

Summary
The polarized nature of neurons as well as the size and density of synapses complicates the manipulation and visualization of cell biological processes that control synaptic function. Here we developed a microfluidic local perfusion (μLP) chamber to access and manipulate synaptic regions and pre- and post-synaptic compartments in vitro. This chamber directs the formation of synapses in >100 parallel rows connecting separate neuron populations. A perfusion channel transects the parallel rows allowing access to synaptic regions with high spatial and temporal resolution. We used this chamber to investigate synapse-to-nucleus signaling. Using the calcium indicator dye, Fluo-4, we measured changes in calcium at dendrites and somata, following local perfusion of glutamate. Exploiting the high temporal resolution of the chamber, we exposed synapses to “spaced” or “massed” application of glutamate and then examined levels of pCREB in somata. Lastly, we applied the metabotropic receptor agonist, DHPG, to dendrites and observed increases in Arc transcription and Arc transcript localization.
Introduction
Neuronal connectivity within the mammalian brain is extensive and complex. The use of dissociated cultured neurons is valuable for investigating the cell biology of neurons and synapses, providing a relatively homogeneous population of neurons, reducing the complex three-dimensional connectivity of brain tissue to two-dimensions, and facilitating access to neurons both visually and pharmacologically. Similar to synapses in vivo, synapses in vitro are functional and can undergo various forms of synaptic plasticity. Unfortunately, the process of dissociating neurons eliminates much of the native circuitry, resulting in a disordered connectivity of neurons. To improve the organization of cultured neurons and their connections, several groups have endeavored to actively polarize cortical/hippocampal neurons using chemically patterned surfaces (Scholl et al., 2000; Stenger et al., 1998; Vogt et al., 2004; Vogt et al., 2005), soluble gradients (Dertinger et al., 2002), or physical structures that guide neuronal growth (Taylor et al., 2005; Taylor et al., 2003; Tooker et al., 2006; Tooker et al., 2004). One of the goals of these approaches is to organize cultured neurons in such a way that they more closely mimic the connectivity found in vivo, enabling them to be studied in a more physiological manner.
Local changes at synapses are critical for plasticity, yet there are limited techniques to investigate local processing within synaptic regions. One technique, glutamate uncaging, which uses photoconversion to yield active glutamate, has revealed much about local processing following neurotransmitter release and has been an important advance for studying mechanisms of synaptic plasticity (Harvey et al., 2008; Lee et al., 2009). Drawbacks of this technique include the limited set of available caged compounds as well as the bath application of the caged molecule to the entire culture/tissue. The recent development of optogenetics to selectively activate or inactivate regions of synapses has also shown great promise (Zhang et al., 2007). These methods require genetically modified neurons to express light-sensitive ion channels. There have also been studies which use small perfusion pipettes to alter local environments at sites of synaptic contact (Song et al., 1997; Sutton et al., 2006; Sutton et al., 2007). While local perfusion allows for the application of any drug or compound, it is cumbersome to set-up and implement, treats a single dendrite or process at a time, and has a relatively slow on-off time for manipulations.
In recent years microfabricated devices have been developed for cell culture applications (El-Ali et al., 2006; Taylor et al., 2005; Taylor et al., 2003). These devices are precisely and reproducibly fabricated with features that are on a similar physical scale to cells. The properties of fluid at the microscale allow the creation of precise microenvironments, such as concentration gradients (Jeon et al., 2002) or fluidically-isolated subcellular compartments (Taylor et al., 2005; Taylor et al., 2003). The most common method for fabricating microfluidic devices for biological applications uses photolithography to create a master or template using the epoxy photoresist, SU-8. SU-8 polymerizes when exposed to ultraviolet light and can be used to create relatively high structures, in the range of hundreds of microns, which are suitable for cell culture. After the template is produced, it can then be used indefinitely to replica mold the microfluidic devices using poly(dimethylsiloxane) (PDMS), a biocompatible, optically transparent polymer. This process, called soft lithography, is cost effective and can be performed in any traditional lab setting (McDonald et al., 2000; Whitesides et al., 2001).
Here we sought to develop a microfluidic device or chamber optimized for synaptic cell biology that allows one to visualize and manipulate synapses and pre- and postsynaptic cell bodies independently. Until now, this has not been possible using other compartmentalized methods, including microfluidic methods. To accomplish this goal, we adapted a previously developed compartmentalized microfluidic chamber used to direct the growth of cortical and hippocampal axons through parallel microgrooves (Taylor et al., 2005; Taylor et al., 2003). Our first goal was to direct the growth of dendrites into microgrooves and verify that functional synapses were formed within the microfluidic chamber when two populations of neurons were grown on either side of the microgrooves. Second, we implemented a small perfusion channel to locally perturb and investigate synaptic regions with high spatial and temporal resolution. As proof of principle, we demonstrate the utility of this chamber to investigate synapse-to-nucleus signaling in multiple ways– using calcium imaging, comparing transcriptional responses to spaced and massed stimulation, and investigating changes in Arc transcription and mRNA localization following the local application of the mGluR1 group I agonist, DHPG.
Results
Dendritic growth within microgrooves
Using the compartmentalized microfluidic chamber, we sought to compartmentalize two distinct populations of neurons, facilitating the visualization of synapses between them. The microfluidic chamber consists of 2 main compartments (1,500 μm wide, 100 μm high) which are connected by ∼150 parallel microgrooves (7.5 μm wide, 900 μm long, 3 μm high) (Figure 1A). Dissociated neurons are plated in each compartment and cultured, during which time processes emanating from the cell bodies grow into the microgrooves and into the adjacent compartment (without the addition of growth factors) (Figure 1B). While axonal growth within microgrooves is well-documented (Taylor et al., 2005), the growth of dendrites within the microgrooves has not been characterized.
Figure 1. Dendrites grow into the microgrooves of microfluidic chambers.
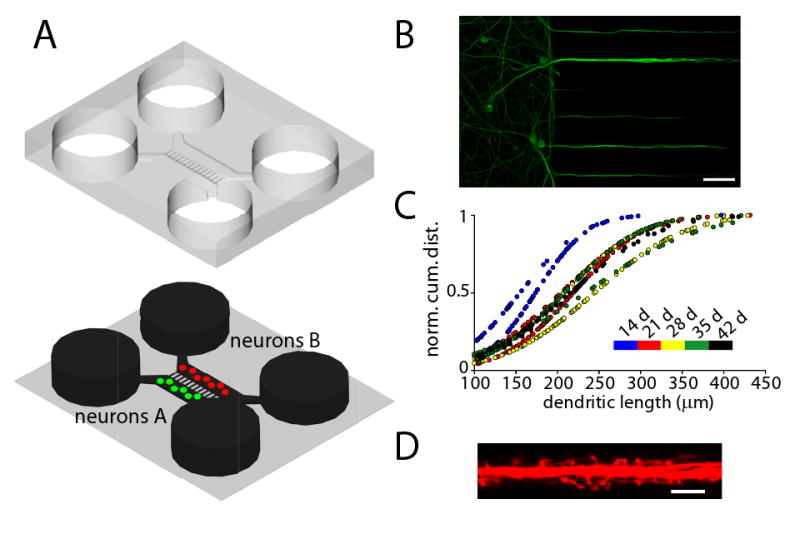
(A) Schematic of a microfluidic chamber. For clarity, the PDMS mold (top) is shown above the glass substrate (bottom). The fluid is shown in black. The four circular wells provide access for the introduction of the neurons and are filled with media to support neuronal growth. Microgrooves (900 μm × 7.5 μm × 3 μm) connect the two rectangular channels (or compartments) that house two independent populations of neurons. (B) Dendrites extend into microgrooves. Fluorescence image of dendrites extending into microgrooves (MAP2 = green). Scale bar = 50 μm. (C) Dendritic length within microgrooves as a function of days in culture. Shown are normal cumulative distribution plots for single chambers (n = 2). (D) Fluorescence image of dendritic spines within microgrooves. Neurons within the microfluidic chamber were infected with an RFP Sindbis virus, which demonstrates the healthy neuronal morphology, including spines. Scale bar = 10 μm.
Here we recorded the growth properties of dendrites in culture from 14 to 42 days in vitro. In general, dendrites are much shorter and thicker than axons and have a tapered appearance. First, we wanted to determine the percentage of microgrooves that possessed dendrites. Although variable and largely dependent on cell plating density, at least 50% of the microgrooves contained dendrites greater than 100 μm in length after 14 days. There was no change in the number of dendrite-filled microgrooves with culture age, indicating that after 14 days in culture the location of the dendrites is well established. Second, we measured the length of dendrites within each chamber for all culture ages and plotted their distribution (Figure 1C). At 14 days in culture, the average dendritic length was 165 μm and at 21 days the average length extended to 214 μm. At longer culture ages, the dendrites did not extend further into the microgrooves, indicating that absolute dendritic growth within the microfluidic chamber slows or stops after 21 days; notably, however, the length did not decrease.
We next examined whether the dendrites present within the microgrooves possess spines, the sites of excitatory synaptic connections. When neurons were infected with an RFP-Sindbis virus, numerous protrusions with spine-like morphologies were evident extending from dendrites within the microgrooves (Figure 1D). Together, these data shows that dendrites grow and extend considerable distance in the microgrooves; many of these dendrites possess spines.
Identification and visualization of synapses within the microfluidic chamber
To test whether synapses between two distinct neuronal populations can form within microgrooves, neurons were plated in the two somatic compartments, cultured for 14 days and then infected with either a GFP or RFP Sindbis virus in separate compartments in order to visualize potential connections (Figure 2A). The compartment-specific expression of either GFP or RFP demonstrates the ability to genetically manipulate each population independently. In these chambers, axons can grow the entire 900 μm extent of the microgrooves, whereas dendrites extend less than 450 μm into the microgrooves (Taylor et al., 2005); therefore, only axons will reach the distal end of the microgrooves. As described above, dendrites are easily identified based on morphological features, and when examined carefully, individual dendrites can be followed back to their somata of origin in the cell compartment. Together, these properties allow us to identify presynaptic processes originating from cell bodies in one compartment and postsynaptic processes originating from cell bodies in the other compartment (Figure 2B).
Figure 2. Synapses form within the microgrooves.
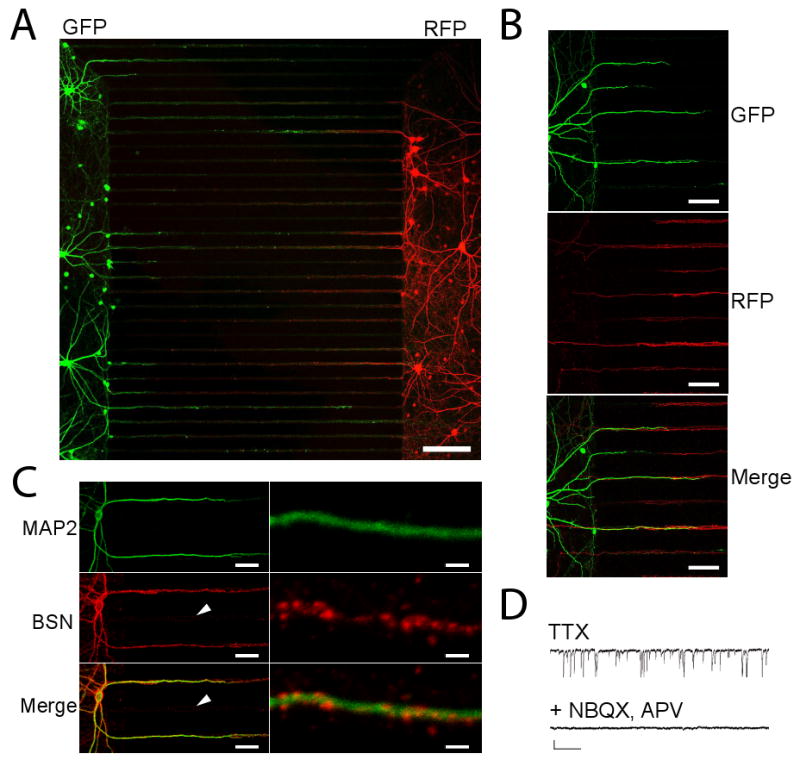
(A) Neuronal processes, extending from compartmentalized neuronal cell bodies, establish contact within the microgrooves. Two sets of neurons were introduced into the left and right compartments. Neurons on the left expressed GFP whereas neurons on the right expressed RFP. Red- and green-labeled processes are evident within the microgrooves. Scale bar = 150 μm. (B) Enlarged image of the left-side of the chamber showing GFP-labeled dendrites entering the microgroove channels (top) and RFP-labeled axons (middle) growing the ∼900 μm extent of the microgrooves. These axons appear to contact the dendrites, as shown in the merged image (bottom). Scale bars = 50 μm. (C) Neurons grown in the chamber for 21 days possess synapses. MAP2-labeled dendrites (green, top) extend into the microgrooves from the left side. Axons (not shown) enter from both the presynaptic and postsynaptic sides. Immunostaining for the presynaptic marker bassoon (red, middle) shows a punctate pattern that decorates the dendrites. Bottom shows merged images. The white arrow shows a microgroove which contains axons (not visible), but no dendrites; axons within this microgroove have minimal bassoon immunoreactivity. Scale bar = 21 and 3 μm for left and right images, respectively. (D) Functional synapses within the microfluidic chamber. Voltage clamp recordings were obtained from a 42 day-old neuron plated in a microfluidic chamber. Top recording shows miniature excitatory postsynaptic currents, recorded in the presence of TTX. NBQX and APV applied locally to one compartment completely abolished the recorded events. Scale bar = 20.5 pA, 256 ms.
Although we were able to identify potential regions of contact between axons and dendrites, it was unclear if bona fide synapses form within the microfluidic chambers. To answer this question, we first used immunocytochemistry to test for and visualize the position of presynaptic puncta (using an anti-bassoon antibody) relative to dendrites (using an anti-MAP2 antibody) (Figure 2C). Bassoon-positive puncta were clustered along the MAP2-positive dendrites, whereas axons in dendrite-free areas of the microgrooves did not contain such bassoon clustering. To detect functional synapses, whole-cell voltage clamp recordings of miniature synaptic currents were obtained from neurons inhabiting one of the compartments. Many synaptic events were visible, indicating abundant synaptic activity in the chambers. Recordings were obtained from neurons cultured up to 42 days. The glutamate receptor antagonists, NBQX and APV were added locally during some recordings, resulting in the immediate elimination of the miniature excitatory postsynaptic currents (Figure 2D). These results show that glutamatergic synapses are functional within the microfluidic chambers. Furthermore, we identified both glutamatergic and inhibitory neurons within the chambers, showing that the cultures mature normally and possess a mix of excitatory and inhibitory neurotransmitters (Figure S1).
Local perfusion using microfluidics
The presence of synapses in microgrooves raises the possibility of focal synaptic stimulation to investigate synaptic function. To implement this feature, we kept the existing compartmentalized structure but added a perfusion channel perpendicular to the microgrooves. In order to access a high number of dendrites, we placed the perfusion channel 75 μm from the edge of one compartment (we refer to this compartment as the “postsynaptic compartment”), and this compartment is connected to the perfusion channel via the microgrooves (which have the same height and width dimensions as the traditional compartmentalized chamber used above). Since the average length of dendrites extending into the microgrooves is 165 μm at 14 days in culture, the location of the perfusion channel allows for a large percentage of the dendrites to be perfused. The other compartment is 500 μm away from the perfusion channel and connected via an additional set of microgrooves. The long length of this set of microgrooves ensures that only axons from this compartment (we refer to this compartment as the “presynaptic” compartment) are able to extend into the perfusion channel. The perfusion channel is 50 μm in width and 100 μm in height (i.e., the same height as the compartments) (Figure 3A). A reservoir (6 mm in diameter) at one end of the channel contains the perfusate and the other end has a smaller opening which is used to connect to efflux tubing and a syringe pump. Withdrawal of the perfusate using the syringe pump results in a negative pressure in the perfusion channel, drawing the perfusate through the channel and preventing diffusion of the perfusate into microgrooves (Figure 3A). To visualize the perfusion, we filled the perfusion reservoir in a neuron-free chamber with Alexa Fluor 488 and withdrew it from the other end using the syringe pump (rate = 100 μl/h) (Figure 3B,C). A uniform fluorescence was rapidly established within the channel; notably, there was no fluorescent signal apparent within the microgrooves. To determine the stability of the perfusion stream and to determine, conclusively, if there is any diffusion from the perfusion stream into the microgrooves, we monitored the fluorescence intensity of Alexa Fluor 488 within the chamber over 60 min. We used single-line z scans within a selected region of interest to determine the signal intensity within the perfusion channel and the neighboring microgroove. The regions of interest (ROI) were limited to 3 μm high (i.e., the height of the microgroove). As shown in Figure 3D, the signal within the perfusion channel was stable over an hour and there was no signal detected within the adjacent microgroove ROI over this same time period. We next examined the on/off kinetics to determine the minimum time required to switch perfusion solutions. We found that the perfusate can be added or removed within approximately 1 min (Figure 3E). These results show that we can locally and rapidly perfuse molecules within a neuron-free chamber.
Figure 3. A stable perfusion stream with rapid on/off kinetics transects the microgrooves.
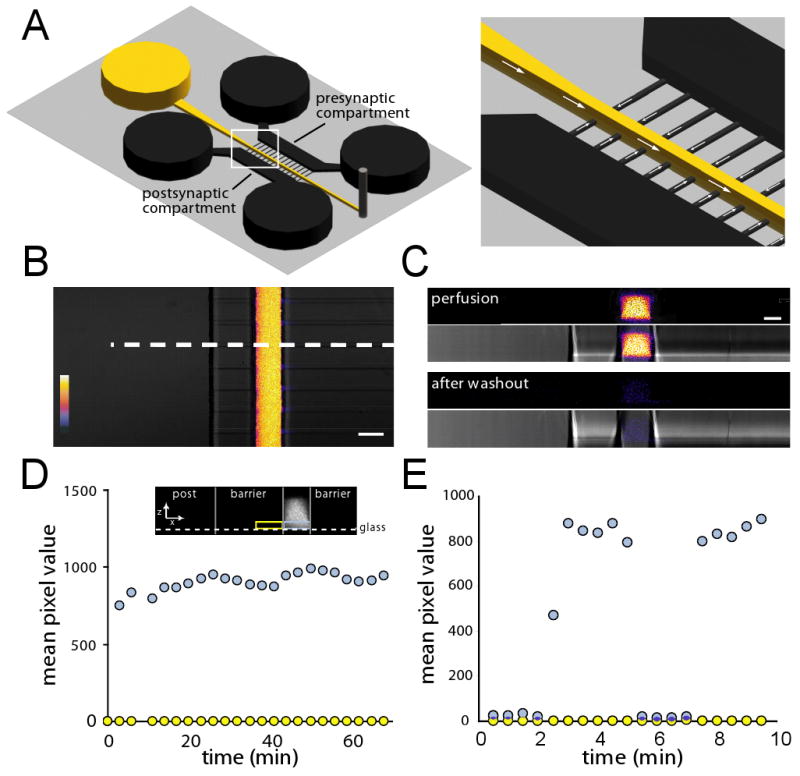
(A) Schematic of a local perfusion chamber showing the perfusion inlet well and channel in yellow. Fluid was withdrawn from the perfusion channel by a syringe pump connected to a tubing outlet (grey peg on right). The PDMS mold is not shown. The enlarged image on the right shows the direction of fluid flow within the local perfusion channel and microgrooves. The slight flow into the perfusion channel from the microgrooves counteracts the diffusion of the perfusate into the microgrooves. (B) A merged fluorescence and DIC image showing the perfusion of the low molecular weight dye Alexa Fluor 488 (2 μM). Color look-up bar shows fluorescence intensity. Scale bar = 50 μm. (C) Fluorescence and DIC depth (z) scans of the white dashed region in (B) showing the profile of the dye within the channel during the perfusion and after wash out. Scale bar = 30 μm. (D) Stability of the perfusion stream. Graph of fluorescence intensity over time for regions of interest within the channel (blue box shown in inset) and proximal microgroove (yellow box shown in inset). A stable fluorescent signal within the channel (light blue circles) was maintained for over an hour while dye in the region immediately adjacent to the channel (yellow circles) remained undetectable. Mean pixel intensity is in arbitrary units. Similar results were obtained in more than 10 experiments. (E) Solutions can be rapidly added and removed from the channel with a response time of less than 1 min. The same regions of interest are used as in D.
Next we wanted to investigate how neurons grow within and across the perfusion-channel chamber and if the flow properties are maintained (as with the neuron-free chamber) when neurons are cultured for extended periods of time. Fourteen days after plating, we observed numerous dendrites extending through the perfusion channel and often extending into the corresponding microgrooves present across the perfusion channel (Figure S2). Dendrites also sometimes branched within the perfusion channel. We then perfused fluorescent dye within the local perfusion channel to determine if we could maintain a distinct perfusion stream, as previously observed in chambers lacking neurons (Fig 3B). We used chambers containing neurons over 14 days in culture and observed that the perfusate often diffused into the microgrooves during the perfusion (Figure S2). In most cases the diffusion occurred in microgrooves which had large accumulations of cellular material, suggesting that the diffusion of perfusate is due to blockage of flow within the microgrooves. These results indicate that the single perfusion stream design is not adequate to prevent diffusion of the perfusate into the microgrooves when neurons are cultured for extended periods of time.
A multi-inlet design prevents diffusion, reduces perfusion spot size, and improves temporal control
To prevent the perfusate from diffusing into the microgrooves, we designed a 3-inlet perfusion channel. In this design, the 2 outer inlets are filled with normal media, resulting in 3 fluid streams entering the perfusion channel. The 2 outer streams serve as virtual fluid barriers preventing diffusion of the perfusate into the microgrooves (Figure 4A). Fluorescence and DIC images of the perfusion are shown in Figure 4B. Another advantage of the 3-inlet design is that the width of the perfusion spot can be reduced considerably, owing to the streams of normal media that flank the perfusate on either side. For example, within microgrooves near the entrance of the perfusion, we were able to restrict the width of the perfusate to a minimum of 10 μm (Figure 4B-C). The z profile of a perfusion spot is shown in Figure 4C. Importantly, perfusion within a neuron-filled chamber shows no evidence of diffusion into the microgrooves. This demonstrates that the microfluidic local perfusion (μLP) chamber incorporating a 3-inlet design prevents diffusion of the perfusate into the microgrooves and can be used to apply pharmacological agents to focal synaptic regions present within the channel.
Figure 4. A multi-inlet microfluidic perfusion chamber narrows the perfusion stream, improves temporal resolution, and allows distinct presynaptic, postsynaptic and synaptic microenvironments.
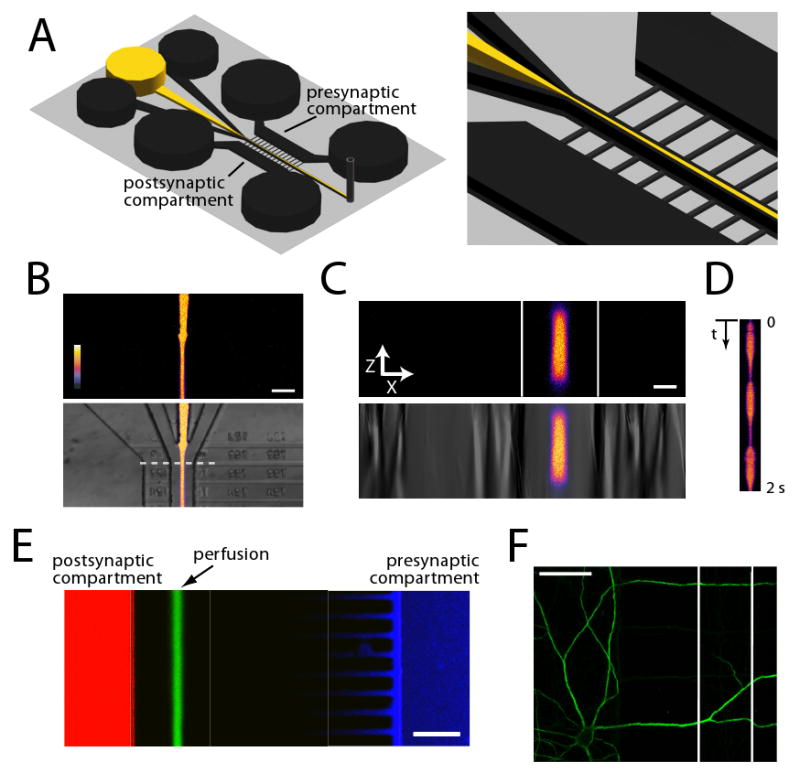
(A) A schematic of the local perfusion chamber with 3 inlet wells. The outer wells that contain normal buffer prevent perfusate from entering the microgrooves and serve to narrow the perfusion stream. (B) Merged fluorescence and DIC images of Alexa Fluor 488 perfusion into the channel, flanked by two buffer channels (color look up table shows intensity). Scale bar = 50 μm. (C) Fluorescence and DIC depth (z) scans of dashed line in (B). Scale bar = 15 μm. (D) Continuous line scans of dashed line in (B) within local perfusion channel shows high temporal resolution. Pulsing was performed by rapidly adding/removing 30-40 μl in the center inlet well. (E) Merged fluorescence micrograph of Alexa Fluor 488 hydrazide (green), Alexa Fluor 568 hydrazide (red) and Alexa Fluor 633 hydrazide (blue) added to perfusate, postsynaptic compartment, and presynaptic compartment respectively after 30 min of perfusion. Fluorescent microenvironments are stable and distinct from one another. The properties of the Alexa Fluor 633 hydrazide make it accumulate at the walls of the PDMS thus the intensity of the dye is not uniform throughout the presynaptic compartment. Scale bar = 100 μm. (F) Example image of a MAP2-immunolabeled neuron (green) following perfusion. There was no observable difference in morphology, illustrating that multiple perfusion patterns (i.e., flow changes) have negligible effect on dendritic morphology. The perfusion channel is between the white lines. Scale bar = 50 μm.
We have shown that we can effect a localized and constant perfusion of small (∼10-50 μm) synaptic regions for minutes up to ∼ 1 hr. This type of treatment mimics the tonic presence of neurotransmitters and modulators. Synaptic events, and many modulatory events, however, occur phasically. Similarly, activation of signaling pathways such as MAPK (Wu et al., 2001) and CREB (Kida et al., 2002) are sensitive to the timing and pattern of stimulation. The 3-inlet design of the μLP chamber allows increased temporal control of the perfusate. Using the advantages of laminar flow, which is defined by a no-slip boundary condition, if there is lower pressure in the center well delivering the perfusate, the outer two wells containing medium will merge resulting in an effective evacuation of the perfusate from the central region. This property can be used to rapidly turn on and off the perfusate. As an initial demonstration of this principle, we fluctuated the pressure in the center well to effect a pulsatile delivery of the Alexa Fluor 488 dye with subsecond resolution (Figure 4D).
An additional advantage of microfluidic devices is the ability to create multiple distinct microenvironments. This is particularly relevant for the study of neurons which have long processes forming synapses in multiple microenvironments or compartments. Using the syringe pump-based flow within the perfusion channels and the virtual boundaries set up with the outer streams of buffer, we can also create distinct microenvironments for post- and pre-synaptic compartments as well as local synaptic regions. To illustrate this feature, we added three different wavelength fluorescent dyes (Alexa Fluor 488, Alexa Fluor 568, and Alexa Fluor 633) to the postsynaptic, perfusion channel and pre-synaptic compartments and imaged them after 30 min of perfusion, showing that the separation of the microenvironments is stable (Figure 4E). We expected that dye from the pre- and postsynaptic compartments would enter the microgrooves but that the buffer streams on either side of the perfusion stream would provide a barrier to maintain the isolation of the fluid microenvironments– this was indeed the case. In addition, the Alexa Fluor 488 dye was restricted only to the postsynaptic compartment. The Alexa Fluor 633 dye was detected only in the presynaptic compartment and the microgrooves proximal to the presynaptic compartment. These data indicate that the μLP chamber can be used to pharmacologically manipulate each of these compartments simultaneously.
Since neurons are susceptible to shear stress, we also wanted to ensure that the perfusions were well tolerated by the neurons. To address this, we immunostained for MAP2 following local perfusion with normal media (Figure 4F). After the perfusion, there was no detectable damage to dendritic morphology of the perfused segments. Together, these data show that we can deliver pharmacological agents to focal synaptic regions with quick response time and with minimal impact on dendritic structure using the μLP chamber.
Do synapses form within the perfusion channel? The ability to target synapses made between cells located in pre- and post-synaptic compartment would be a considerable advancement for experimental manipulations of synapses. To visualize axons from neurons isolated within the presynaptic compartment, we infected neurons in this compartment with a GFP-virus (Figure 5). Many axons were labeled and extended into the microgrooves and through the perfusion channel (not shown). To examine whether there are active presynaptic terminals associated with these axons, we loaded presynaptic terminals within the perfusion channel with the styryl dye, FM5-95 (Figure 5C) by stimulating exo- and endocytosis. We perfused the dye while simultaneously depolarizing the neurons with a high KCl solution. After a couple of wash steps, multiple presynaptic terminals were loaded with the FM dye, which also co-localized with the GFP-labeled axon, showing that active presynaptic terminals are present within the perfusion channel. (Figure 5D).
Figure 5. Synapses, between neurons residing in separate cell body compartments, form within the perfusion channel.
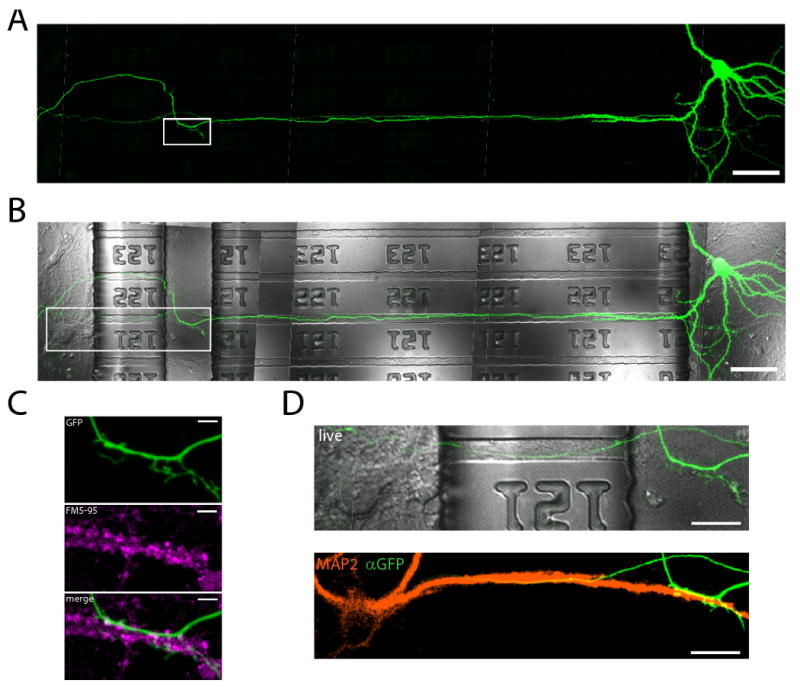
(A) A fluorescent micrograph of a spiny GFP-labeled neuron within the presynaptic compartment extends an axon through the microgrooves and into the local perfusion channel. Scale bar = 50 μm. (B) Merged DIC and GFP images showing the locations of the microgrooves and perfusion channel. (C) Enlarged image of box outlined in (A). Below, FM5-95 was loaded via the postsynaptic compartment and perfusion channel, resulting in the labeling of presynaptic terminals. Multiple presynaptic terminals colocalize with the GFP labeled axon. Scale bar = 5 μm. (D) Enlarged image of box outlined in (B). Below, the postsynaptic neuron is post-hoc labeled for MAP2 (orange) and GFP (green). The dendrite and axon from separated compartments colocalize at the site of FM5-95 loading within the perfusion channel. Scale bar = 20 μm.
Local perfusion leads to changes in dendritic and somatic calcium dynamics
One feature enabled by the μLP chamber is the ability to investigate synapse-to-soma/nucleus signaling. For example, during LTP induction, local synaptic activation at the plasma membrane causes rises in postsynaptic calcium that propagate to the soma and stimulate new transcription. To test the ability to detect such events using our device, we locally perfused glutamate then monitored dendritic and somatic calcium dynamics, using the fluorescent calcium indicator dye, Fluo-4 NW. Monitoring the same neuron over time, we stream acquired images before and after vehicle and glutamate (200 μM) perfusion within the channel; TTX (1 μM) was included to the postsynaptic and presynaptic compartments to prevent neuronal spiking. While vehicle perfusion in the channel did not change calcium levels appreciably (Figure 6A and Movie S1), perfusion with glutamate (∼3 min) on the same dendrite led to a rapid increase in calcium at the perfused dendrite and a slower rise in calcium at the cell body (Figure 6B and Movie S2). Post-hoc immunostaining for MAP2 shows the integrity of the neuron following the perfusion (Figure 6C). Thus, the μLP chamber can also be used to investigate calcium dynamics locally and globally.
Figure 6. Local dendritic perfusion of glutamate rapidly increases local calcium followed by a slower rise of calcium in soma.
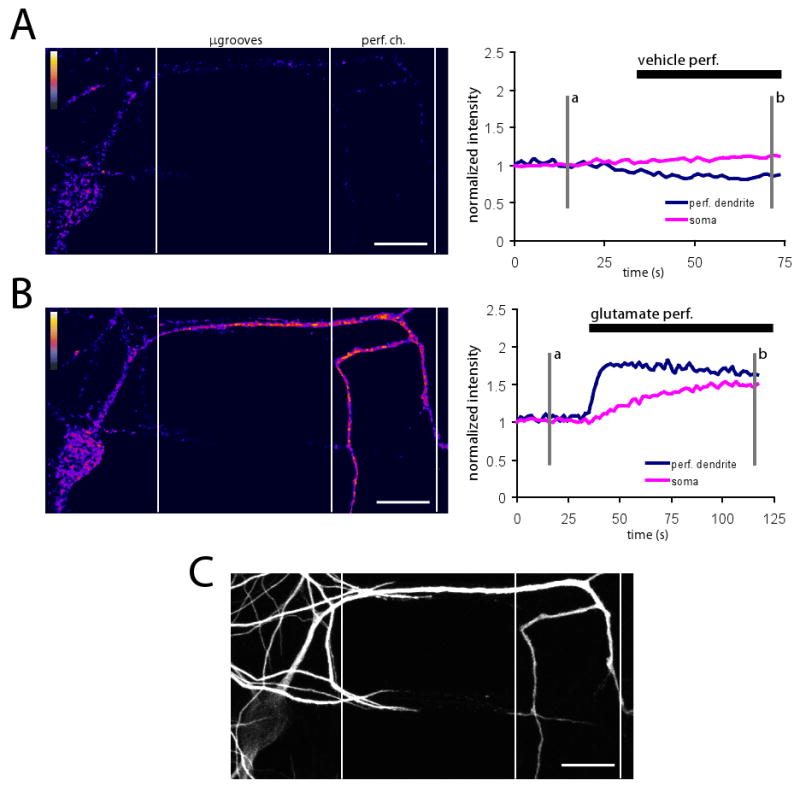
(A) Fluorescence difference image of Fluo-4 signal before and during local vehicle perfusion (left) showing that are no changes in calcium during vehicle perfusion. Fluorescence difference image was created by subtracting image at time point b from image at timepoint a (shown at right). Plot of normalized Fluo-4 intensity over time (right). Fluorescence difference image includes only regions which were MAP2 positive. ‘Fire’ lookup table. (B) Fluorescence difference image of Fluo-4 signal before and during local glutamate perfusion (left). Plot of normalized Fluo-4 intensity over time (right). Imaging was performed immediately following vehicle perfusion shown in (A). During glutamate perfusion, Fluo-4 signal increased rapidly in perfused dendrite, followed by a slower rise in soma. Fluorescence difference image prepared same as in (A). (C) Post-hoc immunostaining for MAP2, showing soma and dendrites of the perfused neuron. Scale bars = 25 μm.
Spaced perfusion of glutamate at dendrites increases pCREB compared to vehicle and massed glutamate perfused dendrites
As described earlier, one benefit of the three-inlet μLP chamber design is the ability to control perfusions with improved temporal resolution. In order to demonstrate this function, we sought to investigate how different patterns of neurotransmitter stimulation alter transcriptional responses in postsynaptic neurons. Previous work in Drosophila and Aplysia has shown that spaced, but not massed, presentation of conditioning stimuli leads to long-term memory formation, mediated by phosphorylation of CREB and new gene expression (Carew et al., 1972; Yin et al., 1994). Here we examined the ability of spaced versus massed applications of glutamate to increase pCREB in neuronal cell bodies (Figure 7). Using the perfusion channel in the μLP chamber we delivered 3 one minute pulses of glutamate to dendrites with 5 minute rest periods between each pulse (spaced condition). In the massed application condition glutamate was applied continuously for 3 minutes. To monitor the kinetics and timing of the perfusion, we included a fluorescent dye (Alexa Fluor 568) together with the glutamate. One hour after the glutamate application, neurons were fixed and immunostained with a pCREB antibody. Analysis of pCREB signal revealed a significant increase in the cell body following spaced application of glutamate. A single, massed application of glutamate for 3 minutes or a spaced application of a vehicle solution did not produce such an increase. These results suggest that in vitro localized spaced stimulation activated new gene expression, paralleling long-term memory formation in whole organisms. These results further demonstrate the benefits of the μLP chamber for studying the cellular mechanisms of synaptic plasticity using more physiologically relevant parameters (i.e., improved spatial and temporal resolution) than traditional culture methods.
Figure 7. Spaced perfusion of glutamate at dendrites increases pCREB at 60 min.
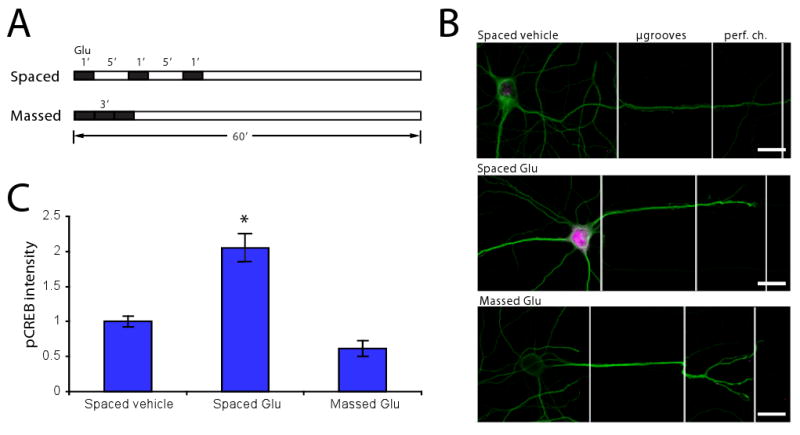
(A) Time course of experiment showing both spaced and massed perfusions. (B) Fluorescence images of μLP chambers immunostained for MAP2 (green) and pCREB (magenta). pCREB intensity increased in soma with dendrites exposed to spaced glutamate perfusion. Scale bar = 20 μm. (C) Quantification of (B) showing that pCREB intensity is significantly higher in soma which had dendrites perfused with spaced glutamate (n = 22) vs. either spaced vehicle (n = 21) or massed glutamate (n = 9) perfusions. pCREB intensities normalized to spaced vehicle. Significance was determined using a two-tailed Student's t-Test; * p < 0.05 vs. spaced vehicle.
Localized DHPG increases Arc transcription and increases the dendritic localization of Arc mRNA
Next, we designed experiments that make use of the perfusion channel to elicit local changes that might prompt synapse-to-nucleus-to-synapse signaling, resulting in the transport of molecules back to the perfused region. Arc is an immediate early gene that has received much attention due to the localization of its mRNA at synapses with sustained high frequency stimulation (Steward et al., 1998), linking gene expression changes with localized synaptic changes. Recent evidence shows that ARC protein expression is required for mGluR-dependent long-term depression (Park et al., 2008; Waung et al., 2008). The group I mGluR agonist DHPG increases dendritic ARC protein levels without a requirement for new transcription, suggesting that preexisting Arc transcripts can be rapidly (within 5 min) locally translated (Park et al., 2008; Waung et al., 2008). An increase in Arc transcription has been reported 20 min following global DHPG application (Park et al., 2008), suggesting that that following rapid translation of Arc, there is also an increase in Arc transcription.
Here we asked if DHPG applied at discrete synaptic regions was sufficient to signal back to the nucleus and increase Arc transcription. We next asked if the recently transcribed mRNA were targeted to the perfused synaptic regions. In μLP chambers housing mature (15-20 DIC) cultured neurons, we perfused DHPG (500 μM) over dendritic segments in the local perfusion channel for 10 min. Twenty minutes following the end of perfusion, we used fluorescence in situ hybridization (FISH) to visualize the location of the Arc mRNA puncta (Figure 8). We saw a significant increase in the number Arc mRNA puncta in the somata of dendrites perfused with DHPG compared to vehicle control. This increase in Arc transcript number was blocked when the group I mGluR antagonist, (RS)-1-Aminoindan-1,5-dicarboxylic acid (AIDA) (1 mM) was co-perfused with DHPG, suggesting that DHPG applied to synaptic regions <50 μm in length is sufficient to signal to the nucleus and increase Arc transcription. To determine if the increase in Arc mRNA was due to new transcription, and not an increase in stability of existing Arc mRNA, we performed the same experiment in the presence of the transcription inhibitor, Actinomycin D (10 μM); this blocked the increase in Arc mRNA indicating a requirement for new transcription (Figure 8C).
Figure 8. Perfusion of DHPG increases the nuclear transcription and localization of Arc mRNA in the dendrites.
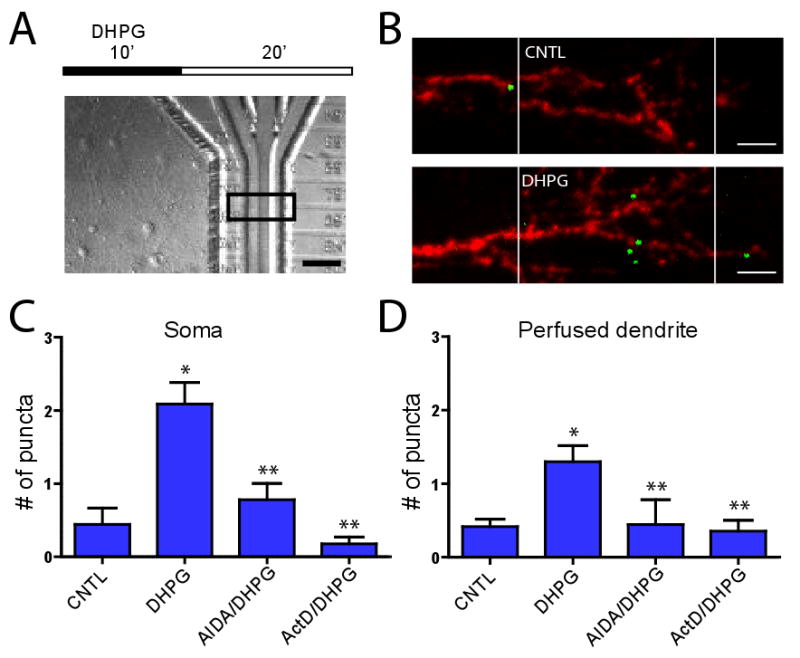
(A) Time course of experiment and example DIC image of perfusion within chamber. Scale bar = 50 μm. (B) Example images showing Arc mRNA (green) and 18S rRNA (red) 20 min following DHPG or control perfusions within the perfusion chambers. The boundaries of the perfusion channel are indicated by the white lines. Multiple Arc mRNA puncta are present within the DHPG-perfused chamber. Scale bar = 10 μm. (C) Mean (±SEM) number of Arc mRNA puncta identified within the soma of perfused dendrites for control (n = 45), DHPG (n = 80), AIDA/DHPG (n = 41), and ActD/DHPG (n = 17). (D) Mean (±SEM) number of Arc mRNA puncta identified within perfused dendrites (normalized to the average number of puncta in control treatment) within 50 μm perfusion channel and proximal 50 μm segment for control (n = 35), DHPG (n = 41), AIDA/DHPG (n = 10), and ActD/DHPG (n = 17). Significance was determined using the two-tailed Mann Whitney Test; * p < 0.05 vs control; ** p < 0.05 vs DHPG.
Next, we examined the number of Arc mRNA puncta within the perfused dendrites to determine if the newly transcribed Arc mRNAs are delivered to the region of synaptic stimulation. We quantified the number of puncta within a 100 μm section of the perfused dendrite (includes the perfusion channel and 50 μm proximal to the channel). We chose this region to quantify because we wanted to include Arc mRNA targeted to the perfusion region, including transcripts which may still be in transit due to the 20 min incubation [Arc mRNA are reported to be transported at a rate of 300 μm/h;(Wallace et al., 1998)]. The results show a significantly higher number of Arc mRNA puncta in the DHPG perfused dendrites over control perfusion, suggesting that following Arc transcription there is a redistribution of Arc mRNA into synaptic regions. Together, these results demonstrate that local group I mGluR activation at synapses triggers a signaling cascade to the nucleus to increase Arc transcription followed by Arc mRNA targeting into the perfused dendritic region.
Discussion
Here we describe and test a novel microfluidic local perfusion chamber for visualizing and manipulating synapses. Importantly, neurons can be cultured within microfluidic chambers for 3 weeks and longer. Both excitatory and inhibitory neurons inhabit the chamber (Figure S1). Furthermore, we demonstrate that we can organize neuronal connectivity between dissociated hippocampal neurons, resulting in the orderly organization of axons and dendrites in parallel rows distal from their cell bodies. The addition of a microperfusion channel that runs orthogonal to the dendrites and axons allows the spatially and temporally controlled addition of various drugs, transmitters etc. The dendrites present in the microgrooves can be readily imaged using Ca2+-indicators and presumably other dyes. As a proof of principle, we demonstrated that the μLP chambers can be used for synapse-to-nucleus signaling. We showed that local dendritic exposure caused increases in local as well as somatic calcium using live calcium imaging and that dendritic stimulation with spaced glutamate increased phospho-CREB in somata over massed stimulation. Lastly, we showed that DHPG applied to discrete (<50 μm) synaptic regions caused increases in Arc transcription within the nucleus and increased Arc mRNA targeted to the perfused dendrites.
An important feature of these microfluidic chambers is the ability to fluidically and genetically manipulate two neuronal populations independently. The genetic manipulation of each compartment facilitates not only the visualization of processes (via the expression of distinct fluorescent proteins) but also the ability to manipulate signaling molecules via dominant negative approaches. Future work could employ neurons from different brain regions to study physiologically relevant circuits. Neurodegenerative diseases often selectively impair certain circuits (e.g., the cholinergic pathway in Alzheimer's disease), making an in vitro model extremely valuable. Given the availability of numerous transgenic disease models, there are many possible configurations using the microfluidic chambers that will enable cell biological investigations of neurodegenerative disease.
This μLP chamber allows spatial and temporal control over neuronal microenvironments. By having multiple perfusion streams fuse into a single perfusion channel, the μLP chambers not only improve the spatial resolution of stimulation but also create thin fluidic barriers which enable the creation of isolated fluidic environments to presynaptic neurons, postsynaptic neurons, and synaptic regions simultaneously. These multiple fluid streams also allow temporal control over duration and frequency of stimulation (Chronis et al., 2007; Unger et al., 2000). Microfluidic technology has the potential to facilitate numerous investigations of synaptic formation, function, and plasticity. Future work will undoubtedly incorporate other microscale components, such as patterned microelectrodes, to create additional tools to study neurons and synapses of the CNS.
Experimental Procedures
Microfluidic chamber fabrication
The microfluidic chambers were fabricated using methods previously described in detail (Taylor et al., 2005; Taylor et al., 2006; Taylor et al., 2003). All microfluidic chambers were replica molded using poly(dimethylsiloxane) (PDMS) from masters which were patterned using the photosensitive epoxy SU-8 (Microchem) [reviewed in (Whitesides et al., 2001)]. All masters consisted of two permanent SU-8 layers on a 3″ silicon wafer and were made in the clean room facility in Michael Roukes' lab (Caltech). The first layer of SU-8 (3 μm depth) contained the microgrooves which were patterned by photolithography using a high resolution chromium mask (5 μm minimum feature size; Advance Reproduction Corp.). The second layer of SU-8 (100 μm depth) contained the compartments and perfusion channels which were patterned by photolithography using a 20,000 dpi printed transparency mask (CAD/Art Services, Inc.). To assess dendritic growth properties and synapse formation, we used previously described chambers with microgrooves 900 μm in length separating the compartments (Taylor et al., 2005). For the μLP chambers, we used the same mask design for the microgrooves, but the second layer mask was redesigned to include the additional perfusion channel and inlet wells and to have a reduced total microgroove length. We have included drawings of both of these masks with exact dimensions (Figure S3). The inlet wells for the perfusion channels were punched using 6 mm diameter sharpened stainless steel tubing. The outlet port was punched using a 15 gauge luer stub adapter (Intramedic).
Neuron cultures and viral infections
Dissociated postnatal (P0-2) rat hippocampal neurons were prepared as described previously (Aakalu et al., 2001). The PDMS replica molded microfluidic chambers were placed onto cover glass coated with poly-D-lysine (354210; BD Biosciences), then neurons were plated into the chambers (Taylor et al., 2005; Taylor et al., 2006; Taylor et al., 2003) at a density of 5×106 neurons per ml with 5 μl of cell suspension added to each well. During the incubation of the cultures, the withdrawal port was sealed to prevent evaporation until tubing was added for perfusion. All wells were filled with Neurobasal A/B27/Glutamax medium (150 μl for 8 mm wells; 100 μl for 6 mm wells). We waited one day before filling the perfusion channel to allow the neurons to attach sufficiently within the somatic compartments. To visualize potential contact between the separate neuron populations, we infected neurons with Sindbis viral vectors encoding either GFP or RFP. For isolating infections to one compartment, virus was added to 50 μl taken from the selected compartment, gently mixed, and then added back into the same compartment. Media within the compartment was recirculated three times to ensure proper mixing. The chambers were used 12 to 24 h after infection. Only chambers cultured longer than 14 days were used.
Microfluidic perfusions
For all perfusions, we used a syringe pump (SP230IW, WPI) with a 1 cc syringe at a withdrawal rate of 100 μL per h using polyethylene tubing (PE100, Intramedic) connected to the outlet port.
For μLP perfusions, we used or replaced medium with either Hibernate A (BrainBits, LLC) containing B27 (1:50; Invitrogen) or HBS. To visualize perfusions, we added 1-2 μM of the low molecular weight Alexa Fluor 488 hydrazide (570 m.w.; A10436; Invitrogen) or Alexa Fluor 568 hydrazide (731 m.w.; A10437; Invitrogen). Alternatively, to monitor perfusion without added fluorescent dyes (in the case of DHPG experiments), we first perfused medium containing 2.5-5% BSA, then added perfusate without BSA to center channel, which allowed us to visualize the start and duration of the perfusion due to the mismatch in refractive indices of the solutions without the use of a fluorescence microscope (see Figure 8A). To show isolation of pre- and post-synaptic compartments, we also used Alexa Fluor 633 hydrazide (1150 m.w.; A30634; Invitrogen).
For FM5-95 labeling, we used procedures similar to those previously published (Branco et al., 2008). First, we replaced the chamber medium with pre-warmed HBS for >30 min. We then perfused the postsynaptic compartment and perfusion channel simultaneously using 10 μM of FM5-95 in a solution containing 90 mM KCl, CNQX (20 μM), and APV (50 μM) for 1 min at room temperature. The postsynaptic compartment and perfusion channel were then rinsed in HBS containing FM5-95 for an additional minute. The postsynaptic compartment and perfusion channel were then washed with HBS containing 0.5 mM Ca2+ and 10 mM Mg2+ to reduce dye loss from spontaneous vesicle release and Advasep-7 (1 mM, Biotium) to facilitate FM dye removal from membranes.
Glutamate (G-5889; Sigma) stock solution (200 mM) was made in HBS and used at a final concentration of 200 μM. For glutamate perfusions, Alexa Fluor 568 (1-2 μM) was added to the glutamate solution to visualize the start and duration of glutamate exposure. We performed experiments in the presence of TTX (1 μM) to eliminate spiking activity which could influence calcium activity. For calcium imaging experiments we used Fluo-4 NW (F36206; Invitrogen) Calcium Assay Kit. We first incubated cell compartments of the chambers with prewarmed Fluo-4 NW dye mix (0.5×) and 1.25 μM probenecid diluted in HBSS with 20 mM HEPES; the 3 inlet wells for the perfusion channel were filled with HBSS and 20 mM HEPES. The chambers were then incubated at 37°C for >20 min. Before beginning the perfusion, the center inlet well was replaced with 200 μM glutamate diluted in HBSS with 20 mM HEPES.
For the spaced treatments, the flow was either reduced to 25 μl/h during the non-glutamate periods or stopped completely. When flow was reduced to 25 μl/h during the glutamate rest periods, the massed treatments received an equivalent amount to time (10 min) with 25 μl/h perfusion immediately after the 3 min glutamate perfusion.
For DHPG perfusions the 8 mm wells which access the neurons were replaced with Hibernate A medium with B27, and the inlet wells were replaced with freshly prepared 2.5-5% BSA. After replacing media, chambers were incubated for >30 min at 37°C. The syringe and tubing were filled with HBS then connected to the chamber. The BSA solution was perfused for ∼5 min prior to treatment, then the solution in the center inlet well was replaced with the pharmacological agent or vehicle control diluted in Hibernate A with B27. We visualized the perfusion of the non-BSA solution using a brightfield tissue culture microscope (Figure 8A).
For DHPG treated chambers, 500 μM DHPG (0342; Tocris Bioscience) solution was perfused for 10 min at 32-37° C. For AIDA controls, 1 mM AIDA (0904; Tocris Bioscience) was first perfused for 5 min, then 1 mM AIDA with 500 μM DHPG was perfused for 10 min. For Actinomycin D controls, 10 μM of Actinomycin D was incubated within the chambers for 30-45 min, followed by DHPG perfusion as described above. The control treated chambers consisted of vehicle (500 μM NaOH). Following DHPG perfusions, all the chambers were incubated for 20 min at 37° C. Chambers were then fixed and processed for FISH (see below).
Electrophysiology
For whole-cell patch-clamp recordings, we removed the top PDMS piece of the microfluidic chamber allowing access to the hippocampal neurons attached to the cover glass surface. The neurons were bathed in HEPES-buffered saline (HBS; containing, in mM: 119 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 30 Glucose, 10 HEPES, pH 7.4) plus 1 μM TTX. We used an Axopatch 200B amplifier. Whole-cell pipette internal solution contained, in mM: 115 cesium gluconate, 20 cesium chloride, 10 sodium phosphocreatine, 10 HEPES, 0.2 EGTA, 2 MgATP, 0.3 NaGTP (pH 7.3). Neurons with pyramidal-like morphology were voltage-clamped at -70 mV, and series resistance was less than 20 MΩ and left uncompensated. mEPSCs were recording for 20 min in the presence of 1 μM TTX. The recording was continued following addition of a 5 μl bolus of 5 mM APV and 2 mM NBQX to block all fast excitatory synaptic transmission.
Immunocytochemistry
Cells were fixed at room temperature for 20-30 min with 4% formaldehyde/4% sucrose in PBS, permeabilized with 0.25% Triton-X in PBS, and blocked with 2% BSA/4% goat serum in PBS. The following primary antibodies were used: mouse anti-microtubule-associated protein 2 (MAP2) monoclonal (1:1000; Sigma; M4403), rabbit anti-MAP2 polyclonal (1:1000; Chemicon; AB5622); mouse anti-bassoon monoclonal (1:1000; Stressgen Bioreagents; VAM-PS003), anti-GAD67 (1:1000; Millipore; MAB5406), chicken anti-GFP polyclonal (1:500; Aves Lab; GFP-1020), and mouse anti-phospho-CREB (Ser133) monoclonal (1:1000; Millipore; 05-807). For secondary antibodies, we used anti-mouse, anti-rabbit, or anti-chicken conjugated Alexa Fluor 488, 568, or 647 (1:500; Invitrogen).
For assessing dendritic growth and synaptic properties within the microfluidic chambers, we focused on dendrites extending greater than 100 μm into the microgrooves since these dendrites are sufficiently removed from cell bodies to enable visualization and manipulation of synapses in isolation. We measured both the total number and length of the MAP2-stained dendrites for two chambers at each culture age.
Fluorescence In Situ Hybridization (FISH)
Fluorescence in situ hybridization procedures (Panomics) used to amplified low copy mRNA targets were used to visualize Arc and 18S mRNA within neurons (Taylor et al., 2009). PDMS top pieces were removed and cells attached to the glass coverslips were fixed immediately using 4% formaldehyde/4% sucrose in PBS for 20 min, followed by ethanol dehydration. After rehydration, cells were permeabilized using a detergent solution (Panomics) for 5 min. We used proteinase K (1:6000; Panomics) to digest proteins for 10 min at room temperature, followed by in situ hybridization using Arc (anti-sense or sense) and 18S probes designed by Panomics, following the manufacturer's instructions. Briefly, probes were diluted 1:100 in hybridization buffer supplied by Panomics, incubated at 40° C (3-5 h), washed, hybridized with pre-amplification oligos (1:100) at 40° C (25 min), washed, hybridized with amplification oligos (1:100) at 40° C (15 min), washed, and finally hybridized with the label oligos (1:100) at 40° C (15 min).
To visualize dendrites in order to quantify the number of Arc mRNA puncta, we either performed simultaneous in situ hybridization with 18S rRNA (which was then conjugated to a 568 wavelength fluorophore) or immunostained for ARC protein following FISH. MAP2 immunostaining was not compatible with FISH.
Microscopy
For live imaging of RFP and GFP infected neurons, a Zeiss LSM 510 Meta laser scanning confocal microscope was used. Ar and HeNe lasers were used for excitation wavelengths of 488 and 546, respectively. We used the following emission filters: LP 505 or BP 565-615. For all other images, we used an Olympus IX-70 laser scanning confocal microscope. For 488 wavelength fluorophores, the Ar ion laser (488 excitation wavelength) was used with long pass (BA510IF) and short pass (BA550RIF) filters. For 546 and 568 fluorophores, a Kr laser (568 and 647 excitation wavelengths) was with long pass (BA585IF) and band pass (605BP) filters. For 647 fluorophores, the Kr laser was used with long pass (BA585IF) and band pass (700BP) filters.
For analysis of flow properties, Alexa Fluor 488 (Invitrogen) was used to visualize perfusion. XZ line scans were taken every 3 min transecting both the local perfusion and a microgroove. Fluorescence intensity was measured from region of interests (ROIs) (50 μm length, 5 μm high) within the microgroove and local perfusion channel close to the glass surface where neurons and their processes attach. Any detectable fluorescence in the microgroove proximal to the postsynaptic cell bodies would indicate diffusion or leakage of perfusate. Mean pixel values were taken from 16-bit images. Fast XT line scans were taken at a rate of 0.00316 seconds per line.
For FM5-95 dye loading, Kr laser excitation at 568 was used with long pass (BA585IF) and band pass (700BP) filters using a 60× water immersion lens (NA 1.2). For Fluo4-NW imaging the confocal aperture was opened to setting 4 and images were acquired continuously every 1.74 s.
Image processing
All image processing was performed using ImageJ (NIH). Mean pixel values were measured from ROIs were taken of a dendritic segment within the perfusion channel and also within the soma. The mean pixel values were normalized to the initial measurement taken at the start of the recording. For the fluorescence difference images, Fluo-4 frames before vehicle and glutamate perfusions were subtracted from frames following perfusions. We used the TurboReg plugin to register the fluorescence difference images with the anti-MAP2 immunostained image. We then used the lowest pixel value calculated by comparing each fluorescence difference image with the MAP2 immunostained image, resulting in the detection of fluorescent intensity changes in MAP2-positive neurons.
Arc puncta were quantified using ImageJ software by first thresholding 8-bit images to 50-70 minimum pixel values to reduce background signal. Particles were counted with 0.2-2 μm2 area and 0.5-1 circularity. These parameters were determined by comparing Arc images with sense control images.
Statistical Analyses
Statistical analyses for Arc mRNA puncta quantification were performed using GraphPad Prism (GraphPad software). Statistical analysis for pCREB data were performed using Excel (Microsoft).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Lin Chen for the hippocampal neuron cultures, Michael Roukes for use of his clean room, Julie Cho for help with preliminary experiments, and Jennifer Hodas for reviewing the manuscript. AMT was partially supported by a Johnson & Johnson fellowship. We acknowledge support from the Weston Havens Foundation, NIH, and Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aakalu G, Smith WB, Nguyen N, Jiang C, Schuman EM. Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron. 2001;30:489–502. doi: 10.1016/s0896-6273(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Branco T, Staras K, Darcy KJ, Goda Y. Local dendritic activity sets release probability at hippocampal synapses. Neuron. 2008;59:475–485. doi: 10.1016/j.neuron.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew TJ, Pinsker HM, Kandel ER. Long-term habituation of a defensive withdrawal reflex in aplysia. Science. 1972;175:451–454. doi: 10.1126/science.175.4020.451. [DOI] [PubMed] [Google Scholar]
- Chronis N, Zimmer M, Bargmann CI. Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat Methods. 2007;4:727–731. doi: 10.1038/nmeth1075. [DOI] [PubMed] [Google Scholar]
- Dertinger SKW, Jiang X, Li Z, Murthy VN, Whitesides GM. Gradients of substrate-bound laminin orient axonal specification of neurons. Proc Natl Acad Sci U S A. 2002;99:12542–12547. doi: 10.1073/pnas.192457199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Ali J, Sorger PK, Jensen KF. Cells on chips. Nature. 2006;442:403–411. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- Harvey CD, Yasuda R, Zhong H, Svoboda K. The spread of Ras activity triggered by activation of a single dendritic spine. Science. 2008;321:136–140. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon NL, Baskaran H, Dertinger SK, Whitesides GM, Van de Water L, Toner M. Neutrophil chemotaxis in linear and complex gradients of interleukin-8 formed in a microfabricated device. Nat Biotechnol. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- Lee SJR, Escobedo-Lozoya Y, Szatmari EM, Yasuda R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature. 2009;458:299–304. doi: 10.1038/nature07842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JC, Duffy DC, Anderson JR, Chiu DT, Wu H, Schueller OJ, Whitesides GM. Fabrication of microfluidic systems in poly(dimethylsiloxane) Electrophoresis. 2000;21:27–40. doi: 10.1002/(SICI)1522-2683(20000101)21:1<27::AID-ELPS27>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, et al. Elongation Factor 2 and Fragile X Mental Retardation Protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl M, Sprössler C, Denyer M, Krause M, Nakajima K, Maelicke A, Knoll W, Offenhäusser A. Ordered networks of rat hippocampal neurons attached to silicon oxide surfaces. J Neurosci Methods. 2000;104:65–75. doi: 10.1016/s0165-0270(00)00325-3. [DOI] [PubMed] [Google Scholar]
- Song Hj, Ming Gl, Poo Mm. cAMP-induced switching in turning direction of nerve growth cones. Nature. 1997;388:275–279. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- Stenger DA, Hickman JJ, Bateman KE, Ravenscroft MS, Ma W, Pancrazio JJ, Shaffer K, Schaffner AE, Cribbs DH, Cotman CW. Microlithographic determination of axonal/dendritic polarity in cultured hippocampal neurons. J Neurosci Methods. 1998;82:167–173. doi: 10.1016/s0165-0270(98)00047-8. [DOI] [PubMed] [Google Scholar]
- Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron. 2007;55:648–661. doi: 10.1016/j.neuron.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Berchtold NC, Perreau VM, Tu CH, Li Jeon N, Cotman CW. Axonal mRNA in uninjured and regenerating cortical mammalian axons. J Neurosci. 2009;29:4697–4707. doi: 10.1523/JNEUROSCI.6130-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Rhee SW, Jeon NL. Microfluidic chambers for cell migration and neuroscience research. Methods Mol Biol. 2006;321:167–177. doi: 10.1385/1-59259-997-4:167. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Rhee SW, Tu CH, Cribbs DH, Cotman CW, Jeon NL. Microfluidic multicompartment device for neuroscience research. Langmuir. 2003;19:1551–1556. doi: 10.1021/la026417v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooker A, Erickson J, Chow G, Tai YC, Pine J. Parylene neurocages for electrical stimulation on silicon and glass substrates. Conf Proc IEEE Eng Med Biol Soc. 2006;1:4322–4325. doi: 10.1109/IEMBS.2006.260472. [DOI] [PubMed] [Google Scholar]
- Tooker A, Meng E, Erickson J, Tai YC, Pine J. Development of biocompatible parylene neurocages. Conf Proc IEEE Eng Med Biol Soc. 2004;4:2542–2545. doi: 10.1109/IEMBS.2004.1403731. [DOI] [PubMed] [Google Scholar]
- Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- Vogt AK, Stefani FD, Best A, Nelles G, Yasuda A, Knoll W, Offenhäusser A. Impact of micropatterned surfaces on neuronal polarity. J Neurosci Methods. 2004;134:191–198. doi: 10.1016/j.jneumeth.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Vogt AK, Wrobel G, Meyer W, Knoll W, Offenhausser A. Synaptic plasticity in micropatterned neuronal networks. Biomaterials. 2005;26:2549–2557. doi: 10.1016/j.biomaterials.2004.07.031. [DOI] [PubMed] [Google Scholar]
- Wallace CS, Lyford GL, Worley PF, Steward O. Differential intracellular sorting of immediate early gene mRNAs depends on signals in the mRNA sequence. J Neurosci. 1998;18:26–35. doi: 10.1523/JNEUROSCI.18-01-00026.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Soft lithography in biology and biochemistry. Annu Rev Biomed Eng. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- Yin JCP, Wallach JS, Del Vecchio M, Wilder EL, Zhou H, Quinn WG, Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.