taverna workflow management and enactment software. BMC Bioinformatics. 2006;7:523. doi: 10.1186/1471-2105-7-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shah SP, He DYM, Sawkins JN, Druce JC, Quon G, Lett D, Zheng GXY, Xu T, Ouellette BFF. Pegasys: software for executing and integrating analyses of biological sequences. BMC Bioinformatics. 2004 Apr;5:40. doi: 10.1186/1471-2105-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bartocci E, Corradini F, Merelli E, Scortichini L. BioWMS: a web-based Workflow Management System for bioinformatics. BMC Bioinformatics. 2007;8(Suppl 1):S2. doi: 10.1186/1471-2105-8-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Aniba MR, Siguenza S, Friedrich A, Plewniak F, Poch O, Marchler-Bauer A, Thompson JD. Knowledge-based expert systems and a proof-of-concept case study for multiple sequence alignment construction and analysis. Briefings in Bioinformatics. 2009 Jan.10:11–23. doi: 10.1093/bib/bbn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Musen M, Shahar Y, Shortliffe E. Biomedical Informatics. Springer; 2006. Clinical Decision Support Systems. [Google Scholar]
- [15].King RD, Rowland J, Oliver SG, Young M, Aubrey W, Byrne E, Liakata M, Markham M, Pir P, Soldatova LN, Sparkes A, Whelan KE, Clare A. The automation of science. Science. 2009 Apr.324:85–89. doi: 10.1126/science.1165620. [DOI] [PubMed] [Google Scholar]
- [16].Roos M, Marshall MS, Gibson AP, Schuemie M, Meij E, Katrenko S, van Hage WR, Krommydas K, Adriaans PW. Structuring and extracting knowledge for the support of hypothesis generation in molecular biology. BMC Bioinformatics. 2009;10(Suppl 10):S9. doi: 10.1186/1471-2105-10-S10-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Barosi G, Magnani L, Stefanelli M. Medical diagnostic reasoning: epistemological modeling as a strategy for design of computer-based consultation programs. Theoretical Medicine. 1993 Mar.14:43–55. doi: 10.1007/BF00993987. [DOI] [PubMed] [Google Scholar]
- [18].Lanzola G, Stefanelli M, Barosi G, Magnani L. NEOANEMIA: a knowledge-based system emulating diagnostic reasoning. Computers and Biomedical Research, an International Journal. 1990 Dec.23:560–582. doi: 10.1016/0010-4809(90)90041-a. [DOI] [PubMed] [Google Scholar]
- [19].Quaglini S, Berzuini C, Bellazzi R, Stefanelli M, Barosi G. Therapy planning by combining AI and decision theoretic techniques. Proceedings of AIME. 1989:147–156. [Google Scholar]
- [20].Keogh K, Sonenberg E. Keeping the patient asleep and alive: Towards a computational cognitive model of disturbance management in anaesthesia. Cognitive Systems Research. 2007 Dec.8:249–261. [Google Scholar]
- [21].Magnani L. Abduction, Reason and Science - Processes of Discovery and Explanation. Springer; 2000. [Google Scholar]
- [22].Ramoni M, Stefanelli M, Magnani L, Barosi G. An epistemological framework for medical knowledge-based systems. IEEE Transactions on Systems, Man and Cybernetics. 1992;22:1375, 1361. [Google Scholar]
- [23].Simon HA. Models of Discovery: And Other Topics in the Methods of Science. Springer; 1977. [Google Scholar]
- [24].Peirce C, Buchler B. Philosophical writings of Peirce. Dover Publications; 1955. Abduction and Induction; pp. 150–156. [Google Scholar]
- [25].Iles MM. What can genome-wide association studies tell us about the genetics of common disease? PLoS Genetics. 2008 Feb.4:e33. doi: 10.1371/journal.pgen.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics. 2006 Aug.38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- [27].Nuzzo A, Segagni D, Milani G, Rognoni C, Bellazzi R. A dynamic query system for supporting phenotype mining in genetic studies. Studies in Health Technology and Informatics. 2007;129:1275–1279. [PubMed] [Google Scholar]
- [28].Nuzzo A, Riva A, Bellazzi R. Phenotypic and genotypic data integration and exploration through a web-services architecture. BMC Bioinformatics. 2009;10(Suppl 2):S5. doi: 10.1186/1471-2105-10-S12-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Malovini A, Rognoni C, Puca A, Bellazzi R. Proceedings of the AMIA Summit on Translational Bioinformatics. San Francisco, CA: 2009. Multi-criteria decision making approaches for quality control of genome-wide association studies. [PMC free article] [PubMed] [Google Scholar]
- [30].Nuzzo A, Riva A. Genephony: a knowledge management tool for genome-wide research. BMC Bioinformatics. 2009;10:278. doi: 10.1186/1471-2105-10-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007 Sep.81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007 Jun.447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jin T, Liu L. The Wnt signaling pathway effector TCF7L2 and type 2 diabetes mellitus. Molecular Endocrinology. 2008 Nov.22:2383–2392. doi: 10.1210/me.2008-0135. [DOI] [PubMed] [Google Scholar]
- [34].Gloyn AL, Braun M, Rorsman P. Type 2 diabetes susceptibility gene TCF7L2 and its role in beta-cell function. Diabetes. 2009 Apr.58:800–802. doi: 10.2337/db09-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]