

Abstract
Novel naphthalene-modified oligonucleotides have been synthesized and characterized with respect to electron transfer chemistry. Using the Sonogashira-coupling reaction, naphthalene can be covalently anchored onto a modified uridine through an ethynyl linkage. This tethering allows for effective electronic coupling with the DNA bases, resulting in a significant red shift of the absorption bands of the naphthalenic chromophore. Modification with this chromophore does not appear to affect the overall stability and structure of the DNA. Upon selective irradiation of the naphthalene moiety at 340 nm, photoreduction of a distal electron trap, 5-bromouridine, embedded in the DNA base stack occurs. This DNA-mediated reduction from a distance was found to be significantly more efficient with substitution of 5-bromouridine towards the 5′-end than towards the 3′-end. These results support a general preference for electron transfer through DNA towards the 5′-end, irrespective of the donor. In addition, differences in efficiency of photoreduction through intrastrand and interstrand pathways are observed. For DNA-mediated reduction, as with DNA-mediated oxidation, significant differences in the charge transfer reaction are apparent that depend upon subtle differences in coupling into the DNA base stack.
Introduction
Inspired by photoinduced enzymatic repair of thymine dimers in DNA,1 intensive studies of reduction of DNA bases and reductive electron transfer (ET) through double stranded DNA have been carried out in several laboratories. An estimate of the reduction potential of DNA bases has been obtained using fluorescence quenching,2 polarography3 or pulse radiolysis experiments.4 The injection of electrons into double stranded DNA upon γ-irradiation was among the first experiments addressing reductive DNA chemistry.5 DNA-mediated reductions on electrode surfaces of pendant probes were also carried out in an effort to construct sensitive DNA-based electrochemical sensors.6 In solution, chemical modifications of oligonucleotides were required for a systematic investigation of ET processes through DNA containing donors and acceptors at given distances.7–15 Using covalently bound photoreductants and electron traps embedded in the DNA base stack, an extensive study of electron migration supported a hopping mechanism with thymine being considered as the primary electron carrier.7 Moreover, a weak distance dependence for long-range reductive DNA chemistry in solution was observed.8 Further investigation with aromatic amines as photoreducing agents gave insight into the distance, sequence and directional dependence of the negative charge migration through DNA.7 The direct comparison between electron and hole transport has also been investigated using organic14 and transition metal complexes.15 Although these processes are fundamentally distinct, they share similar characteristics, a weak distance dependence and sensitivity to perturbations in stacking. Certainly these shared characteristics reflect the dependence of both electron and hole transport on DNA base pair stacking.
Some important mechanistic features of reductive ET through DNA still remain unclear, such as the difference between intrastrand and interstrand electron migration or the influence of the directionality of reductive ET (i.e. from the 3′- to 5′-end or from the 5′- to 3′-end). Few DNA photoreducing agents have been available that efficiently reduce DNA bases and are well coupled into the DNA base stack without overlapping both strands of the duplex. In addition, excitation at wavelengths beyond the spectral region where the DNA bases themselves absorb is required to avoid any direct photolysis of DNA bases.10,16
Here we report on the synthesis and photochemistry of novel naphthalene - DNA conjugates. The naphthalene moiety, known to be a strong photoreducing agent (Eox* = − 2.48 V vs SCE)2, is electronically coupled to a modified deoxyuridine through the Sonogashira coupling reaction. As evidenced for pyrene-modified oligonucleotides,17 the incorporation of a polycyclic aromatic ring via an ethynyl linkage does not perturb the overall B-DNA duplex conformation and allows for a close contact with primarily one DNA strand, hence providing a means of probing both intra- and interstrand ET processes. Moreover, the 5-bromouridine (BrU, Ered ≈ − 2.0 V vs SCE)12, known to be a sensitive probe for DNA-mediated ET,7,8a,10 has been embedded at various positions in the DNA base stack. Upon one electron reduction, this modified pyrimidine ring is irreversibly decomposed on the nanosecond timescale.10 The decomposition of the electron trap therefore represents a signature of an electron migration process.
Based upon measurement of the BrU decomposition as a function of irradiation, then, we apply our novel naphthalene-modified oligonucleotides in exploring the distance- and directional dependence of DNA-mediated ET, as well as in delineating differences between intra- and interstrand electron migration in the DNA duplex.
Results and discussion
Design, synthesis and properties of naphthalene-modified DNA
Selecting a proper electron donor for covalent tethering to DNA is of crucial importance for the investigation of DNA-mediated ET. Indeed, besides having enough reducing power and a sufficiently long excited state lifetime to achieve DNA reduction, the electron injector should be easily derivatized to allow for covalent modification. Importantly, tethering to DNA should not perturb DNA stacking. NADH (nicotinamide adenine dinucleotide)18 derivatives were first considered as potential photoreducing agents in the present study. However, none of them were found to be suitable. The excited state of the NADH moiety is likely too short lived19 to trigger efficient ET. In contrast, the polycyclic aromatic substrates (such as naphthalene or pyrene) are known to be photostable, highly reducing upon irradiation (Eox*naphthalene = − 2.48 V vs SCE; Eox*pyrene = − 2.17 V vs SCE),2 and have long-lived excited states (τnaphthalene = 96 ns20; τpyrene = 322 ns21). A pyrenyl unit had already been connected through an ethynyl linker to the DNA backbone, resulting in a strong electronic coupling between the chromophore and the DNA base-stack.22 This conjugation was shown to have little influence on the stability of DNA conjugate, did not perturb the Watson-Crick base-pairing ability,17 and allowed for a detailed study of the excited state dynamics.23 However, the excited ethynylpyrene chromophore was not sufficiently reducing to trigger electron injection in DNA,17 in contrast to pyrene linked to an oligonucleotide via single C-C bonds.11 Nevertheless, the ethynyl linkage allows for effective electronic communication with DNA bases and its rigidity precludes electron injection beyond the anchoring site, as already observed in other systems.24
In order to gain further insight into the distance and directional dependence of DNA-mediated ET as well as the difference between intra- and interstrand charge migration, we therefore utilized new naphthalene-derivatized oligonucleotides (Figure 1). The naphthalene moiety is expected to be located in the major groove, as it replaces the methyl group naturally occurring in thymidine.25 The electron trap BrU was embedded in an AT tract as thymines appear to provide the most effective paths for reductive CT7,8a,15 For synthetic reasons, we required three different strands; the presence of a nick in the phosphate backbone of the DNA helix has no effect on the CT process.26
FIGURE 1.

Schematic representation of the new naphthalene-modified oligonucleotides used for the study of DNA-mediated ET. Number x represents the position of the BrU from the anchoring site of the naphthalene unit, i.e. for sets I and II, x = 2, 3 or 4 and for sets III and IV, x = 2.
DNA assembly sets I and III were designed to examine ET within the same strand (intrastrand), towards either the 5′-end (set I; 3′-5′ ET) or the 3′-end (set III; 5′-3′ ET) (Figure 1). DNA assembly sets II and IV address interstrand DNA-mediated ET, towards either the 5′-end (set II; 3′-5′ ET) or the 3′-end (set IV; 5′-3′ ET).
The UV-visible absorption spectra of 1-iodonaphthalene and the naphthalene-modified DNA duplex are shown in Figure 2. The naphthalene ring displays π-π* transition bands in the far-UV region (260 – 310 nm) of the spectrum. Upon tethering to DNA (λmax abs DNA = 260 nm) through an ethynyl linker, the absorption bands are significantly red-shifted (310 – 370 nm). This suggests a more extended delocalization of the naphthalene moiety.17 Importantly, this red-shifted absorption allows for selective photoirradiation of the charge injector without irradiation of DNA.
FIGURE 2.

Absorption spectra of 1-iodonaphthalene in 50 mM TRIS-HCl, pH 7.4, containing 3% acetonitrile (red line) and naphthalene-modified DNA (black line; 2.0 μM duplex, 50 mM TRIS-HCl, pH 7.4, 100 mM NaCl). All naphthalene derivatized oligonucleotides showed identical absorption spectra.
Melting temperatures (Tm) of DNA assemblies were obtained by monitoring the characteristic DNA duplex absorption at 260 nm from 90 °C to 15 °C reversibly. The samples were heated at 90 °C for 5 minutes, then cooled slowly by 0.5 °C/min. Cooling the samples (0.5 °C/min) shows the melting curves to be superimposable. All of the synthesized duplexes show Tm values around 46 °C. Table 1 contains the Tm values for several naphthalene-DNA conjugates. Interestingly, there is almost no difference between the modified strands and the unmodified ones, i.e. duplexes not anchored to a naphthalene moiety. It should be noted that in contrast, for the pyrene-modified DNA, the melting curves of the pyrene moiety were consistent with a local structural perturbation.17 For naphthalene-modified DNA, similar changes at 340 nm could not be observed even at higher concentration (data not shown). These results clearly indicate that the presence of the naphthalene-derivatized base does not significantly affect the thermal stability of the oligonucleotides nor likely the modified DNA structure.
TABLE 1.
Melting temperatures (Tm) measured at 260 nm for several naphthalene-modified DNA and unmodified DNA (i.e. containing no naphthalene moiety). Experimental conditions: [naphthalene-modified DNA] = 1.0 μM in 50 mM TRIS-HCl, pH 7.4, 100 mM NaCl.
| Tm, C° (Modified) | Tm, C° (Unmodified) | |
|---|---|---|
| I-2 | 46.7 | 46.7 |
| II-2 | 46.7 | 46.6 |
| III-2 | 45.6 | 45.6 |
| IV-2 | 47.6 | 47.6 |
DNA-mediated ET through naphthalene-modified duplexes
To characterize the photoreduction of the distal electron trap (BrU) resulting from DNA-mediated ET, photolysis experiments on the synthetic duplexes were performed. Selective irradiation of the naphthalenic chromophore (λex = 340 nm) led to significant decomposition of the distal BrU in each set of oligonucleotides. The disappearance of the electron trap was followed by HPLC (Figure 3). A linear relation between the initial decomposition yield and the irradiation time allowed for the determination of the efficiency of BrU decomposition of each sample. In contrast to previously reported experiments with oligonucleotides containing pyrene-derivatized via an ethynyl linkage,17 here the excited state of the naphthalene electronically coupled to the uridine can reduce BrU from a distance. Although it is difficult to estimate the precise Eox* of the naphthalene moiety when tethered to DNA, it is likely that the driving force for the electron injection is higher in the case of naphthalene than with pyrene (Eox*naphthalene = − 2.48 V vs SCE;2 Eox*pyrene = − 2.17 V vs SCE;2 Ered BrU ≈ − 2.0 V vs SCE12). Based on model studies, the naphthalene excited state should also be significantly longer lived.10
FIGURE 3.
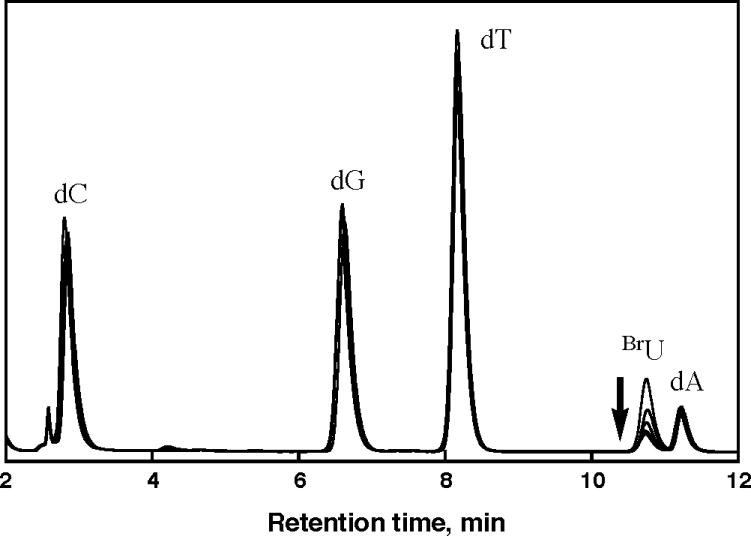
Overlaid HPLC chromatograms (λdetection = 290 nm) of nucleosides obtained after irradiation (0, 15, 30, 45 and 60 min, λex = 340 nm) and subsequent digestion of aliquots (30 μL each) of a naphthalene-modified oligonucleotide assembly. Similar patterns are obtained for each different set of oligonucleotides.
The ease of the electron migration through a specific pathway was also examined. Figure 4 shows the initial BrU decomposition rates (percentage/min) as a function of irradiation obtained for each set of DNA assemblies (Figure 1). The number of base pairs between the naphthalene moiety and the electron trap is kept constant. Considerable differences are observed depending on the position of the BrU relative to the charge injector. High yields are obtained for electron migration towards the 5′-end, with intrastrand migration (set I) being two times more efficient than interstrand migration (set II). Migration towards the 3′-end is, both for interstrand (set IV) and for intrastrand (set III), significantly less efficient. Indeed, intrastrand electron migration 3′ - 5′ versus 5′ - 3′ is 20-fold greater in efficiency. An analogous trend is observed for all other DNA sets, i.e. when the electron trap is placed at a greater distance from the anchoring site of the naphthalene moiety.
FIGURE 4.

(a) Initial decomposition rates (percentage/min) as a function of irradiation of the BrU obtained from photolysis of naphthalene-modified oligonucleotides I-2, II-2, III-2, and IV-2. (b) Schematic representation of the negative charge migration pathway in each different set of naphthalene-modified oligonucleotides.
A similar difference in efficiency as a function of strand orientation has been observed previously for electron migration in diaminonaphthalene-modified DNA assemblies, where more than an 8-fold difference was observed for intrastrand ET towards the 5′-end versus 3′-end.7 Interestingly, for hole transfer studies with photoactivated 2-aminopurine in DNA assemblies, the preferred direction for hole transfer is 5′ - 3′,27 a result consistent with that found here for ET. The asymmetry in overlap of HOMO’s and LUMO’s in the DNA base pair stack would account for these data.27 Importantly, the consistency of results for hole and electron transfer across these different assemblies indicates that this strand asymmetry in transfer is not a function of a particular DNA modification but is instead a general characteristic that depends on the π-stacking in the DNA helix.
Consistent results are not observed for intrastrand versus interstrand transfer for the full family of DNA assemblies. In comparing intrastrand versus interstrand ET towards the 5′ end, where much higher yields are found, we find that intrastrand transfer is more efficient. We would in general expect intrastrand transfer to be more efficient, since in B-form DNA base-base stacking is primarily over the same strand, and no interstrand stacking occurs. A significant difference in rates of intrastrand hole transfer versus interstrand transfer has been observed previously in DNA assemblies containing 2-aminopurine.28
Figure 5 examines the decomposition efficiencies for ET towards the 5′-end as a function of distance. This logarithmic plot of efficiency allows for estimation of the β parameter, representing the exponential decay in yield with distance and gauging the inherent resistivity of a DNA sequence. Here, sets I and II are characterized by a β value of 0.50 Å−1 and 0.34 Å−1, respectively. This shallow distance dependence has been observed repeatedly for hole transfer. More recent studies of DNA-mediated ET have revealed an equally shallow distance dependence. With a tetramethyl diaminonaphthalene as electron donor, a β value of 0.3 Å−1 is obtained.10b When using an Ir(III) complex as electron donor, β = 0.10–0.12 Å−1.15 It appears then that DNA-mediated electron transfer, like hole transfer, may be characterized by a very shallow dependence on distance.
FIGURE 5.

Initial decomposition rates (min−1) of BrU as a function of the distance between the naphthalene-modified uridine (Donor – D) and the BrU (Acceptor – A) for DNA sets I and II. Experimental conditions: λex = 340 nm (Hg-Xe lamp, 1000 W), [naphthalene-modified DNA] = 10 μM, 50 mM TRIS-HCl pH7.4, 100 mM NaCl. Error bars are also given.
Conclusions
Novel naphthalene-modified DNA assemblies have thus been successfully synthesized. The covalent tethering of the naphthalene moiety via a rigid ethynyl linker allowed us to characterize systematically the distance and directional dependence of reductive ET via intrastrand and interstrand pathways. Significantly, substantial differences are apparent in electron transfer towards the 5′-end versus towards the 3′-end. This result represents a general characteristic for DNA-mediated electron transfer, irrespective of the donor modification. Furthermore these results are consistent with the directional asymmetry associated with hole transfer, where the preference is towards the 3′-end. Also, as with hole transfer, DNA-mediated electron transfer shows a shallow distance dependence. These results, taken together, underscore the subtle variations in DNA-mediated electron transfer that depend upon the stacking and helicity of double helical DNA.
Experimental Section
Materials
All phosphoramidites and reagents for DNA synthesis were purchased from Glen Research with the exception of the 5-ethynyluracil phosphoramidite, which was purchased from Berry and Associates. Acetonitrile, dichloromethane, N, N-dimethylformamide, triethylamine, 1-iodonaphthalene, tetrakis(triphenylphosphine)-Pd(0), and copper iodide were purchased from Aldrich in the highest available purity and used as received. All buffers were freshly prepared and filtered using a 0.45 μM filter prior to use.
Oligonucleotide Synthesis
Unmodified oligonucleotides were prepared using standard phosphoramidite chemistry on an ABI DNA Synthesizer. Strands containing a BrU were synthesized by directly placing the commercially available 5-Br-dU phosphoramidite (Glen Research) at the target site. After cleavage from the solid support and deprotection with concentrated ammonium hydroxide, DNA was purified by HPLC on a reversed phase column with acetonitrile and ammonium acetate 50 mM as eluents. The purified products were characterized by UV-visible spectroscopy and MALDI-TOF mass spectrometry.
Synthesis of Naphthalene-modified Oligonucleotides
The modification of DNA with naphthalene was carried out as depicted in Scheme 1. DNA containing a 5-ethynyldeoxyuridine at the target position for modification was synthesized on a solid support (PS beads) under ultramild conditions. After DNA synthesis, the beads were placed in an oven-dried flask, with 2.5 mL of DMF/Et3N (3.5:1.5). 1-iodonaphthalene (250 μmol) and CuI (52 μmol) were added to the flask, and the solution was flushed with argon. Pd(PPh3)4 (64 μmol) was then added to the flask, and the solution was again flushed with argon. The mixture was stirred at ambient temperature for 10–16h under argon. Subsequently, the beads were successively washed with a 5% (w/v) EDTA solution, DMF, CH3CN, and CH2Cl2 to remove any excess of reagents. After 12 hours incubation at room temperature in 1 mL of 0.05 M K2CO3 methanol solution leading to simultaneous cleavage from the solid support and deprotection, the DNA was purified twice (with the DMT protecting groups on and off, respectively) by HPLC on a reversed phase column with acetonitrile and 50 mM ammonium acetate as eluents. The desired products were characterized by UV-visible spectroscopy and MALDI-TOF mass spectrometry.
SCHEME 1.

Melting Temperatures
Melting temperatures (Tm) of all duplexes were measured using a Beckman DU 7400 spectrophotometer with a temperature control attachment. Absorption at 260 nm (A260) of equimolar DNA complements (1.0 μM in 100 mM NaCl, 50 mM TRIS-HCl, pH7.4) were measured every 0.5 °C from 90 °C to 15 °C with rate 0.5 °C/min. The reverse temperature traces were measured under the same conditions to confirm the reversibility of the DNA annealing process. The data were fit to a sigmoidal curve to determine the Tm. The error of Tm over at least three sets of individual experiments was less than 1°C.
Photoreduction Experiments
Aliquots (10 μM DNA, 100 mM NaCl, 50 mM TRIS-HCl, pH 7.4, total volume 30 μL) for irradiation were prepared by annealing equimolar amounts of the desired DNA complements on a DNA thermal cycler (Perkin Elmer Cetus) from 90 °C to 15 °C over a period of 2.5 hours. Aliquots were then transferred to a lucent cell sealed with a rubber septum and deoxygenated with Argon for 20 min. Subsequent irradiation of the naphthalene-tethered duplexes was achieved with a 1000W Hg/Xe lamp equipped with a 320 nm LP filter and a monochromator. After irradiation at 340 nm, duplex samples were digested by 37 °C incubation with phosphodiesterase I (USB) and alkaline phosphatase (Roche) for 1 h in order to yield the free nucleosides, and the samples were analyzed by reversed phase HPLC (Chemcobond 5-ODS-H, 4.6×100 mm). The percentage decomposition of BrU was determined by subtracting the ratio of the area under the BrU peak in an irradiated sample from that in a non-irradiated sample using guanine as an internal standard for all HPLC traces. The decomposition rate (min−1) was obtained from the plot of the initial BrU decomposition as a function of the irradiation time. Irradiations were repeated three times and the results averaged. Actinometry was performed using a 6 mM ferrioxalate standard29 to allow for comparison between experiments performed on separate days.
Supplementary Material
Acknowledgments
We are grateful to the NIH (GM49216) for financial support. M.T. thanks the Japan Society for the Promotion of Science for a postdoctoral fellowship.
Footnotes
Supporting information available
Purification data (HPLC) and characterization data (UV-vis and MS) of naphthalene-modified oligonucleotides. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Mees A, Klar T, Gnau P, Hennecke U, Eker APM, Carell T, Essen LO. Science. 2004;306:1789–1793. doi: 10.1126/science.1101598. [DOI] [PubMed] [Google Scholar]; (b) Sancar A. Chem Rev. 2003;103:2203–2237. doi: 10.1021/cr0204348. [DOI] [PubMed] [Google Scholar]
- 2.(a) Yeh SR, Falvey DE. J Am Chem Soc. 1992;114:7313–7314. [Google Scholar]; (b) Scannell MP, Fenick DJ, Yeh S-R, Falvey DE. J Am Chem Soc. 1997;119:1971–1977. [Google Scholar]
- 3.Seidel CAM, Schulz A, Sauer MHM. J Phys Chem. 1996;100:5541–5553. [Google Scholar]
- 4.(a) Steenken S. Chem Rev. 1989;89:503–520. [Google Scholar]; (b) Steenken S, Telo JP, Novais HM, Candeis LP. J Am Chem Soc. 1992;114:4701–4709. [Google Scholar]; (c) Steenken S. Free Radical Res Commun. 1992;16:349–379. doi: 10.3109/10715769209049187. [DOI] [PubMed] [Google Scholar]
- 5.(a) Razskazovskii Y, Swarts SG, Falcone JM, Taylor C, Sevilla MD. J Phys Chem B. 1997;101:1460–1467. [Google Scholar]; (b) Messer A, Carpenter K, Forzley K, Buchanan J, Yang S, Razskazovskii Y, Cai Z, Sevilla MD. J Phys Chem B. 2000;104:1128–1136. [Google Scholar]; (c) Cai Z, Xifeng L, Sevilla MD. J Phys Chem B. 2002;106:2755–2762. [Google Scholar]
- 6.Boon EM, Drummond TG, Ceres D, Hill MG, Barton JK. Nature Biotechnology. 2000;18:1096–1100. doi: 10.1038/80301. [DOI] [PubMed] [Google Scholar]
- 7.Ito T, Rokita SE. Angew Chem Int Ed. 2004;43:1839–1842. doi: 10.1002/anie.200353038. [DOI] [PubMed] [Google Scholar]
- 8.(a) Manetto A, Breeger S, Chatgilialoglu C, Carell T. Angew Chem Int Ed. 2006;45:318–321. doi: 10.1002/anie.200502551. [DOI] [PubMed] [Google Scholar]; (b) Behrens C, Carell T. Chem Commun. 2003;14:1632–1633. doi: 10.1039/b303805j. [DOI] [PubMed] [Google Scholar]; (c) Breeger S, Hennecke U, Carell T. J Am Chem Soc. 2004;126:1302–1303. doi: 10.1021/ja038358t. [DOI] [PubMed] [Google Scholar]
- 9.(a) Lewis FD, Liu X, Miller SE, Hayes RT, Wasielewski MR. J Am Chem Soc. 2002;124:11280–11281. doi: 10.1021/ja026751d. [DOI] [PubMed] [Google Scholar]; (b) Lewis FD, Wasielewski MR. Top Curr Chem. 2004;236:45–65. [Google Scholar]
- 10.(a) Ito T, Rokita SE. J Am Chem Soc. 2004;126:15552–15559. doi: 10.1021/ja045637n. [DOI] [PubMed] [Google Scholar]; (b) Ito T, Rokita SE. J Am Chem Soc. 2003;125:11480–11481. doi: 10.1021/ja035952u. [DOI] [PubMed] [Google Scholar]; (c) Ito T, Kondo A, Terada S, Nishimoto S. J Am Chem Soc. 2006;128:10934–10942. doi: 10.1021/ja061304+. [DOI] [PubMed] [Google Scholar]
- 11.(a) Amann N, Pandurski E, Fiebig T, Wagenknecht HA. Chem Eur J. 2002;8:4877–4883. doi: 10.1002/1521-3765(20021104)8:21<4877::AID-CHEM4877>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (b) Raytchev M, Mayer E, Amann N, Wagenknecht HA, Fiebig T. Chem Phys Chem. 2004;5:706–712. doi: 10.1002/cphc.200301205. [DOI] [PubMed] [Google Scholar]; (c) Kaden P, Mayer-Enthart E, Trifonov A, Fiebig T, Wagenknecht HA. Angew Chem Int Ed. 2005;44:1636–1639. doi: 10.1002/anie.200462592. [DOI] [PubMed] [Google Scholar]
- 12.Wagner C, Wagenknecht HA. Chem Eur J. 2005;11:1871–1876. doi: 10.1002/chem.200401013. [DOI] [PubMed] [Google Scholar]
- 13.Giese B, Carl B, Carl T, Carell T, Behrens C, Hennecke U, Schiemann O, Feresin E. Angew Chem Int Ed. 2004;43:1848–1851. doi: 10.1002/anie.200353264. [DOI] [PubMed] [Google Scholar]
- 14.Valis L, Wang Q, Raytchev M, Buchvarov I, Wagenknecht HA, Fiebig T. Proc Natl Acad Sci U S A. 2006;103:10192–10195. doi: 10.1073/pnas.0600957103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Shao F, Barton JK. J Am Chem Soc. 2007;129:14733–14738. doi: 10.1021/ja0752437. [DOI] [PubMed] [Google Scholar]; (b) Elias B, Shao F, Barton JK. J Am Chem Soc. 2008;130:1152–1153. doi: 10.1021/ja710358p. [DOI] [PubMed] [Google Scholar]; (c) Elias B, Genereux JC, Barton JK. Angew Chem Int Ed. 2008;47:9067–9070. doi: 10.1002/anie.200803556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagenknecht H-A, editor. Charge Transfer in DNA. Wiley-VCH; Weinheim, Germany: 2005. [Google Scholar]
- 17.(a) Mayer E, Valis L, Wagner C, Rist M, Amann N, Wagenknecht HA. Chem Bio Chem. 2004;5:865–868. doi: 10.1002/cbic.200300845. [DOI] [PubMed] [Google Scholar]; (b) Trifonov A, Raytchev M, Buchvarov I, Rist M, Barbaric J, Wagenknecht HA, Fiebig T. J Phys Chem B. 2005;109:19490–19495. doi: 10.1021/jp052108c. [DOI] [PubMed] [Google Scholar]; (c) Wagenknecht HA. Curr Org Chem. 2004;8:251–266. [Google Scholar]
- 18.Tanaka M, Ohkubo K, Fukuzumi S. J Am Chem Soc. 2006;128:12372–12373. doi: 10.1021/ja065073i. [DOI] [PubMed] [Google Scholar]
- 19.Buxton GV, Greenstock CL, Helman WP, Ross AB. J Phys Chem Ref Data. 1988;17:513. [Google Scholar]
- 20.Berlman IB. Handbook of Fluorescence Spectra of Aromatic Molecules. Academic; New York: 1971. [Google Scholar]
- 21.Murov SL, Carmichael I, Hug GL. Handbook of Photochemistry. Marcel Decker Inc; New York: 1993. [Google Scholar]
- 22.(a) Korshun VA, Prokhorenko IA, Gontarev SV, Skorobogatyi MV, Balakin KV, Manasova EV, Malakhov AD, Berlin YuA. Nucleosides Nucleotides. 1997;16:1461–1464. [Google Scholar]; (b) Malakhov AD, Malakhova EV, Kuznitsova SV, Grechishnikova IV, Prokhorenko IA, Skorobogatyi MV, Korshun VA, Berlin YuA. Russ J Bioorg Chem. 2000;26:34–44. [PubMed] [Google Scholar]
- 23.(a) Gaballah ST, Vaught JD, Eaton BE, Netzel TL. J Phys Chem B. 2005;109:12175–12181. doi: 10.1021/jp044457x. [DOI] [PubMed] [Google Scholar]; (b) Gaballah ST, Hussein YH, Anderson N, Lian TT, Netzel TL. J Phys Chem A. 2005;109:10832–10845. doi: 10.1021/jp053359o. [DOI] [PubMed] [Google Scholar]
- 24.Ito T, Hayashi A, Kondo A, Uchida T, Tanabe K, Yamada H, Nishimoto S. Org Lett. 2009;11:927–930. doi: 10.1021/ol802896y. [DOI] [PubMed] [Google Scholar]
- 25.(a) Freier SM, Altmann KH. Nucl Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) De Mesmaeker A, Haener R, Martin P, Moser HE. Acc Chem Res. 1995;28:366–374. [Google Scholar]
- 26.Liu T, Barton JK. J Am Chem Soc. 2005;127:10160–10161. doi: 10.1021/ja053025c. [DOI] [PubMed] [Google Scholar]
- 27.O’Neill MA, Barton JK. Proc Nat Acad Sci USA. 2002;99:16543–16550. doi: 10.1073/pnas.012669599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelley SO, Barton JK. Science. 1999;283:375–381. doi: 10.1126/science.283.5400.375. [DOI] [PubMed] [Google Scholar]
- 29.Hatchard CG, Parker CA. Proc R Soc Lon Ser A. 1956;235:518–536. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.