

SUMMARY
Toll-like receptors (TLRs) have previously been shown to play critical roles in the activation of innate immunity. Here, we describe that T cell expression of TLR2 regulates T helper 17 (Th17) cell responses. Stimulation with TLR2 agonists promoted Th17 differentiation in vitro and led to more robust proliferation and Th17 cytokine production. Using the experimental autoimmune encephalomyelitis (EAE) model, we found that TLR2 regulated Th17 cell-mediated autoimmunity in vivo and that loss of TLR2 in CD4+ T cells dramatically ameliorated EAE. This study thus reveals a critical role of a TLR in the direct regulation of adaptive immune response and pathogenesis of autoimmune diseases.
INTRODUCTION
Toll-like receptors (TLRs) function in the “first line of defense” against various infectious agents. As evolutionarily conserved pattern recognition receptors, TLRs recognize distinct microbial products. Upon binding to their cognate ligands, TLRs activate the innate immune response, leading to the production of pro-inflammatory cytokines and chemokines (Kawai and Akira, 2007). These receptors also stimulate maturation of dendritic cells and the expression of costimulatory molecules on antigen-presenting cells (APCs) to facilitate the activation of adaptive immunity (Janeway and Medzhitov, 2002). Recently, TLRs have been found to directly activate the innate function of γδ T cells, resulting in the production of the cytokines interleukin-17 (IL-17) and IL-22 (Martin et al., 2009). Although generally thought to regulate adaptive immunity indirectly through the activation of innate responses, TLRs have been also reported to modulate the function of T lymphocytes as well (reviewed in (MacLeod and Wetzler, 2007). TLR2, in particular has been shown by multiple groups to be expressed by and to function in CD4+ T lymphocytes. For example, TLR2 was found to provide a costimulatory signal on activated CD4+ T helper cells (Komai-Koma et al., 2004). Furthermore, TLR2 engagement in CD8+ T cells enhanced proliferation, activation, and memory cell formation even under suboptimal activation conditions (Cottalorda et al., 2006) (Mercier et al., 2009). TLR2 has also been shown to be functional directly on regulatory T cells (Chen et al., 2009; Liu et al., 2006). Thus αβ T cells appear to directly express TLRs; however, the physiological and pathological significance of TLR expression in antigen-specific T cells remains unclear.
Naïve CD4+ T helper (Th) cells, upon activation by the innate immune system, differentiate into distinct effector lineages depending on the environmental signals present during activation (Dong and Flavell, 2000; Glimcher and Murphy, 2000). Recently, Th17 cells have been identified and characterized as a distinct T cell lineage mediating tissue inflammation, especially in autoimmunity (Dong, 2006; Weaver et al., 2006). In addition to activation through T cell receptor (TCR) and co-stimulatory receptors, Th17 cells require distinct factors for their development and maintenance, including transforming growth factor beta (TGFβ), IL-6, IL-21, IL-1β, and IL-23 (Chung et al., 2009) (Dong, 2008). Th17 cells and IL-17 activity has been attributed to the pathogenesis of a variety of autoimmune disorders, including experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS) (Langrish et al., 2005) (Park et al., 2005) (Yang et al., 2008a).
Previously, TLR2 function has been linked to central nervous system (CNS) inflammation. During EAE, TLR2 along with poly (ADP-ribose) polymerase 1 expression on macrophages and astrocytes was found to promote CNS inflammation (Farez et al., 2009). In this study, we investigated the direct role of TLR2 in the generation and function of Th17 cells in vitro and in vivo. We found that TLR2 activation during Th17 differentiation enhanced proliferation and Th17 cytokine production. More importantly, deficiency of TLR2 in CD4+ T cells substantially impaired Th17 responses in vivo and the pathogenesis of EAE. Therefore, TLR2 directly regulates Th17 responses and Th17-mediated autoimmune disease.
RESULTS
TLR2 signaling in T cells enhances Th17 differentiation
In an attempt to identify genes that are differentially expressed in Th1 and Th17 cells, we performed microarray analysis and found that TLR2 was more highly expressed by Th17 cells (GEO accession number GSE11924). To further investigate TLR expression in T cell subsets, we performed real-time RT-PCR on Th1, Th2, and Th17 subsets using bone marrow-derived macrophages and RFP+ γδ T cells from IL-17F-RFP reporter mice as positive controls. It has previously been shown that TLR2 signaling requires heterodimerization with TLR1 or TLR6 (Schumann and Tapping, 2007). In accordance, TLR1, 2, and 6 mRNAs were expressed to the highest degree in Th17 cells compared to Th1 and Th2 subsets (Figure 1A and Figure S1A available online). TLR4 expression was also found to be enhanced in Th17 cells, whereas TLR9 expression was similar between Th2 and Th17 cells. To further analyze TLR expression in Th subsets, we stained various effector T cells (either left untreated or activated through TLR2) with anti-TLR2 for flow cytometry. Th17 cells exhibited positive staining, although to a much lower degree compared to a macrophage control (Figure S1B available online). Treatment with a TLR2 -TLR1 ligand, Pam3Cys-Ser-(Lys)4-trihydrochloride (Pam3Cys), however, induced TLR2 expression in Th2 cells. Recently, the expression of TLR2 and 4 was found to be enhanced in IL-17-producing γδ T cells (Martin et al., 2009). Although we consistently observed upregulated TLR expression in Th17 cells compared to other types of Th cells, there was much greater expression of various TLRs in IL-17-producing γδ T cells. Thus, our results suggest that similar to these innate T cells, Th17 cells may be able to respond to pathogen-associated molecules.
Figure 1. TLR expression in Th17 cells.

(a) Expression of various TLRs on Th1, Th2, and Th17 cells following 4-day differentiation. Bone marrow-derived macrophages and IL-17-producing γδ T cells were utilized as positive controls. Relative mRNA expression was compared by setting the lowest expression amount to an arbitrary value of 1. (b) Expression of TLRs on αβ and γδ T cells following IL-23 timepoint stimulation. 20 ng/ml IL-23 was added to sorted splenic CD4+ and γδ T lymphocytes and cells were lysed at 0, 2, 4, 6, 8, 16, and 24 hrs post IL-23 stimulation. Unstimulated cells served as the expression baseline. For all experiments, data are presented as mean + SD and are a representative of at least two independent experiments. All gene quantities were normalized to the expression of the reference gene β-actin.
Because IL-23 is important in IL-17 responses in both αβ and γδ T cells (Langrish et al., 2005) (O’Brien et al., 2009), we next investigated whether TLR2 can influence IL-23 expression and vice versa. TLR2 activation of bone marrow-derived macrophages and dendritic cells resulted in substantially increased IL-23 p19 mRNA expression (Figure S1C available online). Moreover, IL-23 stimulation of γδ, but not αβ T cells led to enhanced TLR1, 2, and 4 mRNA expression in a time-dependent manner (Figure 1B). As we will address later (Figure 4B), IL-23 is not sufficient by itself to drive γδ T cell proliferation, so IL-23 was unlikely expanding a subpopulation of IL-17-producing γδ T cells in these experiments.
Figure 4. The effect of TLR2 activation in combination with IL-23 on αβ and γδ T cells.

(a) Overnight anti-CD3 activation of Th17 cells with various TLR ligands following 4-day differentiation. The production of IL-17 (top) and IL-22 (bottom) was assessed in triplicate by ELISA. * = Students t test, p < 0.05. * = significant comparison of samples without IL-23 to untreated control. ** = significant comparison of samples treated with IL-23 to untreated IL-23 samples and untreated controls. (b) CFSE labeling of sorted naïve CD4+, memory CD4+, and γδ T cells following 2-day stimulation with TLR2 agonist or IL-23. Data are representative of three individual experiments. (c) CFSE labeling of sorted total CD4+ T cells (left) and γδ T cells (right) derived from WT (top) and Tlr2−/− (bottom) animals. Data are a representative of three independent experiments. Numbers shown in each gate are indicative of mean fluorescent intensity.
Considering the TLR2 expression observed in Th17 cells, we next investigated if TLR2 activation in CD4+ T cells in vitro could modulate Th17 differentiation and/or function. FACS-sorted naïve CD4+ T cells were cultured with plate-bound anti-CD3 and anti-CD28 in the presence of TGFβ and IL-6 along with blocking antibodies against IFNγ and IL-4. A TLR2 ligand, Pam3Cys, was added to the differentiation media in some cases as well. Cells differentiated in the presence of Pam3Cys exhibited a ~50% increase in IL-17 single positive and IL-17-IL-17F double positive cells (Figure 2A). IFNγ was not detected in either group (data not shown). ELISA analysis of primary culture supernatants as well as those from effector Th17 cells restimulated with anti-CD3 overnight revealed a similar enhancement of IL-17 and IL-17F production in cells cultured in the presence of TLR2 stimuli (Figure 2B). We further examined whether this effect was TLR2-specific by using TLR2-deficient naïve CD4+ T cells. TLR2-deficient (Tlr2−/−) T cells did not exhibit obvious differences in Th17 polarization in the absence of exogenous TLR2 ligands compared to WT controls, however; the number of IL-17-producing cells did not substantially increase following stimulation with the Pam3Cys ligand (Figure 2C).
Figure 2. Enhanced Th17 differentiation following TLR2 activation.

(a) Intracellular cytokine staining following four-day Th17 differentiation in the absence (left) or presence (right) of Pam3Cys. Each panel represents cells derived from an individual mouse, 2 mice were used for each treatment group. (b) ELISA analysis of supernatants from primary four-day differentiation (left) or overnight anti-CD3 only restimulated (right) cultures. All groups are analyzed in triplicate and are presented here as the mean + SD from three individual mice per group. (c) Th17 differentiation was performed as in (a) using naïve T cells derived from WT and Tlr2−/− animals. (d) mRNA expression of naïve and Th17 cells differentiated in the absence or presence of Pam3Cys. Each bar represents mean + SD values obtained from T cells of three individual mice. Naïve T cell gene expression was used as the baseline of expression and all gene quantities were normalized to the expression of β-actin. * = Students t test; p < 0.05. All data are a representative of at least three individual experiments.
We additionally considered the possibility that contaminating APCs may be the cause of increased Th17 differentiation upon TLR2 ligand treatment; dendritic cells or macrophages activated through TLR2 could produce such factors as IL-23 that could, in turn, amplify the Th17 response. To address this, we performed Th17 differentiation of naïve CD4+ T cells derived from WT and IL-23-deficient (Il23a−/−) animals that were first depleted of APCs and then selected for CD4+ expression. The production of IL-17 and IL-17F and the frequency of Th17 cells were increased in the cultures that were stimulated with TLR2 ligands, whereas no real differences were observed between T cells derived from Il23a−/− animals compared to WT controls. Furthermore, IL-1β or IL-23 was not detected above background in all Th17 culture conditions analyzed (Figure S2A and B available online). Moreover, CD11c expression was not detectable above background in our purified T cells (Figure S2C available online). Finally, we performed Th17 differentiations in the absence or presence of increasing concentrations of LPS and speculated that if TLR2 ligand treatment was in fact activating contaminating APCs rather than naïve T cells, then LPS treatment would do the same. The production of IL-17 and IL-17F remained unchanged after LPS treatment compared to untreated controls (Figure S2D available online), ruling out contamination of APCs.
TLR2 activation induces Th17-related genes
To assess the mechanism whereby TLR2 enhances Th17 cytokine production, we compared the gene expression of CD4+ T cells differentiated under Th17 conditions in the absence or presence of Pam3Cys. Consistent with the protein results, upregulation of Th17-associated mRNA (IL-17, IL-17F, IL-21, and CCR6) was also detected (Figure 2D). The transcription factors RORγ and RORα are critical in regulating the Th17 cell genetic programming (Yang et al., 2008c). Moreover, IRF-4 and AhR have recently been identified as Th17-promoting transcription factors (Brustle et al., 2007) (Veldhoen et al., 2008). Analysis of these genes revealed an enhancement of RORγ, RORα, and IRF-4 mRNA expression upon TLR2 engagement (Figure 2D), suggesting a direct role of TLR signaling in the expression of critical Th17-regulating transcriptional factors. It seems that the amounts of T-bet mRNA in this experiment were higher in the naïve T cell population than in the test population; however, if compared directly to Th1 cells, naïve cells actually expressed miniscule amounts of T-bet. To determine if increased Th17 cytokine expression by TLR2 activation was a result of an increase in the number of Th17 cells, we performed a similar experiment using our IL-17F-RFP reporter mice (Yang et al., 2008b). Naïve T cells from these mice were differentiated in the presence or absence of Pam3Cys. RFP+ cells were then sorted, and mRNA quantification was performed. RFP+ cells that were activated through TLR2 exhibited similar increases in expression of Th17-related factors (Figure S2E available online), suggesting that TLR2 signaling enhances Th17 differentiation on a per cell basis rather than simply expanding the Th17 population.
TLR2 signaling enhances Th17 proliferation
Previous studies have demonstrated that both activated CD4+ and CD8+ T cells proliferate more extensively upon TLR2 activation (Cottalorda et al., 2006) (Komai-Koma et al., 2004). Indeed, developing Th17 cells proliferated 2-fold more extensively when Pam3Cys was included in the differentiation media than those cultured without Pam3Cys by total cell counts following four days of differentiation (Figure 3A). Likewise, using carboxyfluorescein diacetate succinimidyl ester (CFSE) to determine proliferation of differentiating Th17 cells, we demonstrated that more cells were in the later stages of division after TLR2 activation compared to cells differentiated without TLR2 ligand (Figure 3B). Taken together, our data suggest that TLR2 can directly regulate the differentiation of Th17 cells and their expansion in vitro.
Figure 3. Proliferation of Th17 cells following TLR2 engagement.
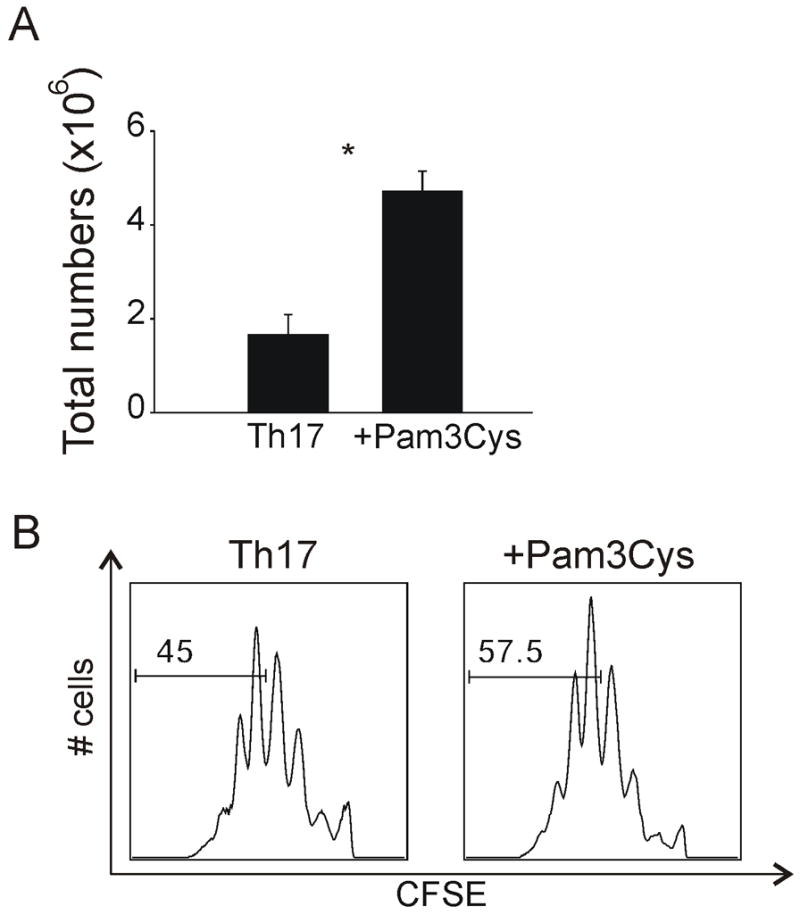
(a) Assessment of total cell numbers following Th17 differentiation in the presence or absence of Pam3Cys. Each bar represents T cells derived from 2 individual mice. * = Students t test; p < 0.05. (b) CFSE staining of Th17 cells following 4 days of differentiation in the absence (left) or presence (right) of Pam3Cys. Numbers shown in each gate are indicative of mean fluorescent intensity. Data are a representative of four independent experiments.
TLR2 signaling enhances proliferation and IL-17 production by effector, memory and γδ T cells
Because TLR2 activation enhanced Th17 cytokine production and proliferation of naïve CD4+ T cells when they were polarized to Th17 cells, we next investigated whether TLR2 stimulation could influence the function of other types of T lymphocytes. To address this possibility, we first investigated TLR2 activity on fully differentiated Th17 cells. Following four day in vitro differentiation, Th17 cells were restimulated in the absence or presence of various TLR ligands in the absence of TCR activation. IL-17 and IL-22 production was then assessed by ELISA. We found that treatment of fully differentiated Th17 cells with most TLR ligands alone (except for polyI:C) enhanced both IL-17 and IL-22 production compared to untreated cells (Figure 4A). Moreover, IL-23 and TLR agonists synergistically enhanced the production of these Th17 cytokines. Of all the conditions tested, the combination of TLR2 and IL-23 was one of the strongest in inducing IL-17 and IL-22 production. TLR4 activation, in combination with IL-23, has recently been shown to induce IL-17 in γδ T cells (Martin et al., 2009). Here we observed a similar effect of LPS in the promotion of IL-17, but not IL-22, production in αβ T cells both in the presence or absence of IL-23.
The TLR2-IL-23 synergy observed on Th17 cells was further extended to other T cell subsets. As shown in a previous report (Martin et al., 2009), we found that γδ T cells, in the absence of TCR stimulation, exhibited a strong IL-17-specific response when treated with IL-23 alone (data not shown); however, very little IL-17 production was observed in cells treated with only Pam3Cys. Moreover, similar to differentiated Th17 cells, γδ T cells treated with a combination of IL-23 and Pam3Cys exhibited the highest degree of IL-17 production. TLR2-deficient γδ T cells failed to exhibit this synergy and their IL-17 production in response to IL-23 alone was reduced as well. Additionally, although not on the same scale as γδ T cells, memory T cells also produced IL-17 in a synergistic fashion following Pam3Cys and IL-23 treatment in the absence of TCR stimulation (Figure S3 available online).
We next investigated the contributions of TLR2 and IL-23 on cellular proliferation in the absence of TCR stimulation. TLR2 stimulation alone was found to be sufficient for the proliferation of γδ T cells; ~50% of γδ T cells divided following two-day stimulation (Figure 4B). Naïve and memory cells were also expanded in response to Pam3Cys alone without TCR activation, albeit to a reduced degree compared to γδ T cells, possibly due to their reduced TLR2 expression. The proliferative effect of the Pam3Cys observed here was directly dependent on TLR2 signaling as TLR2-deficient T lymphocytes failed to expand (Figure 4C). Surprisingly, IL-23 stimulation had little to no effect on the proliferation of all T cells tested, suggesting that TLR2 stimulation may drive proliferation, where IL-23 is only essential for regulating IL-17 differentiation programs.
We have previously shown that Th17 cells derived from naïve TLR2-deficient T cells do not have intrinsic defects in IL-17 production (Figure 2C). Next, we decided to examine if the same were true for γδ T cells. Interestingly, we found that TLR2-deficient γδ T cells exhibited decreased IL-17 expression in response to IL-23 alone in adult γδ T cells from peripheral lymphoid tissues (data not shown). Therefore, to examine the possibility that TLR2-deficient γδ T cells may have an intrinsic defect in IL-17 production, we harvested both peripheral γδ T cells and embryonic thymocytes derived from WT and TLR2-deficient animals, stimulated with PMA and ionomycin, and then measured IL-17 production (Figure 5A). Adult γδ T cells derived from the spleen of TLR2-deficient animals on a C57BL/6 background exhibited a reduced capacity for IL-17 production; however, no difference was observed in embryonic thymus between the 2 groups, indicating that TLR2 γδ T cells develop normally and have full IL-17 responsiveness at the time of thymic maturation. Likewise, IL-23-deficient γδ T cells on a C57BL/6 × 129 mixed background derived from spleen or lung exhibited greatly decreased IL-17 production as well upon PMA plus ionomycin restimulation (Figure 5B). Although IL-23 has been shown to be important in IL-17 expression by γδ T cells (Martin et al., 2009), we investigated if these cells are competent in IL-17 expression at the time of thymic maturation. We found that γδ T cells derived from IL-23-deficient embryonic thymus had amounts of IL-17-expressing cells comparable to that derived from WT. Thus, TLR2 and IL-23 are not critical for early IL-17-producing γδ T cell development but appear to be critical in proliferation or maintenance of IL-17-producing γδ T cells.
Figure 5. Analysis of WT, Tlr2−/−, and Il23a−/− γδ cells.

(a) Splenic (left) or embryonic (right) γδ T cells derived from WT (top) or Tlr2−/− (bottom) thymus were stimulated with PMA plus ionomycin for five hours and stained for IL-17 and IFNγ production. Embryos were harvested from timed breeders following day 18.5 post vaginal plug observation. (b) IL-17 production was investigated in γδ T cells derived from WT or Il23a−/− spleens (left), lungs (center), and fetal thymi (right). Data presented is representative of multiple experiments. γδ T cells were isolated and intracellular cytokine staining was assessed following five-hour PMA plus ionomycin stimulation. Data are a representative of at least two experiments, using at least two mice per group.
TLR2 deficiency reduces Th17 generation in vivo and confers protection against EAE
To evaluate the direct role of TLR2 signaling in the regulation of Th17 cells in vivo, Rag1−/− mice were reconstituted with WT or TLR2-deficient bone marrow and EAE was induced. In this manner, we were able to assess the role of TLR2 directly in the lymphoid compartment. Mice reconstituted with WT bone marrow cells exhibited substantially higher disease severity compared to animals reconstituted with Tlr2−/− bone marrow; however, disease incidence was similar between the two groups as all mice developed disease (Figure 6A). Additional examination of CNS tissues revealed reduced brain and spinal cord infiltration with concomitant decreases of CD4 and CD11b positive cells in animals reconstituted with Tlr2−/− bone marrow cells (Figure 6B). Interestingly, the result obtained here is not in entire accordance with two other reports (Chen et al., 2009) (Prinz et al., 2006) that utilized germline TLR2-deficient animals in their EAE experiments. These authors did not observe differences in EAE severity or incidence in Tlr2−/− animals compared to WT controls. Because all of our mice that were reconstituted with Tlr2−/− bone marrow did, in fact, develop disease, these studies are not altogether different except for our observed decrease in disease severity. Additional effects of TLR2 on non-lymphoid cells may account for this discrepancy.
Figure 6. Assessment of EAE in Tlr2−/− bone marrow-reconstituted mice.

(a) Clinical score data comparing EAE mice reconstituted with WT or TLR2-deficient bone marrow. Data are representative of 3 individual experiments, n = 3–5 mice per group per experiment. (b) CNS infiltration analysis of the bone marrow-reconstituted mice presented in (a). Total infiltrating CNS and splenic cell counts are presented in the left panel. Analysis of CNS-infiltrating CD4+, CD11b+, IL-17+, and IFNγ+ are presented in the right panel following five-hour PMA plus ionomycin restimulation. (c) Representative CNS intracellular cytokine staining of WT and Tlr2−/− bone marrow-reconstituted mice following five-hour PMA plus ionomycin restimulation (left). The right panel is a comparison of the percentage of cytokine-positive cells of all the mice analyzed, n = 5 per group. (d) Splenic MOG recall assays. Splenocytes derived from each group were restimulated with MOG peptide for three days and cytokine production was assessed by ELISA (left and center). Proliferation was examined following three-day MOG restimulation and an eight-hour 3H-thymidine pulse (right). Data are presented as mean + SD for triplicate determinations. * = Students t test; p < 0.05.
IL-17 plays a major role in pathogenesis of EAE (Dong, 2006). To investigate the role of TLR2 in the generation of Th17 along with other Th subsets in vivo, we restimulated CNS-infiltrating CD4+ T cells with PMA plus ionomycin. CNS tissues derived from WT mice exhibited higher numbers of IL-17 single and IL-17-IFNγ double positive cells (Figure 6B). However, in terms of frequency, only IL-17+ cells were substantially decreased in Tlr2−/− reconstituted mice (Figure 6C), suggesting that Th17, not Th1, function is more dependent on TLR2 activity. Splenocytes derived from EAE mice were also evaluated for the generation of antigen-specific T cell responses by MOG recall assays (Figure 6D). We found that IL-17 production was substantially decreased in Tlr2−/− splenocytes compared with WT cells following MOG restimulation in a dose-dependent manner. However, as was observed in the CNS, no differences were observed in IFNγ production. Proliferation was assessed upon MOG restimulation as well. We found that TLR2 deficiency resulted in a reduction of antigen-specific T cell division following MOG immunization. Taken together, our data indicate that Th17 generation in vivo was impaired in the absence of functional TLR2 signaling in lymphoctes.
TLR2 deficiency in CD4+ T cells impairs pathogenesis of EAE
Our previous results demonstrated a role of the TLR2 pathway in the development of Th17 responses during autoimmunity. To directly delineate the function of TLR2 specifically on CD4+ T cells in vivo, we transferred total CD4+ T cells from WT and TLR2-deficient animals into Rag1-deficient animals and induced EAE. As observed with bone marrow-reconstituted animals, we found that mice receiving Tlr2−/− CD4+ T cells exhibited greatly reduced disease severity compared to mice receiving WT CD4+ T cells (Figure 7A). In contrast to bone marrow-reconstituted EAE mice where there was no difference in disease incidence, mice transferred with Tlr2−/− CD4+ T cells exhibited little to no disease across multiple experiments.
Figure 7. Assessment of EAE development and Th17 differentiation by Tlr2−/− CD4+ T cells.

(a) Clinical scores of EAE mice that were transferred with WT or Tlr2−/− CD4+ T cells. Data are a representative of two individual experiments with five mice utilized per group. (b) Examination of CNS infiltration following EAE induction. The top panel represents total cell infiltration, where the bottom panel represents the total numbers of cytokine-positive cells following five-hour PMA plus ionomycin reactivation. (c) mRNA analysis of CNS tissues at day 17 following EAE initiation. Three mice per group were analyzed at day 17 and gene expression was normalized to the expression of the reference gene β-actin. (d) Rag1-deficient mice were reconstituted with CD4+ T cells derived from C57BL/6 (WT) or TLR2-deficient mice. Clinical scores were monitored daily following EAE induction with LPS emulsified in IFA and MOG. Fractions on the right represent mice developing EAE out of total mice analyzed. (e) IL-17- and IFNγ-positive cells in the draining lymph nodes of Rag1−/− mice transferred with WT or Tlr2−/− CD4+ T cells following seven day KLH immunization and three day KLH reactivation. (f) IL-17 and IFNγ cytokine production was measured by ELISA in bone marrow chimeric mice following seven day KLH immunization. CD45.1 (WT) versus CD45.2 (Tlr2−/−) CD4+ T cells were sorted and cultured with irradiated WT splenocytes and 50μg/ml KLH for 3 days prior to cytokine assessment. * = Students t test; p < 0.05. Data are a representative of at least two individual experiments.
Next, we investigated CNS infiltration and cytokine production in EAE mice. We found substantial decreases in total, CD4+, and CD11b+ infiltrates into the CNS tissues of animals receiving Tlr2−/− CD4+ T cells (Figure 7B). Both IL-17 and IFNγ positive cells were decreased in mice with Tlr2−/− T cells; however, no difference was observed in total IL-17-IFNγ double-positive populations. Recent publications have indicated the importance of γδ T cells in the development of EAE (reviewed in (Blink and Miller, 2009). Therefore, we tested spleen tissues of CD4+ T cell-transferred mice; however, γδ T cells as well as B and CD8+ T cells were completely absent in the spleen of both sets of animals (data not shown), which rules out a possible role of γδ T cell TLR activation in this experiment.
To further investigate the mechanisms of EAE protection observed in mice reconstituted with TLR2-deficient CD4+ T cells, we analyzed mRNA expression in the CNS tissues of 3 individual mice from each group. Almost all Th17-related genes were downregulated in the CNS of mice reconstituted with TLR2-deficient CD4+ T cells, compared to those reconstituted with WT cells, with a substantial reduction observed for IL-17 and IL-17F transcripts (Figure 7C). Additionally, WT cell-reconstituted mice had higher expression of chemokines with established roles in EAE, such as CCL2 and CCL20, than those with TLR2-deficient T cells, providing a mechanistic basis for the reduction in CD4+ and CD11b+ infiltrates observed in TLR2-deficient reconstituted mouse CNS. Interestingly, IFNγ mRNA was greatly reduced in TLR2-deficient cell-reconstituted CNS tissues, which may be secondary to the Th17 defect in these mice, as Th17 cells are also important for migration of Th1 cells into CNS (Yamazaki et al., 2008). Moreover, no differences were observed in CNS cell staining for Foxp3 or in Foxp3 mRNA expression (data not shown). IL-4 and IL-10 were undetectable in the CNS tissues of both groups of mice (not shown), indicating that TLR2 deficiency might not alter Th2 or regulatory T cell behavior.
To address the possibility that EAE pathogenesis may be influenced directly by endogenous TLR2 ligands generated in vivo, we induced EAE in Rag1-deficient mice reconstituted with WT and Tlr2−/− CD4+ T cells through the use of LPS rather than CFA as an adjuvant (Figure 7D). We found that mice receiving Tlr2−/− T cells still exhibited a pronounced decrease in EAE severity and incidence with only 55.5% of Tlr2−/−-reconstituted mice developing disease compared to 100% of WT CD4-reconstituted mice. However, this difference in EAE incidence and severity was moderate compared to EAE using CFA as an adjuvant (Figure 7A). Thus, endogenous TLR2 ligands may be important in the EAE pathogenesis, although TLR2 activation may not be required for the initial activation and maintenance of antigen-specific T cells in this model.
To further assess the direct role of TLR2 in Th17 cell differentiation in vivo, we immunized Rag1-deficient mice with keyhole limpet hemocyanin (KLH) reconstituted with both total WT or Tlr2−/− CD4+ T cells and WT B cells as we recently described (Nurieva et al., 2009). Seven days later, draining lymph nodes were then harvested and stained for IL-17 following culture with KLH. WT T cell-transferred mice exhibited higher numbers of IL-17 positive cells compared to those receiving Tlr2−/− T cells (Figure 7E). No statistically significant differences were observed in IFNγ–positive cells. In addition, we also performed a competitive experiment by immunizing WT and Tlr2−/− mixed bone marrow chimera mice with KLH. WT and Tlr2−/− T cells were then purified based on their congenic markers and assessed for their cytokine expression following antigen restimulation. Although no differences were observed in the homeostatic proliferation of WT or Tlr2−/− T cells before immunization (not shown), KLH restimulation revealed a decrease of Tlr2−/− IL-17-producing cells in draining lymph nodes (Figure 7F). Consistent with previous results, the production of IFNγ was not statistically different between the two groups. Thus, TLR2 seems to regulate early Th17 differentiation in vivo.
DISCUSSION
TLRs play crucial roles in the regulation of innate immune responses. The enhancement of TLR2 expression in Th17 cells suggests an unexpected role of these evolutionarily conserved receptors in direct regulation of adaptive immunity. TLR2, a pattern recognition receptor that binds a wide variety of bacterial products such as lipopeptides (Kawai and Akira, 2007), thus became the focus of this investigation; however, other TLRs were increased in this subset as well. Both TLR1 and TLR6 are dependent on TLR2 for their cell surface expression (Schumann and Tapping, 2007), so their increased expression in Th17 cells may allow increased TLR2 downstream activity in T cells. TLR4 expression was also upregulated in Th17 cells, suggesting a functional relationship with IL-17 production that was previously discovered in γδ T cells (Martin et al., 2009). Indeed, fully differentiated Th17 cells restimulated with LPS and IL-23 exhibited increased IL-17 responses as well. Thus, TLR4 signaling may represent another pathway modulating the function of T helper cells.
Our data show that TLR2 stimulation of highly purified CD4+ T cells during the Th17 differentiation process resulted in increased proliferation and IL-17 production. Previous work has suggested that TLR2 enhances the proliferative capacity of both activated and memory CD4+ and CD8+ T cells (Cottalorda et al., 2006) (Komai-Koma et al., 2004) (Mercier et al., 2009). Herein, we have demonstrated that even in the absence of TCR activation, naïve T cells are strongly influenced by TLR2 signals as well. The proliferative effect of TLR2 activation observed on Th17 cells in vitro could in theory affect homeostasis and maintenance of Th17 and other IL-17-producing cells in vivo. This idea is supported by the reduced numbers of IL-17+ γδ T cells observed in Tlr2−/− mice. Alternatively, TLR2 activation is not sufficient to influence IL-17 production by itself in naïve CD4+ T cells. In the case of Th17 differentiation, TCR activation along with TGFβ and IL-6 are also necessary for lineage commitment. Likely, the increased IL-17 production observed in Th17 cells differentiated in the presence of a TLR2 agonist is due to a costimulatory effect in the differentiation of Th17 cells. This additional TLR2 signal, in conjunction with TCR activation, may also directly target the Th17-related transcription factors RORγ, RORα, or IRF-4 to aid in the production of IL-17. Interestingly, no major effect was observed on other Th subset-related transcription factors, such as T-bet and GATA-3, suggesting that the TLR2 signaling selectively targets factors crucial for Th17 cells rather than inhibiting the alternative Th subsets. The effect of TLR2 activation was assessed on Th1 and Th2 cells as well; however, whereas proliferation differences were observed, there was no effect on the production of IFNγ or IL-4, respectively (data not shown).
IL-17 production by CD4+ T cells was found to be strongly affected by TLR2, even after the early stages of lineage commitment. Fully differentiated Th17 cells treated with a TLR2 agonist were still found to produce increased amounts of IL-17 and IL-22 proteins following TLR2 ligand restimulation even when the TCR stimulus was removed. However, the addition of IL-23 in combination with Pam3Cys resulted in a synergistic increase of both IL-17 and IL-22 compared to Pam3Cys-treated alone. The synergy we observed between TLR2 and IL-23 on Th17 cells suggested that these mediators may be sufficient and TCR activation may not be required for fully committed Th17 memory cells to produce IL-17. Indeed, we found that treatment of CD4+ memory cells with Pam3Cys and IL-23 induced a small amount of IL-17 expression even without TCR stimulation. Additionally, naïve and memory CD4+ cells both proliferated in response to Pam3Cys alone. Thus, our results suggest a unique pathway for maintenance of CD4+ Th17 populations, one that requires only external or endogenous TLR2 stimuli.
γδ T cells can produce substantial amounts of IL-17 depending on TCR specificity and the environment (O’Brien et al., 2009). Moreover, during Mycobacterium tuberculosis (TB) infection, γδ T cells were found to be a predominant source of IL-17 in both humans and mice (Lockhart et al., 2006) (Peng et al., 2008). Due to the prevalence of TLR ligands during TB infection, coupled with the TLR2-IL-23 effect observed on αβ T cells, we investigated the role of TLR2 on IL-17-producing γδ T cells. In concurrence with a recent report (Martin et al., 2009), we found that although TLR2 activation alone did not induce IL-17 production from γδ T cells, the addition of IL-23 induced enhanced IL-17 responses compared to cells treated with IL-23 alone. Likewise, as was observed for naïve and memory CD4+ T cells, TLR2 activation was sufficient to induce proliferation whereas IL-23 had little to no effect on γδ T cells. Taken together, our results illustrate a mechanism of γδ T cell IL-17 production. γδ T cells have the ability to rapidly respond to TLR2 signals and produce IL-17 when IL-23 is present in the environment. IL-23, in turn, can be produced by innate cells such as dendritic cells and macrophages after TLR2 activation, setting up a scenario in which the same TLR2 signal could potentially activate antigen presenting cells as well as γδ T cells, which may provide a sufficient environment for a rapid IL-17 response against invading pathogens.
Rag1−/− mice reconstituted with TLR2-deficient bone marrow exhibited decreases in EAE disease severity and in vivo Th17 generation in the EAE model, demonstrating the importance of TLR2 expression during autoimmunity by lymphoid cells. A recent report found that TLR2 in conjunction with poly(ADP-ribose) polymerase 1 expression by macrophages promoted CNS inflammation and IL-17 production during EAE (Farez et al., 2009). However, it was not clear in our model whether these effects were due to TLR2 expression on innate or adaptive immune cells. Therefore, most importantly in this study, we tested the EAE model in Rag1-deficient mice reconstituted with only CD4+ T cells. Interestingly, we found an even greater degree of protection against EAE in mice reconstituted with TLR2-deficient CD4+ T cells compared to mice receiving total Tlr2−/− bone marrow. In the CD4+ transfer model of EAE, we observed reduced IFNγ production as well in the Tlr2−/− cell-transferred animals. We previously reported that in IL-17-deficient animals, there was also a reduced Th1 response (Yang et al., 2008a), indicating a role of IL-17 in Th1 priming. However, we cannot completely rule out a role of TLR2 in Th1 cells. The results presented here clearly demonstrate that TLR2 signaling in CD4+ cells is crucial in the pathogenesis of autoimmune inflammatory disorders.
Of great interest was the result that demonstrated that Rag1−/− mice reconstituted with Tlr2−/− CD4+ T cells developed greatly decreased EAE disease. However, Rag1−/− mice reconstituted with Tlr2−/− bone marrow developed EAE normally, although with less severity, compared to WT controls. We do not know the exact mechanism to explain this discrepancy. Our current theory is that TLR2 activation in non-CD4+ cells in certain cases may actually inhibit the development of Th17 responses through the production of mediators such as IL-12 or even IL-10. Indeed, in some cases it has been shown that TLR2 activation can suppress rather than promote CNS inflammation (Stenzel et al., 2008). Therefore, in germline Tlr2−/− or Tlr2−/− bone marrow-reconstituted animals, lack of TLR2 activation in non-CD4+ cells may actually promote Th17-dependent autoimmunity. So even though T cells in these mice have a deficiency in Th17 differentiation and expansion, the lack of these inhibitory signals allows these mice to initiate EAE. When CD4+ T cells are transferred into Rag1−/− mice though, this defect in Th17 generation exhibited by Tlr2−/− T cells is amplified; Tlr2−/− T cells have a greatly reduced capacity expanding and differentiating into Th17 cells compared to their B6 counterparts.
Overall, our results demonstrate a function of TLR2 in T cells in the regulation of Th17 responses. TLR2, although traditionally associated with rapid innate-type responses, can act to enhance both the proliferation and IL-17 production by Th17, memory, and γδ T cells. Thus, our observations described here have broadened the function of TLRs to the regulation of adaptive immunity. Although whether human multiple sclerosis is initiated through TLR2-dependent mechanisms is unknown, there are possible endogenous TLR2 ligands present that may have an effect on Th17 generation, function, and maintenance. Further studies are needed to examine if antagonistic targeting of TLR2 pathway may lead to the treatment and prevention of IL-17-related disorders, such as that seen in MS, arthritis, or colitis.
Experimental Procedures
Mice
C57BL/6, TLR2−/−, B6SJL, and Rag1−/− mice were purchased from The Jackson Laboratory. Il23a−/− mice came from NIH Mutant Mouse Regional Resource Centers. All mice were housed under SPF conditions at the University of Texas M.D. Anderson Cancer Center, Houston, Texas. All experiments were performed with mice 6–10 weeks old with protocols approved by the Institutional Animal Care and Use Committee of the M.D. Anderson Cancer Center.
T cell isolation and differentiation
Naïve CD4+ T cells were purified, activated and analyzed as previously described (Yang et al., 2008b). All antibodies utilized were purchased from Bio X Cell. Additionally, Pam3Cys-Ser- (Lys)4-trihydrochloride (Pam3Cys) (Alexis Biochemicals) or FSL-1 (Pam2CGDPKHPKSF) (Invivogen) was added to some of the treatment groups. Memory CD4+ T cells (CD4+CD62LloCD44hiCD25−) were derived from spleen and lymph nodes and then sorted on a FACSAria. γδ T cells were harvested from spleen, lung, and lymph nodes and further purified using a γδ T cell isolation kit and autoMACS (Miltenyi). Thymus-derived, embryonic γδ T cell were isolated in a similar manner from timed breeders at g18.5. Gene expression analysis was performed with an iCycler Optical System with IQ™ SYBR green from Bio-Rad. Gene expression was normalized to the Actb reference gene. Specific primer sequences can be found in supplementary experimental procedures.
Bone marrow reconstitution
Bone marrow was harvested from WT (C57BL/6 or B6SJL) and Tlr2−/− mice, filtered, and washed 3X in PBS. Cells were then counted and 5 × 106 cells were injected in the tail veins of sub-lethally irradiated Rag1-deficient animals. Six weeks after injection, mice were checked for reconstitution by blood analysis.
CD4+ T cell transfer
Total CD4+ T cell populations were isolated from the spleens and lymph nodes of WT and TLR2-deficient animals. CD4+ T cells were further purified using CD4 microbeads and autoMACS (Miltenyi). Cells were counted and washed 3X in PBS and then injected i.v. into tail veins of Rag1−/− mice at 5 × 106 cells per animal. EAE was induced or KLH was immunized the day following CD4+ transfer.
Induction of EAE
EAE disease was induced using CFA and analyzed as previously described (Nurieva et al., 2007). For EAE induction using LPS, CD4+ cells were transferred to Rag1−/− recipients. On the following day mice were immunized with MOG emulsified in IFA and LPS (Sigma, Escherichia coli O111:B4). From this emulsion, each mouse received a total of 150μg MOG peptide and 30μg LPS. Pertussis toxin and a second MOG immunization were administered as in the CFA adjuvant model. Clinical scores were as follows: 0 = no signs of disease, 1 = tail paralysis, 2 = wobbly gait, 3 = hind limb paralysis, 4 = forelimb paralysis, and 5 = moribund or dead. 0.5 gradations were utilized on mice exhibiting symptoms falling between two of the above listed scores.
Proliferation analysis
For Th17 differentiation, cells were stained with CFSE (CFDA; Invitrogen) prior to stimulation. Following four-day differentiation, CFSE dilution was assessed by flow cytometry. For MOG recall, splenocytes were incubated with 1, 5, and 25μg/ml MOG (Synbiosci) three days prior to eight-hour pulse with 3H-thymidine (Amersham/GE Healthcare). Analysis (cpm) was performed on a Perkin Elmer 1450 Microbeta Liquid Scintillation Counter.
ELISA
Supernatants from primary differentiation, MOG restimulated, or KLH restimulated cultures were harvested on days four and three, respectively. Restimulated cultures were harvested after overnight incubation. IL-17, IL-17F, and IFNγ cytokine production was assessed by ELISA kits (BD Biosciences and R & D Systems).
KLH immunization
For CD4+ T cell transfer experiments, total CD4+ T cells and B220+ B cells were prepared from the lymph nodes and spleen of C57BL/6 and Tlr2−/− mice by autoMACs. Cells were washed 3X in PBS and 5×106 WT or Tlr2−/− CD4+ T cells were injected i.v. along with 5×106 B cells. On the following day mice were immunized with KLH (0.5 mg/ml) emulsified in CFA (0.5 mg/ml) at the base of the tail (100 μl each mouse). Seven days after immunization, these mice were sacrificed and draining lymph nodes were plated in the presence of 50μg/ml KLH. Following three-day incubation, cells were treated with Golgi Stop (BD Biosciences) for five hours prior to intracellular cytokine staining for IL-17 and IFNγ. For the bone marrow reconstitution experiments, B6SJL (CD45.1) and Tlr2−/− (CD45.2) bone marrow was harvested and injected at a 1:1 ratio i.v. into sub-lethally irradiated Rag1-deficient mice. Six weeks after reconstitution, mice were immunized with KLH as described above. Draining lymph nodes were then harvested and CD45.1+ or CD45.2+ CD4+ T cells were sorted and then stimulated with 50μg/ml KLH for three days in the presence of irradiated WT APCs. Supernatants were then assayed for ELISA (BD-Pharmingen).
Supplementary Material
Acknowledgments
We thank the flow cytometry core facility at the M.D. Anderson Cancer Center and the entire Dong lab for their help and suggestions. This work is supported by research grants from the National Institutes of Health (NIH; to C.D. and R.I.N.). J.M.R. is a recipient of a T32 training grant from the National Cancer Institute (NCI). B.P.P. received an Odyssey Fellowship from the M.D. Anderson Cancer Center. G.J.M. was a Schissler Foundation M.D. Anderson Cancer Center Fellow in cancer research. R.I.N. was a recipient of a Scientist Development Grant from the American Heart Association (AHA). C.D. is a Leukemia & Lymphoma Society Scholar and a Trust Fellow of the MD Anderson Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blink SE, Miller SD. The contribution of gammadelta T cells to the pathogenesis of EAE and MS. Curr Mol Med. 2009;9:15–22. doi: 10.2174/156652409787314516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, Arpaia E, Mak TW, Kamradt T, Lohoff M. The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol. 2007;8:958–966. doi: 10.1038/ni1500. [DOI] [PubMed] [Google Scholar]
- Chen Q, Davidson TS, Huter EN, Shevach EM. Engagement of TLR2 does not reverse the suppressor function of mouse regulatory T cells, but promotes their survival. J Immunol. 2009;183:4458–4466. doi: 10.4049/jimmunol.0901465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottalorda A, Verschelde C, Marcais A, Tomkowiak M, Musette P, Uematsu S, Akira S, Marvel J, Bonnefoy-Berard N. TLR2 engagement on CD8 T cells lowers the threshold for optimal antigen-induced T cell activation. Eur J Immunol. 2006;36:1684–1693. doi: 10.1002/eji.200636181. [DOI] [PubMed] [Google Scholar]
- Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6:329–333. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- Dong C, Flavell RA. Cell fate decision: T-helper 1 and 2 subsets in immune responses. Arthritis Res. 2000;2:179–188. doi: 10.1186/ar85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farez MF, Quintana FJ, Gandhi R, Izquierdo G, Lucas M, Weiner HL. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat Immunol. 2009;10:958–964. doi: 10.1038/ni.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 2000;14:1693–1711. [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. TLR2 is expressed on activated T cells as a costimulatory receptor. Proc Natl Acad Sci U S A. 2004;101:3029–3034. doi: 10.1073/pnas.0400171101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Komai-Koma M, Xu D, Liew FY. Toll-like receptor 2 signaling modulates the functions of CD4+ CD25+ regulatory T cells. Proc Natl Acad Sci U S A. 2006;103:7048–7053. doi: 10.1073/pnas.0601554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- MacLeod H, Wetzler LM. T cell activation by TLRs: a role for TLRs in the adaptive immune response. Sci STKE 2007. 2007:pe48. doi: 10.1126/stke.4022007pe48. [DOI] [PubMed] [Google Scholar]
- Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Mercier BC, Cottalorda A, Coupet CA, Marvel J, Bonnefoy-Berard N. TLR2 engagement on CD8 T cells enables generation of functional memory cells in response to a suboptimal TCR signal. J Immunol. 2009;182:1860–1867. doi: 10.4049/jimmunol.0801167. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, Dong C. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien RL, Roark CL, Born WK. IL-17-producing gammadelta T cells. Eur J Immunol. 2009;39:662–666. doi: 10.1002/eji.200839120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng MY, Wang ZH, Yao CY, Jiang LN, Jin QL, Wang J, Li BQ. Interleukin 17-producing gamma delta T cells increased in patients with active pulmonary tuberculosis. Cell Mol Immunol. 2008;5:203–208. doi: 10.1038/cmi.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, Piesche M, Schroers R, Weiss E, Kirschning CJ, et al. Innate immunity mediated by TLR9 modulates pathogenicity in an animal model of multiple sclerosis. J Clin Invest. 2006;116:456–464. doi: 10.1172/JCI26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann RR, Tapping RI. Genomic variants of TLR1--it takes (TLR-) two to tango. Eur J Immunol. 2007;37:2059–2062. doi: 10.1002/eji.200737604. [DOI] [PubMed] [Google Scholar]
- Stenzel W, Soltek S, Sanchez-Ruiz M, Akira S, Miletic H, Schluter D, Deckert M. Both TLR2 and TLR4 are required for the effective immune response in Staphylococcus aureus-induced experimental murine brain abscess. Am J Pathol. 2008;172:132–145. doi: 10.2353/ajpath.2008.070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, Martin-Orozco N, Kang HS, Ma L, Panopoulos AD, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol. 2008;181:8391–8401. doi: 10.4049/jimmunol.181.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008a;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008b;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008c;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.