

Abstract
Naturally occurring strains of Newcastle disease virus (NDV) are currently being investigated in multiple clinical trials for oncolytic cancer therapy in the United States and abroad. We have previously reported, for the first time, the development of recombinant NDVs designed for enhanced cancer therapeutic efficacy. Specifically, we have shown that NDV engineered to express interleukin-2 (IL-2) generates a robust therapeutic response associated with increased tumor-specific T-cell infiltration after intratumoral administration in mice. We have now demonstrated that this therapeutic response is dependent on T cells and we have investigated the potential to focus the NDV-induced immune response toward a tumor-associated antigen (TAA) to enhance the inherent therapeutic efficacy of NDV further. We found that intratumoral treatments of tumor-bearing mice with recombinant NDV expressing a model TAA elicited an enhanced tumor-specific response, resulting in a significant increase in the number of complete tumor regressions compared with control NDV. Additionally, coadministration of NDV expressing a model TAA with NDV expressing IL-2 enhanced the TAA-directed response and led to more complete tumor regressions. Our results show that TAA-directed immunotherapy by oncolytic recombinant NDV alone or in combination with IL-2 results in an enhanced therapeutic efficacy and warrant consideration in the development of cancer therapies based on the use of oncolytic NDV.
Introduction
As a result of genetic alterations, tumors express unique antigens that can be recognized by T cells. Tumor-associated antigens (TAAs) can be tumor-specific antigens, shared antigens specific from particular cell lineages, differentiation antigens, mutated proteins, antigens derived from oncogenes or viral antigens.1 These antigens are processed and presented by major histocompatibility complex class I and major histocompatibility complex class II molecules for priming and activating CD4+ and CD8+ T cells.2,3,4 Numerous studies indicate that the immune system has the potential of eliciting a T cell–mediated tumor-specific immune response capable of large tumor destruction. Despite this fact, cancer cells, even when expressing these antigens, may not elicit a robust immune response. In some cases, cancer cells escape immune detection because they originate from normal cells and do not activate antigen-presenting cells.5 Nonactivated antigen-presenting cells that present unique cancer antigens without co-stimulation could lead to tolerance.6,7 Specifically, T cells that encounter TAAs in the absence of co-stimulation may become ignorant, anergic, or apoptotic.8,9 Therefore, although a robust TAA-specific immune response is capable of tumor eradication, the tolerance generated during oncogenic development may lead to large tumor growth.10
Although TAA tolerance presents a significant challenge to cancer immunotherapy, this barrier can be overcome. Interestingly, several viruses are strong inducers of an immune response and offer an attractive strategy for cancer therapy by stimulating the patient's immune system within the tumor in order to overcome these immunologic barriers. The ability of oncolytic viruses to induce tumor-specific immune responses has been well documented, tested in clinical trials,11 and was also recently reported by our group when intratumoral injections of a recombinant Newcastle disease virus (NDV) caused complete tumor regression in tumor-bearing mice, and protected these mice against subsequent tumor challenge with the same parental tumor cells.12 In our previous report, we found that intratumoral injection of NDV/F3aa resulted in complete regression in 20% of mice bearing subcutaneously implanted CT26 autologous colon carcinoma cells. In addition, recombinant expression of interleukin-2 (IL-2) by NDV/F3aa enhanced the therapeutic efficacy of this treatment and resulted in 60% of the mice undergoing complete regression, consistent with previous reports using HSV-expressing IL-2 (refs. 13,14). Mice that underwent complete regression in either treatment were protected from subsequent tumor rechallenge highlighting the induction of a potent and long-lasting antitumor adaptive immune response. Mice treated with NDV/F3aa-IL-2 also had increased T-cell infiltration and enhanced tumor-specific adaptive immune responses, correlating with the enhanced therapeutic response. We have now investigated whether expression of a TAA by a genetically engineered NDV could focus the NDV-induced adaptive T-cell response toward the TAA and enhance the NDV therapeutic efficacy.
The quality of the immune response to an antigen is dependent on many interrelated factors including tissue distribution, pre-existing tolerance, T-cell repertoire, and immune activation. To study the ability of NDV to induce a TAA-specific response we have used a tumor model system in which β-galactosidase (β-gal) is expressed by murine CT26 tumor cells. In this model, expression of a β-gal-derived major histocompatibility complex class I H-2 Ld epitope (amino acid sequence TPHPARIGL) by NDV allowed the evaluation of a single CD8 T-cell antigenic determinant in enhancing the regression of established CT26 tumors by NDV. This CD8 T-cell epitope has previously been reported to be an effective TAA target for the generation of T cells against CT26 colon carcinoma cells expressing β-gal.15 Because β-gal is a xenogeneic antigen, this model is an example of tumors that express unique nonself antigens such as viral antigens, mutated self–antigens,16 or frame-shift mutations.17
Results
Generation of a recombinant NDV expressing a TAA epitope (NDV/F3aa-minigal)
A complementary DNA encoding the β-gal-specific CD8 T-cell epitope TPHPARIGL (minigal) was cloned between the NDV P/V and M genes of a full-length infectious clone of NDV (NDV/F3aa) (Figure 1 ). We chose the NDV/F3aa backbone, containing a multibasic cleavage site in the F protein of the virus, based on our previous studies showing increased antitumor efficacy of this virus compared with the wild-type parental NDV (B1 strain). For proper expression by the NDV replication/transcription machinery, the minigal epitope was flanked by NDV-specific transcriptional start and stop signals.18 In addition, the epitope was expressed after an endoplasmic reticulum–insertion sequence which was known to enhance the immunogenicity of the encoded epitope by directing the epitope to the endoplasmic reticulum, most likely enhancing antigen presentation.19,20,21 Endoplasmic reticulum–targeted CD8 T-cell epitopes are at least as immunogenic as the full-length proteins from which they are derived.22 The recombinant NDV/F3aa-minigal was rescued as previously described,18 plaque purified, and the growth kinetics of this virus in A549 cells were comparable to the previously reported recombinant NDV/F3aa-IL-2 (Figure 1b).
Figure 1.
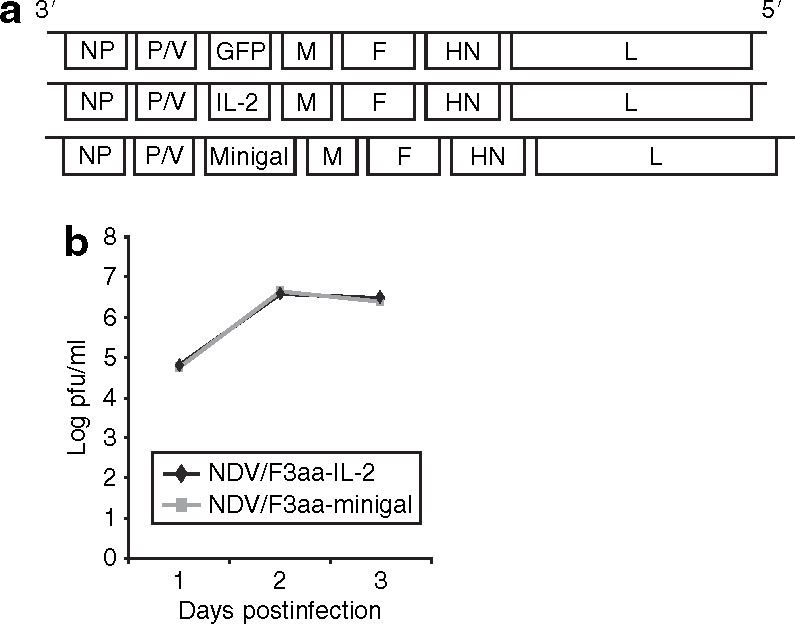
Recombinant Newcastle disease virus (NDV) used in our studies. (a) Schematic representation of NDV genomes. Green fluorescent protein (GFP), interleukin-2 (IL-2), and minigal were inserted into the Xba I site created between the P and M genes of pT7NDV/F3aa as an extra transcriptional unit. The minigal tumor-associated antigen corresponding to the major histocompatibility complex class I H-2 Ld epitope of β-gal (amino acid sequence TPHPARIGL) was expressed after an endoplasmic reticulum–insertion sequence in NDV/F3aa-minigal (not drawn to scale). (b) Growth kinetics of NDV/F3aa-minigal and NDV/F3aa-IL-2 was tested in A549 cells. A549 cells were infected at a multiplicity of infection of 0.1 and supernatants were collected on days 1, 2, and 3 after infection. pfu, plaque-forming units.
NDV oncolytic efficacy is T-cell dependent
We previously reported that oncolytic therapy with recombinant NDV/F3aa expressing IL-2 associated with an increase in the number of T-cell tumor infiltrates and in the adaptive antitumor immune response.12 Based on our previous results, recent publications,23 and because IL-2 is a known T-cell growth factor, we investigated whether NDV/F3aa-IL-2 oncolytic therapy is dependent on T cells. For this purpose, we tested the therapeutic efficacy of recombinant NDV/F3aa-IL-2 in a nude mouse model. Nude mice are severely deficient in T cells and produce only small numbers of T cells due to a genetic mutation in the Foxn1 gene that causes a developmental deterioration of the thymus.24 Nude mice maintain normal numbers of macrophages, natural killer (NK) cells, antigen-presenting cell functions, and normal complement activity. BALB/c nude mice were subcutaneously injected with the 5 × 105 syngeneic CT26 colon carcinoma cells. The injected cells were allowed to establish solid tumors until tumors reached between 5 and 8 mm in diameter (~12 days). Mice were then treated with 1 × 107 plaque-forming units (pfu) of recombinant NDV every other day for a total of four intratumoral injections. As seen in Figure 2 , nude mice treated with NDV/F3aa-IL-2 did not undergo a single complete regression and tumor growth was not impaired as compared to phosphate-buffered saline–treated nude mice. Same results were obtained in nude mice treated with NDV/F3aa-minigal virus, which in this case was used as a control virus, because the used CT26 cells did not express β-gal. This is in contrast to our previously reported results where 6/10 immunocompetent BALB/c mice underwent complete regression following NDV/F3aa-IL-2 intratumoral treatments and 2/10 mice following NDV/F3aa treatment.12 These results highlight the dependency for T-cell function in both treatment groups and illustrate that T cells are critical for therapeutic efficacy.
Figure 2.
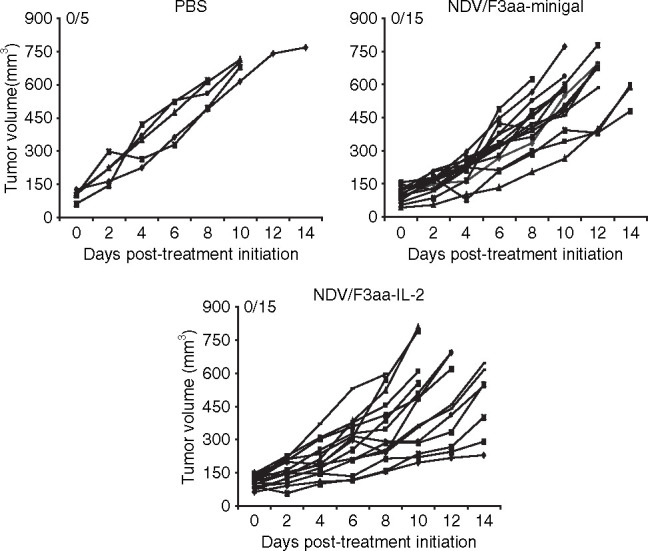
Treatment of tumor-bearing nude mice with Newcastle disease virus (NDV). Nude mice were subcutaneously implanted with 5 × 105 CT26 cells. When tumors reached a size of 5–8 mm in diameter (~12 days), mice were intratumorally treated with phosphate-buffered saline (PBS), 1 × 107 plaque-forming units of NDV/F3aa-minigal, or NDV/F3aa-IL-2 every 2 days for a total of four injections. Tumor volumes were monitored every 2 days and mice were killed when tumor diameters were more than 18 mm in any dimension. Numbers on graphics indicate number of mice undergoing complete tumor regression versus total number of mice. IL-2, interleukin-2.
Therapeutic antitumor efficacy of NDV/F3aa-minigal in mice bearing CT26 tumors expressing β-gal
Knowing that the oncolytic properties in vivo of NDV/F3aa are T-cell dependent, we next investigated whether expression of a model TAA by NDV/F3aa could enhance its antitumor efficacy after intratumoral injections of immunocompetent wild-type BALB/c mice. BALB/c mice were subcutaneously implanted with 5 × 105 CT26.CL25 or CT26.wt syngeneic cancer cells. CT26.CL25 cells represent a tumor cell line derived from CT26.wt cells but expressing β-gal under the long-terminal repeat of the Moloney murine leukemia virus complementary DNA.25 After ~12 days when tumors were between 5 and 8 mm in diameter, mice were intratumorally treated with 1 × 107 pfu of NDV/F3aa-GFP, NDV/F3aa-minigal, NDV/F3aa-IL-2 or phosphate-buffered saline every other day for a total of four injections. As seen in Figure 3 , intratumoral injection of NDV/F3aa-minigal resulted in higher percentage of complete regressions in mice harboring CT26.CL25 tumors (60%) compared to treatment with NDV/F3aa-GFP control virus (20%) (P = 0.0253). This enhanced response is absent in CT26.wt tumor-bearing mice treated with NDV/F3aa-minigal. Importantly, all of the mice that had complete tumor regressions were protected when rechallenged on the contralateral flank 60 days post-tumor treatment initiation with 5 × 105 of the same tumor cells as they originally bore (data not shown). Both NDV/F3aa-minigal- and NDV/F3aa-GFP-treated CT26.wt tumor-bearing mice that had underwent complete tumor regressions were capable of inducing protective immunity against CT26.wt cells suggesting that inherent tumor-specific antigens present on this cell line are capable of mediating adaptive immune recognition and challenge protection. This result highlights the induction of a potent adaptive immune response capable of rejecting tumor challenge.
Figure 3.

Treatment of tumors expressing a model tumor-associated antigen (TAA) with Newcastle disease virus (NDV). Five BALB/c mice per group were subcutaneously implanted with either 5 × 105 CT26.wt or CT26.CL25 cells. When tumors reached a size of 5–8 mm in diameter (~12 days), mice were intratumorally treated with phosphate-buffered saline (PBS), 1 × 107 plaque-forming units of NDV/F3aa-GFP, NDV/F3aa-minigal, or NDV/F3aa-IL-2 every 2 days for a total of four injections. Tumor volumes were monitored every 2 days during the experimental period. Numbers on graphics indicate number of mice undergoing complete tumor regression versus total number of mice. GFP, green fluorescent protein; IL-2, interleukin-2.
NDV/F3aa-minigal induces tumor-specific T-cell responses
To assess the induction of a tumor-specific cellular immune response, three mice per group were treated as in Figure 3, and the tumor-draining lymph node (LN) cells were harvested and co-cultured with irradiated CT26.CL25 cells. Supernatants from these co-cultures were assayed for interferon-γ (IFN-γ) release and measured by enzyme-linked immunosorbent assay. The results show that after 3 days of co-culture with CT26.CL25 cells, LN cells from NDV/F3aa-minigal-treated mice produced significantly higher levels of IFN-γ than LN cells from phosphate-buffered saline or NDV/F3aa-GFP-treated mice (Figure 4a ). However, LN cells from mice treated with NDV/F3aa-IL-2 produced more IFN-γ when co-cultured with CT26.CL25 cells than LN cells from mice treated with NDV/F3aa-minigal. The IFN-γ response correlated with the increased percentage of complete tumor regressions following NDV/F3aa-IL-2 treatment compared with NDV/F3aa-minigal treatment (Figure 3).
Figure 4.
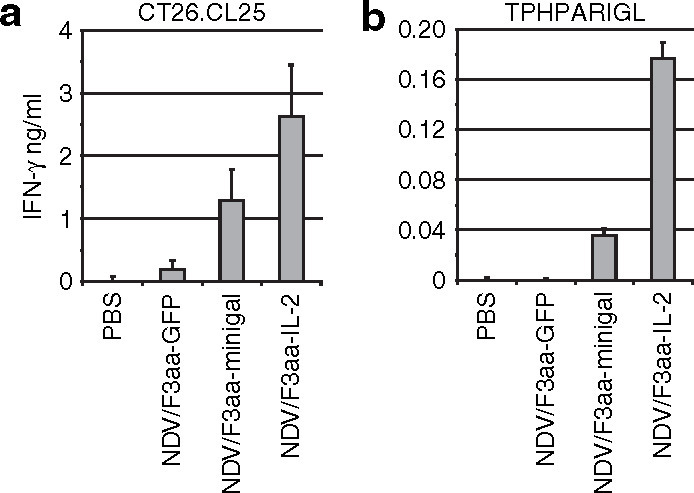
Induction of interferon-γ (IFN-γ)-secreting T cells in Newcastle disease virus (NDV)-treated tumor-bearing mice. Animals bearing CT26.CL25 tumors were treated as in Figure 3. Tumor-draining lymph nodes were pooled from three mice per group on day 14 post-treatment initiation. (a) Lymph node (LN) cells were dissociated and co-cultured with irradiated CT26.CL25 cells or plated alone and assayed for IFN-γ production 3 days later (mean ± SD, n = 3). LN cells in media alone did not produce detectable IFN-γ (data not shown). (b) LN cells were cultured with 5 µg/ml of minigal peptide or control peptide and assayed for IFN-γ production 3 days later. IFN-γ production from control peptide was undetectable (data not shown). GFP, green fluorescent protein; PBS, phosphate-buffered saline.
In a separate experiment, LN cells were cultured with the β-gal TAA peptide minigal (TPHPARIGL) or with an irrelevant control peptide (AMQMLKETI), and IFN-γ production was measured from supernatants after 3 days of incubation. As seen in Figure 4b, LN cells from NDV/F3aa-minigal-treated mice had a minigal-specific induction of IFN-γ production. This response was undetectable when cultured with the control peptide (data not shown). Again, LN cells from CT26.CL25 tumor-bearing mice that were treated with NDV/F3aa-IL-2 also produced a minigal-specific immune response which was more potent than this in NDV/F3aa-minigal-treated mice.
Co-administration of NDV/F3aa-minigal and NDV/F3aa-IL-2 to CT26.CL25 tumor-bearing mice results in 90% of tumor regression
Next, we wanted to determine whether co-administration of NDV/F3aa-minigal and NDV/F3aa-IL-2 had a synergistic therapeutic effect against CT26.CL25 tumors expressing β-gal. Ten mice per group were implanted with CT26.CL25 tumor cells and treated with NDV/F3aa-minigal alone or in combination with NDV/F3aa-IL-2. As control virus, we used NDV/F3aa-GFP. As shown in Figure 5 , treatment of CT26.CL25 tumor–bearing mice with NDV/F3aa-minigal again resulted in a statistically significant number of tumor regressions than treatment with control NDV/F3aa-GFP (P = 0.0015). Moreover, co-treatment with NDV/F3aa-minigal and NDV/F3aa-IL-2 resulted in 9/10 mice undergoing complete regressions compared with 5/10 mice treated with NDV/F3aa-minigal alone or 5/10 in the NDV/F3aa-GFP and NDV/F3aa-IL-2 control co-treatment group (P = 0.0114). All of the mice that had complete tumor regressions were protected when rechallenged on the contralateral flank 60 days post-tumor treatment initiation with 5 × 105 CT26.CL25 cells (Figure 5b). On day 140, five mice were challenged with a syngeneic renal cell carcinoma cell line (Renca) as a control for specificity to CT26 cells. All of the CT26.CL25-protected mice (5/5) and naive control mice (5/5) developed visible tumors between 15 and 20 days after Renca cell implantation (data not shown).
Figure 5.

Newcastle disease virus (NDV) combination therapy in immunocompetent BALB/c mice. Ten mice per group were subcutaneously implanted with 5 × 105 CT26.CL25 cells. When tumors reached a size of 5–8 mm in diameter, mice were intratumorally treated with phosphate-buffered saline (PBS), 1 × 107 plaque forming units of NDV/F3aa-GFP, NDV/F3aa-minigal, NDV/F3aa-minigal + NDV/F3aa-GFP, NDV/F3aa-GFP + NDV/F3aa-IL-2, or NDV/F3aa-minigal + NDV/F3aa-IL-2 every 2 days for a total of four injections. (a) Tumor volumes were monitored every 2 days during experimental period. (b) Percentage of complete regressions per treatment group is indicated. Surviving mice were challenged with CT26.CL25 60 days post-treatment initiation and percentage protection is also shown. GFP, green fluorescent protein.
Co-administration of NDV/F3aa-minigal and NDV/F3aa-IL-2 to CT26.CL25 tumor–bearing mice increases the TAA-specific T-cell response
To assess the induction of a tumor-specific immune response by co-administration of NDV/F3aa-minigal and NDV/F3aa-IL-2, LN cells from treated mice were harvested and co-cultured with either CT26.wt or CT26.CL25 cells and supernatants were measured for IFN-γ release. The results show that after 3 days of coculture with β-gal-expressing tumor cells (CT26.CL25), LN cells from mice that were treated with NDV/F3aa-minigal and NDV/F3aa-IL-2 together produced significantly more IFN-γ than treatment with one of these viruses plus the control NDV/F3aa-GFP alone. This synergistic effect was specific for the minigal antigen, as it was not seen when CT26.wt cells were used as stimulator cells (Figure 6a ) and it was also observed when LN cells were stimulated only by minigal peptide (Figure 6b). To negate the contribution of IFN-γ by NK cells, we repeated this assay with LN cells depleted of NK cells by magnetic cell sorting and separation and observed similar results (Figure 6c). Additionally, by fluorescence-activated cell-sorting analysis we observed internal IFN-γ cytokine staining from CD3+CD8+ T cells 14 hours after co-culturing with CT26.CL25 cells (Figure 6d). Our findings suggest that the T-cell response to CT26.CL25 tumor cells induced by NDV treatment can be increased by expression of IL-2 and of a known CD8 T-cell antigen present on the tumors, resulting in enhanced tumor regression.
Figure 6.

Induction of interferon- γ (IFN-γ)-secreting T cells after Newcastle disease virus (NDV) combination therapy. Animals were treated as in Figure 5. Tumor-draining lymph nodes were pooled from three mice per group on day 14 post-treatment initiation. (a) Lymph node (LN) cells were dissociated and co-cultured with irradiated CT26.wt or CT26.CL25 cells or plated alone and assayed for IFN-γ production 3 days later. LN cells in media alone did not produce detectable IFN-γ (data not shown). (b) LN cells were cultured with 5 µg/ml of minigal peptide or control peptide and assayed for IFN-γ production 3 days later. IFN-γ production from control peptide was undetectable (data not shown). (c) Tumor-draining lymph nodes were pooled and depleted of natural killer (NK) cells using anti-NK (DX5) MicroBeads according to manufacture's protocol. NK-depleted LN cells were co-cultured with irradiated CT26.CL25 cells and assayed for IFN-γ production 3 days later. (d) Tumor-draining LN cells were pooled and co-cultured with irradiated CT26.CL25 cells. After 14 hours of incubation, cells were stained with anti-CD3, anti-CD8, and anti-IFN-γ antibodies, and analyzed by fluorescence-activated cell-sorting.
Discussion
Here, we investigate the use of reverse genetics to enhance the therapeutic antitumor efficacy of NDV by eliciting a TAA-focused immune response. We first demonstrate the dependence of T cells for NDV oncolytic therapy in the CT26 murine colon carcinoma tumor model. We previously reported the induction of an adaptive immune response against tumor cells resulting in 20% of CT26 tumor-bearing mice undergoing complete tumor regression following intratumoral injections of NDV/F3aa. Importantly, the induced antitumor adaptive response in surviving treated mice was capable of protecting from subsequent tumor rechallenge.12 This is in contrast to the results in Figure 2 where nude mice subcutaneously implanted with CT26 cells did not undergo tumor regressions upon NDV oncolytic therapy (0/15 mice). Additionally, we found that tumor-bearing nude mice treated with NDV/F3aa expressing the T-cell growth factor IL-2 were also unresponsive to treatment therapy (0/15). Although we cannot exclude that the role of T cells in these experiments might be supportive of other cellular functions different from direct lysis of tumor cells, we conclude that the presence of T cells is necessary for antitumor efficacy by NDV, and highlights the importance of T cells in NDV oncolytic therapy. This is in contrast to other virus oncolytic agents, such as vesicular stomatitis virus, which can mediate destruction of tumors in mice in the absence of T cells.26 This is probably due to the more robust cytolytic activity and higher levels of replication of vesicular stomatitis virus in tumor cells as compared with NDV.
Next, we investigated the potential to enhance the T cell–mediated oncolytic efficacy of NDV by expression of a TAA in NDV, which would result in overexpression of this TAA in NDV-infected cells. This approach resulted in enhanced therapeutic efficacy of NDV against CT26 tumors expressing the same TAA. Specifically, intratumoral inoculations of oncolytic NDV expressing a model TAA (minigal) induced an enhanced TAA-specific immune response resulting in an increased percentage of regressions of tumors expressing the same TAA (CT26.CL25) (5 of 10 mice, compared with 2 of 10 mice in NDV/F3aa-GFP control-treated mice). Furthermore, tumor-bearing mice that did not express the model TAA (CT26.wt tumor-bearing mice) responded similar to the treatment with NDV/F3aa-minigal or with NDV/F3aa-GFP, demonstrating the specificity of the response for the TAA. We also found that co-expression of the T-cell growth factor IL-2 and TAA by NDV significantly improved the T cell–directed therapeutic response, resulting in 9 of 10 mice undergoing complete tumor regression (P = 0.0114). These mice had a significant increase in their tumor-specific T-cell immune response and an increased T cell–specific response to the β-gal TAA peptide. This enhanced therapeutic response might be a result of an increase in T-cell infiltration within the tumor of mice treated with NDV/F3aa-IL-2, resulting in increased antitumor-specific adaptive responses, as we reported previously12 or of a reverse of anergy in T cells upon IL-2 signaling.27 Nonetheless, the increased TAA-specific adaptive response upon IL-2 treatment translates into an increased therapeutic response by NDV, consistent with a previous report observed using poxvirus TAA-directed therapy.28 Interestingly, high levels of IFN-γ after incubation with CT26.wt cells were produced by LN cells derived from CT26.CL25 tumor–bearing mice treated with NDV/F3aa-minigal. We speculate that this high level of IFN-γ production can be explained by a broadening effect that others have observed when immunizing against a specific antigen. For example, Markiewicz et al. have shown that cured mice vaccinated with p815 tumor peptide rejected a tumor-derived cell line that did not express the vaccine peptide.29 Also, mice vaccinated with DNA encoding her-2/neu rejected her-2/neu negative tumors in 50% of mice.30 This phenomenon, referred to as antigen cascade, has also been observed in several other preclinical and clinical reports.31,32,33,34
NDV possesses numerous characteristics making it an ideal vaccine vector and is recognized as an effective vaccine vector capable of eliciting a potent immune response targeted to the encoded vaccine antigens. The ability of recombinant NDV vectors to be used as a prophylactic vaccine vector has been harnessed by several groups and used for vaccination against respiratory syncytial virus,35 highly pathogenic avian influenza virus,36,37,38 simian immunodeficiency virus,39 human parainfluenza virus type 3,40 infectious bursal disease virus,41 as well as severe acute respiratory syndrome–associated coronavirus.42 These studies have found that NDV vectors expressing foreign antigens are capable of inducing protective immunity against several pathogens in multiple animal models including murine, avian, and nonhuman primates. Importantly, NDV vaccine vectors induced both humoral and cellular immunity against expressed recombinant antigens. Recombinant virus–based immunotherapy is recognized as one of the most potent inducers of TAA-directed CTLs.43 These tumor-specific CTL-mediated responses are traditionally sought in therapeutic cancer development as well as cancer vaccines, and are important in mediating antitumor responses in vivo. 44 Oncolytic NDV has demonstrated therapeutic efficacy in several clinical trials and administration of ex vivo NDV-infected oncolysates to cancer patients results in an increased tumor-specific CD8+ T-cell response and is associated with prolonged survival.11 While generating an antigen-specific immune response may more easily translate into prophylactic therapies for the prevention of a number of cancers, the necessity for a therapeutic agent given subsequent to cancer diagnosis is traditionally the focus of cancer therapy. Here, we demonstrate the ability to direct a targeted TAA immune response by recombinant NDV encoding a model TAA. Virus-based cancer vaccines targeting specific TAAs for clinical cancer therapy have entered clinical trials and have reported statistically significant therapeutic responses.45 However, the presence of pre-existing neutralizing antibodies to some of these viral vectors is a major concern for their future development. This concern is less important in NDV-TAA vaccine therapy due to the low prevalence of prior exposure in humans to this avian paramyxovirus. NDV is antigenically distinct from common human pathogens and vaccines, making it immunogenic to the general human population without the concern of pre-existing neutralizing antibodies. However, NDV causes hemagglutination and binds to sialic acid, thereby possibly limiting the effectiveness of intravenous delivery. For this reason, we investigated intratumoral injections of oncolytic NDV as our delivery route. Nonetheless, intravenous delivery of NDV has demonstrated therapeutic efficacy in both preclinical and clinical studies, and may be a possible route of delivery for some forms of cancer.
Previously, we reported for the first time the use of reverse genetics to enhance the cancer therapeutic efficacy of oncolytic NDV. We have now investigated the potential to enhance the therapeutic efficacy of recombinant NDV by TAA-directed therapy. NDV encoding a model CD8 T-cell TAA epitope elicited a targeted TAA-directed adaptive immune response resulting in a statistically significant increase in therapeutic efficacy. Additionally, this response could be further enhanced by co-treatment with NDV expressing IL-2, resulting in 90% of mice undergoing complete tumor regression in our tumor model system. The results presented here are the first to demonstrate the ability of recombinant NDV expressing a TAA to be used as a therapeutic cancer vaccine vector.
Materials and Methods
Cell culture. The CT26.wt and CT26.CL25 cell lines were generously provided by Nicholas P. Restifo (National Cancer Institute, National Institutes of Health, Bethesda, MD) and were maintained in Rosewell Park Memorial Institute-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 100 µg/ml streptomycin, 100 µg/ml penicillin, 0.03% L-glutamine, and 400 µg/ml G418. A549 and Madin-Darby bovine kidney cells are maintained in Dulbecco's modified Eagle's medium and rich organic medium, respectively. Chicken embryo fibroblasts cells are prepared as previously described and maintained in minimum essential medium. Dulbecco's modified Eagle's medium, minimum essential medium, and rich organic medium are supplemented with 10% fetal bovine serum, 100 µg/ml streptomycin, and 100 µg/ml penicillin.
Recombinant NDV. The NDV complementary DNA sequence was derived from the Hitchner B1 lentogenic strain, which is commonly used as a live attenuated vaccine in chickens and recently used in a phase I/II oncolytic clinical trial.46 The recombinant NDV viruses were generated as previously described47 and sequenced by reverse transcription PCR for fidelity. Viruses are plaque purified and grown in 10-day old pathogen-free chicken eggs (Charles River Laboratories, SPAFAS). Virus stock preparations are tested for contamination by streaking them on sheep blood agar plates and incubating them at 37 °C overnight (data not shown). We engineered rNDV/F3aa to express the model TAA, GFP, and IL-2 from a gene cassette insert as diagramed in Figure 1. Virus preparations were titrated by plaque purification in Madin-Darby bovine kidney cells, and growth curves were assessed by immunofluorescence in A549 cells using a polyclonal antibody against NDV.
Animal studies. All procedures involving animals followed National Institutes of Health guidelines and were approved by and performed according to specific guidelines of the Institutional Animal Care and Use Committee of Mount Sinai School of Medicine. Six-week-old female BALB/c mice were purchased from Taconic Farms and housed in a pathogen-free environment. BALB/c wild type or BALB/c nude mice were subcutaneously implanted with either 5 × 105 CT26.wt or CT26.CL25 cells. When tumors reached a size of 5–8 mm in diameter (~12 days), mice were intratumorally treated with phosphate-buffered saline or 1 × 107 pfu of the indicated virus. Tumor volumes were monitored every other day using a digital caliper in two dimensions. Tumor volumes were calculated using the following formula: tumor volume (V) = 4/3 × π × S2/2 × L/2, where S is the smallest measured diameter and L is the larger diameter. Mice were intratumorally injected with 1 × 107 pfu of recombinant NDV (in 100 µl) every other day for four injections total. Mice in the co-treatment group received 1 × 106 pfu of the NDV/F3aa-IL-2 or NDV/F3aa-GFP, supplemented with 9 × 106 pfu of the co-treatment virus in a total volume of 100 µl. Animals were killed when tumor size reached 18 mm in any dimension or at defined experimental time points.
IFN-γ response assay. Tumor-draining LNs were dissected from three mice per group on day 14 post-treatment initiation and manually dissociated into a single-cell suspension. Approximately 250,000 LN cells were then seeded into 96 wells in triplicate and co-cultured with irradiated CT26.wt or CT26.CL25 cells at a ratio of 5:1. Supernatants were collected after 3 days of incubation and release of IFN-γ was assayed by enzyme-linked immunosorbent assay kit (R&D Systems) following the supplier's protocol. Peptide-specific IFN-γ response was measured by incubating 250,000 LN cells per group with 5 µg/ml of either the minigal-specific peptide (TPHPARIGL) or a control peptide (AMQMLKETI from human immunodeficiency virus). Supernatants were collected after 3 days of incubation and IFN-γ was assayed by enzyme-linked immunosorbent assay kit (R&D Systems) following the supplier's protocol. Where indicated, LN cells from two mice per treatment group were depleted of NK cells by MicroBeads separation using DX5 anti-NK Abs according to manufacture's protocol (Miltenyi Biotec) and assayed for IFN-γ upon co-culture with irradiated CT26.CL25 cells for 3 days.
Intracellular IFN-γ staining. Purified LN cells from two mice per treatment group were co-cultured with irradiated CT26.CL25 cells for 14 hours in the presence of GolgiStop (BD Pharmingen). At the end of the incubation, cells were collected, stained with CD3 and CD8 (17A2 and 53-6.7, respectively, BD Pharmingen), and then permeabilized with Cytofix/Cytoperm (BD Pharmingen) followed by anti-IFN-γ staining (XMG1.2, eBiosciences). Data were analyzed by flow cytometry in a Cytomics FC500 machine (Beckman Coulter).
Acknowledgments
This work was supported in part by the National Institutes of Health (NIH) fellowship F31CA110209 and F31AI056678 (to A.V. and M.A.C., respectively), the NIH training grant T32AI07647 (to A.V.), NIH grants (to A.G.-S.), and the National Institute of Allergy and Infectious Diseases grant U19AI62623 (Center to Investigate Viral Immunity and Antagonism). We thank Nicholas P. Restifo for the CT26.wt and CT26.CL25 cell lines and Richard Cadagan for excellent technical assistance.
Footnotes
published online 19 August 2008
References
- 1.Van den Eynde BJ, van der Bruggen P. T cell defined tumor antigens. Curr Opin Immunol. 1997;9:684–693. doi: 10.1016/s0952-7915(97)80050-7. [DOI] [PubMed] [Google Scholar]
- 2.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188:2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golumbek PT, Lazenby AJ, Levitsky HI, Jaffee LM, Karasuyama H, Baker M. Treatment of established renal cancer by tumor cells engineered to secrete interleukin-4. Science. 1991;254:713–716. doi: 10.1126/science.1948050. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs EJ, Matzinger P. Is cancer dangerous to the immune system. Semin Immunol. 1996;8:271–280. doi: 10.1006/smim.1996.0035. [DOI] [PubMed] [Google Scholar]
- 6.Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 9.Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: turning on newborn T cells with dendritic cells. Science. 1996;271:1723–1726. doi: 10.1126/science.271.5256.1723. [DOI] [PubMed] [Google Scholar]
- 10.Greten TF, Jaffee EM. Cancer Vaccines. J Clin Oncol. 1999;17:1047–1060. doi: 10.1200/JCO.1999.17.3.1047. [DOI] [PubMed] [Google Scholar]
- 11.Batliwalla FM, Bateman BA, Serrano D, Murray D, Macphail S, Maino VC. A 15-year follow-up of AJCC stage III malignant melanoma patients treated postsurgically with Newcastle disease virus (NDV) oncolysate and determination of alterations in the CD8 T cell repertoire. Mol Med. 1998;4:783–794. [PMC free article] [PubMed] [Google Scholar]
- 12.Vigil A, Park MS, Martinez O, Chua MA, Xiao S, Cros JF. Use of reverse genetics to enhance the oncolytic properties of Newcastle disease virus. Cancer Res. 2007;67:8285–8292. doi: 10.1158/0008-5472.CAN-07-1025. [DOI] [PubMed] [Google Scholar]
- 13.Tung C, Federoff HJ, Brownlee M, Karpoff H, Weigel T, Brennan MF. Rapid production of interleukin-2-secreting tumor cells by herpes simplex virus-mediated gene transfer: implications for autologous vaccine production. Hum Gene Ther. 1996;7:2217–2224. doi: 10.1089/hum.1996.7.18-2217. [DOI] [PubMed] [Google Scholar]
- 14.Carew JF, Kooby DA, Halterman MW, Kim SH, Federoff HJ, Fong Y. A novel approach to cancer therapy using an oncolytic herpes virus to package amplicons containing cytokine genes. Mol Ther. 2001;4:250–256. doi: 10.1006/mthe.2001.0448. [DOI] [PubMed] [Google Scholar]
- 15.Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J Immunol. 1993;151:3971–3980. [PubMed] [Google Scholar]
- 16.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 17.Townsend A, Ohlen C, Rogers M, Edwards J, Mukherjee S, Bastin J. Source of unique tumour antigens. Nature. 1994;371:662. doi: 10.1038/371662a0. [DOI] [PubMed] [Google Scholar]
- 18.Nakaya T, Cros J, Park MS, Nakaya Y, Zheng H, Sagrera A. Recombinant Newcastle disease virus as a vaccine vector. J Virol. 2001;75:11868–11873. doi: 10.1128/JVI.75.23.11868-11873.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685–4692. [PMC free article] [PubMed] [Google Scholar]
- 20.Minev BR, McFarland BJ, Spiess PJ, Rosenberg SA, Restifo NP. Insertion signal sequence fused to minimal peptides elicits specific CD8+ T-cell responses and prolongs survival of thymoma-bearing mice. Cancer Res. 1994;54:4155–4161. [PMC free article] [PubMed] [Google Scholar]
- 21.Ciernik IF, Berzofsky J, Carbone DP. Mutant oncopeptide immunization induces CTL specifically lysing tumor cells endogenously expressing the corresponding intact mutant p53. Hybridoma. 1995;14:139–142. doi: 10.1089/hyb.1995.14.139. [DOI] [PubMed] [Google Scholar]
- 22.Eisenlohr LC, Bacik I, Bennink JR, Bernstein K, Yewdell JW. Expression of a membrane protease enhances presentation of endogenous antigens to MHC class I-restricted T lymphocytes. Cell. 1992;71:963–972. doi: 10.1016/0092-8674(92)90392-p. [DOI] [PubMed] [Google Scholar]
- 23.Zhao H, Janke M, Fournier P, Schirrmacher V. Recombinant Newcastle disease virus expressing human interleukin-2 serves as a potential candidate for tumor therapy. Virus Res. 2008;136:75–80. doi: 10.1016/j.virusres.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 24.Fogh J, Giovanella BC. The Nude Mouse in Experimental and Clinical Research. 1978 [Google Scholar]
- 25.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J Immunol. 1995;154:4685–4692. Academic: New York. [PMC free article] [PubMed] [Google Scholar]
- 26.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz RH. Models of T cell anergy: is there a common molecular mechanism. J Exp Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bronte V, Tsung K, Rao JB, Chen PW, Wang M, Rosenberg SA. IL-2 enhances the function of recombinant poxvirus-based vaccines in the treatment of established pulmonary metastases. J Immunol. 1995;154:5282–5292. [PMC free article] [PubMed] [Google Scholar]
- 29.Markiewicz MA, Fallarino F, Ashikari A, Gajewski TF. Epitope spreading upon P815 tumor rejection triggered by vaccination with the single class I MHC-restricted peptide P1A. Int Immunol. 2001;13:625–632. doi: 10.1093/intimm/13.5.625. [DOI] [PubMed] [Google Scholar]
- 30.Pilon SA, Kelly C, Wei WZ. Broadening of epitope recognition during immune rejection of ErbB-2-positive tumor prevents growth of ErbB-2-negative tumor. J Immunol. 2003;170:1202–1208. doi: 10.4049/jimmunol.170.3.1202. [DOI] [PubMed] [Google Scholar]
- 31.Chakraborty M, Abrams SI, Coleman CN, Camphausen K, Schlom J, Hodge JW. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004;64:4328–4337. doi: 10.1158/0008-5472.CAN-04-0073. [DOI] [PubMed] [Google Scholar]
- 32.Cavacini LA, Duval M, Eder JP, Posner MR. Evidence of determinant spreading in the antibody responses to prostate cell surface antigens in patients immunized with prostate-specific antigen. Clin Cancer Res. 2002;8:368–373. [PubMed] [Google Scholar]
- 33.Gulley JL, Arlen PM, Bastian A, Morin S, Marte J, Beetham P. Combining a recombinant cancer vaccine with standard definitive radiotherapy in patients with localized prostate cancer. Clin Cancer Res. 2005;11:3353–3362. doi: 10.1158/1078-0432.CCR-04-2062. [DOI] [PubMed] [Google Scholar]
- 34.Butterfield LH, Ribas A, Dissette VB, Amarnani SN, Vu HT, Oseguera D. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clin Cancer Res. 2003;9:998–1008. [PubMed] [Google Scholar]
- 35.Martinez-Sobrido L, Gitiban N, Fernandez-Sesma A, Cros J, Mertz SE, Jewell NA. Protection against respiratory syncytial virus by a recombinant Newcastle disease virus vector. J Virol. 2006;80:1130–1139. doi: 10.1128/JVI.80.3.1130-1139.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park MS, Steel J, Garcia-Sastre A, Swayne D, Palese P. Engineered viral vaccine constructs with dual specificity: avian influenza and Newcastle disease. Proc Natl Acad Sci USA. 2006;103:8203–8208. doi: 10.1073/pnas.0602566103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ge J, Deng G, Wen Z, Tian G, Wang Y, Shi J. Newcastle disease virus-based live attenuated vaccine completely protects chickens and mice from lethal challenge of homologous and heterologous H5N1 avian influenza viruses. J Virol. 2007;81:150–158. doi: 10.1128/JVI.01514-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veits J, Wiesner D, Fuchs W, Hoffmann B, Granzow H, Starick E. Newcastle disease virus expressing H5 hemagglutinin gene protects chickens against Newcastle disease and avian influenza. Proc Natl Acad Sci USA. 2006;103:8197–8202. doi: 10.1073/pnas.0602461103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakaya Y, Nakaya T, Park MS, Cros J, Imanishi J, Palese P. Induction of cellular immune responses to simian immunodeficiency virus gag by two recombinant negative-strand RNA virus vectors. J Virol. 2004;78:9366–9375. doi: 10.1128/JVI.78.17.9366-9375.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bukreyev A, Huang Z, Yang L, Elankumaran S, St Claire M, Murphy BR. Recombinant newcastle disease virus expressing a foreign viral antigen is attenuated and highly immunogenic in primates. J Virol. 2005;79:13275–13284. doi: 10.1128/JVI.79.21.13275-13284.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Z, Elankumaran S, Yunus AS, Samal SK. A recombinant Newcastle disease virus (NDV) expressing VP2 protein of infectious bursal disease virus (IBDV) protects against NDV and IBDV. J Virol. 2004;78:10054–10063. doi: 10.1128/JVI.78.18.10054-10063.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DiNapoli JM, Kotelkin A, Yang L, Elankumaran S, Murphy BR, Samal SK. Newcastle disease virus, a host range-restricted virus, as a vaccine vector for intranasal immunization against emerging pathogens. Proc Natl Acad Sci USA. 2007;104:9788–9793. doi: 10.1073/pnas.0703584104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adamina M, Daetwiler S, Rosenthal R, Zajac P. Clinical applications of recombinant virus-based cancer immunotherapy. Expert Opin Biol Ther. 2005;5:1211–1224. doi: 10.1517/14712598.5.9.1211. [DOI] [PubMed] [Google Scholar]
- 44.Boon T, Cerottini JC, Van den Eynde B, van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 45.Borysiewicz LK, Fiander A, Nimako M, Man S, Wilkinson GW, Westmoreland D. A recombinant vaccinia virus encoding human papillomavirus types 16 and 18, E6 and E7 proteins as immunotherapy for cervical cancer. Lancet. 1996;347:1523–1527. doi: 10.1016/s0140-6736(96)90674-1. [DOI] [PubMed] [Google Scholar]
- 46.Freeman AI, Zakay-Rones Z, Gomori JM, Linetsky E, Rasooly L, Greenbaum E. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13:221–228. doi: 10.1016/j.ymthe.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 47.Nakaya T, Cros J, Park MS, Nakaya Y, Zheng H, Sagrera A. Recombinant Newcastle disease virus as a vaccine vector. J Virol. 2001;75:11868–11873. doi: 10.1128/JVI.75.23.11868-11873.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]