

Abstract
Current treatment for Duchenne Muscular Dystrophy (DMD) is chronic administration of the glucocorticoid prednisolone. Prednisolone improves muscle strength in boys with DMD, but the mechanism is unknown. The purpose of this study was to determine how prednisolone improves muscle strength by examining muscle contractility in dystrophic mice over time and in conjunction with eccentric injury. Mdx mice began receiving prednisolone (n=23) or placebo (n=16) at 5-wks of age. Eight wks of prednisolone increased specific force of the EDL muscle 26%, but other parameters of contractility were not affected. Prednisolone also improved the histological appearance of muscle by decreasing the number of centrally-nucleated fibers. Prednisolone treatment did not affect force loss during eccentric contractions or recovery of force following injury. These data are of clinical relevance, because the increase in muscle strength in boys with DMD taking prednisolone does not appear to occur via the same mechanism in dystrophic mice.
Keywords: Duchenne Muscular Dystrophy, skeletal muscle function, glucocorticoids
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a progressive neuromuscular disorder affecting 1 out of 3500 live male births 11. This disease arises from a mutation in the dystrophin gene. Lack of this important structural protein that links the cytoskeleton to the extracellular matrix leads to muscle weakness and atrophy, limited mobility, and joint contractures. Boys with DMD eventually rely completely on wheelchairs for locomotion and ultimately succumb to the disease in their late to mid-twenties via failure of the respiratory muscles.
While there is no known cure, the current pharmacological treatment for DMD patients is the glucocorticoid prednisone or prednisolone. This paper will use the term prednisolone, because it is the active form of the drug. While prednisolone’s mechanism of action in dystrophic muscle is not known, glucocorticoids are typically used to suppress inflammation. This class of drugs reduces inflammation by binding to glucocortiocoid receptors which (1) bind to and enhance the transcription of anti-inflammatory genes and (2) bind to and inhibit pro-inflammatory factors, such as nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) 2, 9. A recent meta-analysis on prednisolone use in boys with DMD shows positive effects on strength and muscle performance 27. Prednisolone treatment at 0.75 mg/kg body weight per day is the most common dose used for optimal increases in strength in boys, which occur in as little as 10 days to 1 month of treatment 3, 17, 29. These improvements continue for 2 to 3 months, but eventually strength gains plateau after 3 to 6 months of treatment 3, 17, 29, 38. It is important to note that prednisolone can only delay disease progression, such as delaying the time until a patient becomes wheelchair dependent, and it is not sufficient to reverse the course of the disease. Nonetheless, these transient increases in muscle strength and performance are consistently seen with prednisolone in the DMD population and are functionally meaningful to the patient.
To understand physiological and molecular responses associated with various treatment strategies for DMD, investigators have turned to the mdx mouse. The mdx mouse has a single point mutation in the dystrophin gene and these mice demonstrate many characteristics seen in boys with DMD including muscle weakness 39. Multiple studies have assessed voluntary strength or performance of mdx mice in response to prednisolone treatment, and results have been conflicting, with some studies detecting improved performance 10, 13, 16, 18, 22 and others failing to detect an effect 10, 15, 36. One limitation to these previous studies is the use of voluntary measurements of strength or performance in rodents. More precise and sensitive methodologies for examining muscle contractile function may provide additional insight into the mechanism of prednisolone’s clinically-meaningful action in dystrophic muscle.
In addition to losses in strength, DMD patients and mdx mice are extremely susceptible to contraction-induced injury 37, 47. Markers of accumulated muscle damage include centrally-nucleated fibers and high creatine kinase (CK) activity in the circulation. Fewer centrally-nucleated fibers have been detected in muscles from boys with DMD, 19 and mdx mice receiving prednisolone 36. However, other studies have failed to find this association 1, 12, 45, 48. Serum CK levels are typically not affected by prednisolone treatment in the mdx mouse 1, 13, 15, 16, 36, 48 or in boys with DMD 19. Both utrophin and dystrophin are important structural proteins that prevent eccentric contraction-induced injury. Prednisolone can also increase utrophin and dystrophin protein expression in healthy and DMD primary muscle cultures 8, 33, 40, as well as in wild-type and dystrophic mouse muscle cells 34. However, other studies have failed to find this association in both rodents and humans 6, 12, 21. While prednisolone did not affect force loss during eccentric injury to the diaphragm in mdx mice 49, it is unknown if prednisolone can prevent eccentric force loss in hindlimb skeletal muscles. There is also evidence that prednisolone can benefit certain aspects of skeletal muscle regeneration following injury. For example, prednisolone can increase the number of regenerating fibers in boys with DMD 19 and induce myogenesis in culture 30, 34, 40. However, the effect of prednisolone on the ability of skeletal muscle to produce force following physiological injury in the mdx mouse has not been investigated.
There are multiple lines of evidence that prednisolone improves muscle strength and physical abilities in boys with DMD. The evidence that prednisolone improves muscle in the mdx model is equivocal, and all previous studies have measured voluntary strength or performance, not muscle contractile function. The primary purpose of this study was to determine if prednisolone can improve parameters of muscle contractility in the mdx mouse which would result in better overall muscle performance. We hypothesized that prednisolone would improve parameters of skeletal muscle contractility related to force-generating capacity. The secondary purpose of this study was to determine if prednisolone can prevent the loss of force during injury and improve the recovery of contractile function from that injury. We hypothesized that prednisolone would not affect the initial damage but would hasten the recovery process.
MATERIALS AND METHODS
Animals and study design
Male mdx mice aged 4 wk (n=39) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were group housed and on a 12:12-h light:dark cycle. Mice were provided food and water ad libitum. After 1-wk of acclimation, mdx mice were randomly assigned to receive 60-d time-released prednisolone or placebo pellets for a total of 8 wks. Mice were anesthetized with fentanyl citrate (10 mg/kg BW), droperidol (0.2 mg/kg BW) and diazepam (5 mg/kg BW), and an ~3-mm incision was made with a trochar on the dorsal side of the neck for pellet implantation. Two studies were completed to address the overall purpose of the study (See Figure 1). Mice were sacrificed under sodium pentobarbital anesthesia after 8 wks of prednisolone administration. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Minnesota.
Figure 1.
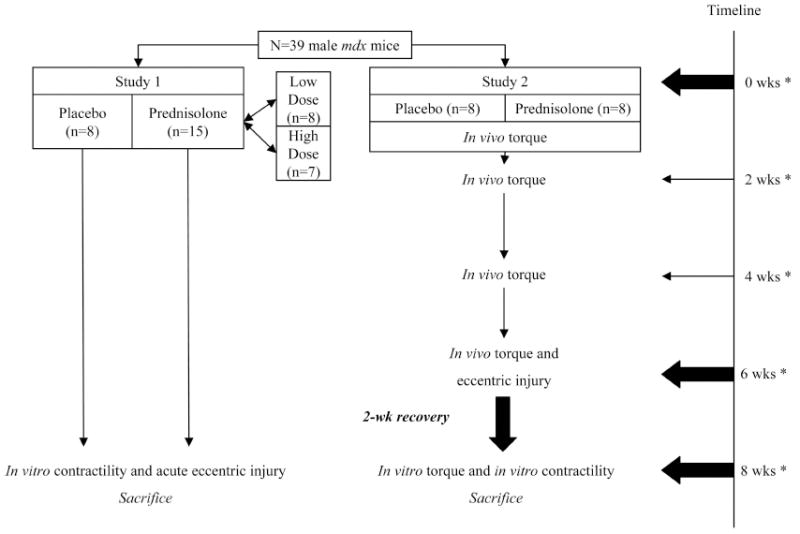
A total of 39 5-wk-old male mdx mice were used in two different studies. In the first study, mice were randomly assigned to receive placebo (n=8), low-dose prednisolone (n=8), or high-dose prednisolone (n=7). T-tests were performed between low and high prednisolone for all variables, and no differences were found. These groups were combined into one group (n=15). After 8 wks of prednisolone treatment, EDL muscles were assessed in vitro for contractile function and susceptibility to an eccentric contraction-induced injury. In the second study, mice received placebo (n=8) or prednisolone (low-dose only; n=8). Torque was measured in vivo every 2 wks. After 6 wks of prednisolone treatment, physiological eccentric injury was induced in the left anterior crural muscles. Mice were allowed 2 wks of recovery. In vivo torque was then re-assessed as well as in vitro contractility of EDL muscles. *Denotes when active torque measurements were made.
Study 1
Mice were implanted with either a low dose (1.5 mg prednisolone; approximately 0.8-1.3 mg/kg BW/d; n=8), a high dose (2.5 mg prednisolone; approximately 1.4-2.1 mg/kg BW/d; n=7), or placebo pellet (n=8) at age 5-wk (Innovative Research of America; Sarasota, FL). These pellets release a constant dose of the hormone up to 60 days using the Matrix-Driven Delivery pellet system. The matrix of the pellet consists of cholesterol, cellulose, lactose, phosphates and stearates and is fused with prednisolone. The prednisolone is released at a constant dose by both erosion of the pellet and diffusion of the prednisolone. These doses were chosen, because they are similar to what boys with DMD receive in the clinic and mimic the 1-2 mg/kg BW/d dose that has typically been used in mdx mice to improve voluntary measures of muscle performance 10, 13, 16, 18, 22. These prednisolone pellets have been used in other studies and are sufficient to induce bone loss over 28 days, body mass loss within the first 7 days of implantation, and do not alter food intake 24, 46. The placebo pellets contained the same matrix as the prednisolone pellets, but did not contain any hormone. Mice were euthanized after 8 wks of placebo or prednisolone treatment. These mice were used to study the functional and catabolic effects of prednisolone on extensor digitorum longus (EDL) muscles and also its acute effect on muscle injury.
Study 2
In the second study, only the lower dose of prednisolone (n=8) and placebo (n=8) pellets were implanted in mice at age 5-wk. The lower dose was selected, because the 1.5 mg dose of prednisolone was released over a 60-d period, and this dosage approximated the 0.75 mg/kg body weight per day that is common for DMD patients. Also, there was no further improvement with the higher dose in Study 1. Contractile function of the anterior crural muscles was measured in vivo every 2 wk from the age of 5 to 11 wk in order to determine if prednisolone affected disease progression. The effects of prednisolone on the induction of an in vivo injury and then the subsequent recovery from that injury were also determined (Figure 1). Physiological injury was induced in vivo via eccentric contractions (See In vivo torque measurements and injury protocol) after 6 wks of placebo or prednisolone administration. Mice were euthanized 2 wks later, and muscle recovery was assessed at this time point. Recovery from injury was assessed by in vitro and in vivo muscle function, serum CK, and centrally-nucleated fibers. The right leg served as an uninjured control muscle for assessing the effects of prednisolone on muscle size and the percentage of centralized nuclei in the absence of injury. Body mass and food intake were also monitored throughout this 8-wk study. Food intake was measured every 2 wk for 3 consecutive days for each cage of mice (4 mice/cage) and was averaged for a weekly assessment.
In vitro contractility measurements and injury protocol
The EDL muscles were assessed for baseline contractile function as previously described 32. Mice were anesthetized with sodium pentobarbital (100 mg/kg BW). Muscles were mounted on a dual-mode muscle lever system (300B-LR; Aurora Scientific Inc., Aurora, ON, Canada) with 6-0 suture in a 0.38-ml bath assembly filled with Krebs Ringer bicarbonate buffer that was maintained at 25 °C. Muscles were adjusted to their anatomic optimal length (Lo) based on resting tension. Muscle length was then measured from myotendonous junction to myotendonous junction using digital calipers. Muscles remained quiescent in the bath for 10 min before beginning a protocol for testing contractile function. First, passive stiffness of the muscle was determined by stretching the muscle sinusoidally from 97.5% Lo to 102.5% Lo at 0.5 Hz while measuring the resulting force 14, 41. Thirty seconds later peak twitch force was elicited by stimulating the muscle with a 0.5-ms pulse at 150 V (Grass S48 stimulator delivered through a SIU5D stimulus isolation unit; Grass Telefactor, Warwick, RI). A second twitch was elicited 30 s later. A maximal isometric tetanic contraction (Po) was next performed by stimulating muscles for 400 ms at 180 Hz. Two minutes later, a second tetanic isometric contraction was elicited, and at peak force a sinusoidal length oscillation of 0.01% Lo at 500 Hz was imposed to determine active stiffness 14, 41. This contraction protocol was used for both Study 1 and Study 2.
For the first study, following baseline contractility measures, an eccentric contraction injury protocol was performed on EDL muscles as previously described 31 with the following modifications. The injury protocol consisted of 5 eccentric contractions. For these contractions, muscles were passively shortened from Lo to 0.95 Lo over 3 s, stimulated tetanically for 133 ms as the muscle lengthened to 1.05 Lo at 0.75 Lo/s, and then passively returned to Lo. Each eccentric contraction was separated by 3 min of rest. This protocol has been validated so that fatigue does not occur between each eccentric contraction, and force does not return to baseline values after 2 hours post-injury 26. Three minutes following the final eccentric contraction, Po was measured again to assess the degree of injury. Lactate dehydrogenase (LDH) released into the bath was also assayed as a marker of membrane damage as previously described 31. Five μl of Krebs buffer was withdrawn immediately before the first and following the final eccentric contraction to determine the change in LDH activity. LDH assays were conducted as described by Warren and coworkers 43 except the assays were done at 25°C.
For both studies, muscles were removed from the bath assembly at the end of the contractile protocol, trimmed, blotted, weighed, and frozen in liquid nitrogen. Muscle weight and length data, using a fiber length-to-muscle length ratio of 0.44 was used to calculate physiological cross-sectional area for determination of specific Po 5, 44. The EDL was cut at the mid-belly. One half was mounted in optimum cutting temperature (OCT) compound for histology, and the other half was frozen in liquid nitrogen.
For the second study, tibialis anterior (TA) muscles were cut at the mid-belly. One half was mounted in OCT and frozen in liquid nitrogen-cooled isopentane for histology, while the other half was frozen in liquid nitrogen. All muscles were stored at -80°C until further analysis.
In vivo torque measurements and injury protocol
In vivo maximal isometric torque was measured as previously described 20. Mice were given a mixture of fentanyl citrate (10 mg/kg BW), droperidol (0.2 mg/kg BW) and diazepam (5 mg/kg BW). Once a mouse was sedated, the hair on the left hindlimb was shaved and removed with depilatory cream. Betadine and ethanol were applied to clean the skin. The mouse was positioned on its right side on a Plexiglas chamber that was heated to 39 °C (Temperature Controller, model 89810-04, Cole-Parmer, Vernon Hills, IL). The left foot of the mouse was taped to a metal foot plate which was connected to the shaft of the servomotor (model 300B-LR, Aurora Scientific, Aurora, Ontario, Canada). The center of rotation for the ankle was aligned with the longitudinal axis of the servomotor shaft. The forelimbs and right foot were also taped to the chamber to secure the mouse. The left knee was stabilized with a knee clamp so that the tibia was perpendicular to the foot.
Two sterilized platinum subdermal needle electrodes (model E2-12, Grass Technologies, West Warwick, RI, USA) were inserted through the skin 1-2 mm on either side of the left common peroneal nerve. One electrode was inserted just distal to the fibular head, while the other electrode was inserted approximately 1 cm distal to the first electrode. The peroneal nerve was stimulated via the platinum electrodes with a stimulator and stimulus isolation unit (models S48 and SIU5, respectively, Grass Technologies, West Warwick, RI, USA) to cause contraction of the anterior crural muscles (TA, EDL, and extensor hallucis longus muscles). The parameters on the stimulator were set at a 200-ms contraction duration consisting of 0.5-ms square-wave pulses at 300 Hz to produce maximal isometric tetanic torque at the ankle. The voltage on the stimulator started at 3.0 V and was adjusted between 1.0 and 8.0 V until torque no longer increased. Peak torque was remeasured every 2 wks by the same investigator.
The in vivo eccentric contraction protocol was performed as previously described 20. The stimulator settings were kept the same during the eccentric contraction protocol, except for the voltage. The voltage was gradually increased 0.2 V every 20 contractions. If a greater torque was produced than the previous contraction, the voltage remained 0.2 V greater for the next 20 contractions. If torque did not improve with the increased voltage, the voltage was returned to the previous setting. The servomotor passively dorsiflexed the foot 20° over a duration of 3 s, and the nerve was stimulated while the foot was plantar flexed 40° at a velocity of -1200 °/s. After the stimulation, the foot was passively dorsiflexed 20° to its original resting position over a 3-s period. One contraction was performed every 12 s for a total of 150 eccentric contractions. After a 2 min rest at the end of the protocol, maximum isometric torque was reassessed. Preliminary data confirmed that isometric torque remained depressed >50% 3 days after injury, verifying that this protocol does induce injury rather than fatigue (data not shown). Isometric torque results are presented as the raw data, per kg of body mass, and for the final time point, per g of muscle mass (sum of TA and EDL muscle masses).
Voluntary cage activity
After 5 wks of treatment, all mdx mice from Study 2 were monitored for 24-hr cage activities using activity chambers from Med Associates Inc. (St. Albans, Vermont) as previously described 23. Briefly, mice spent 24 h in a mock chamber to acclimate them to the testing environment. After the acclimation period, mice were placed in the activity chambers where ambulation distance was measured for 24 h. All data were acquired using Activity Monitor version 5 software (Med Associates Inc.) on a PC computer.
Serum creatine kinase
Approximately 75 μl of blood was obtained from the retrooribital sinus under isoflurane anesthesia every 2 wks in conjunction with the in vivo torque measurements. Blood was allowed to clot at room temperature for 15 min and centrifuged at 10,000 g for 10 min at 4 °C. Serum was stored at -80°C until analysis. Ten μl of serum sample was dropped onto a Vitros CK/CPK slide and was run on a spectrophotometer (Ortho Clinical Vitros DT 60 II Dry Slides and System) at 37°C and at wavelength 670 nm. Activity was then reported in U/l. The active range for the Ortho Clinical Vitros DT 60 II System is 20 – 1600 U/l, so samples were diluted 1:10 in sterilized phosphate buffered saline to fit within the range.
Muscle fiber cross-sectional area and central nuclei
Transverse sections (10 μm) were cut from the mid-belly of the TA and EDL muscles on a cryostat at -20°C. Hematoxylin and eosin staining was performed on the sections to measure fiber cross-sectional area. Preliminary work was done on 4 samples to determine the number of fibers necessary to measure. It was determined that the mean and SD of each individual sample plateaued after 150 fibers. Digital photographs were taken from each section at a 200X magnification with a digital camera (Micropublisher 5.0, QImaging, Surrey, British Columbia, Canada) mounted to a microscope (DM2000, Leica Microsystems Inc., Bannockburn, IL) using QCapture software (QImaging, Surrey, British Columbia, Canada). To insure at least 150 fibers were sampled per section, 2 pictures were taken for the EDL, and 3 pictures were taken for the TA. Care was taken to insure that the entire section was represented, and the photographer was blinded to the treatments while imaging the slides. Approximately 300 fibers from both the EDL (range: 224-481 fibers) and TA (range: 227-506 fibers) were assessed per session by the same investigator who was also blinded to the treatment groups. Fiber area was traced with imaging software (ImageJ, Frederick, MD) and a digital pen tablet (Intuos, Wacom Technology Coporation, Vancouver, WA).
The same images that were used for fiber CSA were also used to count fibers with centralized nuclei, a marker of fiber regeneration. A centralized nucleus was defined as a nucleus that was closer to the center of the fiber than the periphery. Data are expressed as the percentage of centrally-nucleated myofibers from the total myofibers assessed.
Statistics
Data are presented as means ± SE. For study 1, independent t-tests were performed between low (n=8) and high (n=7) prednisolone-treated mice on body weight (P=0.838), twitch force (P=0.907), Po (P=0.505), and specific force (P=0.944). Because there were no differences in any of these measures, data were collapsed for the prednisolone groups in study 1. Chi-square analysis was used to determine differences in fiber size distribution. A repeated measures two-way ANOVA was used to determine differences in CK activity and body mass between placebo-and prednisolone-treated mice with time as the repeated factor. If an interaction existed, Tukey post-hoc measures were used to determine differences. All other data were analyzed with independent t-tests comparing placebo- and prednisolone-treated mice within each study. Significance was set at P<0.05.
RESULTS
Study 1: The effects of prednisolone on muscle size and contractility
Prednisolone is a classical catabolic hormone that induces body mass loss in rodents 24, 46. Body mass was not different between placebo (16.6 ± 0.5 g) and prednisolone (17.9 ± 0.6 g; P=0.145) groups prior to treatment. Prednisolone-treated mice weighed 7-17% less than placebo-treated mice after 2, 3, 4, and 5 wks of treatment, and they weighed 6% less at the end of the study (28.8 ± 0.5 vs. 30.7 ± 0.7 g; P=0.038; Figure 2). Similar catabolic effects were found in the EDL muscle (Table 1). Eight wks of prednisolone administration resulted in 15% smaller EDL muscles. To determine if muscles from prednisolone-treated mice were losing water, muscles were lyophilized to assess dry mass (Table 1). There were no differences between prednisolone and placebo treatment on EDL dry mass, but the dry:wet mass ratio was greater in prednisolone-treated mice.
Figure 2.
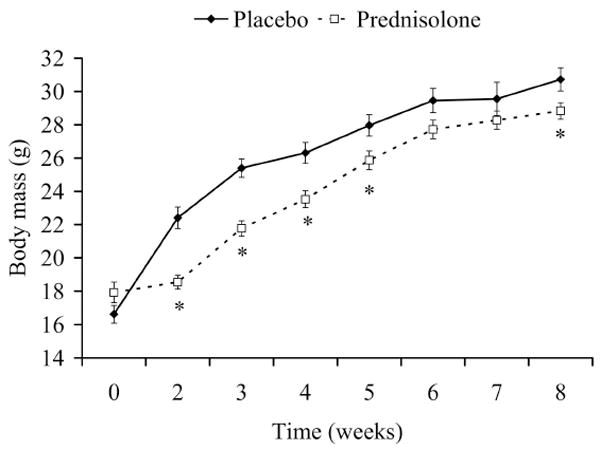
Body mass over 8 wks of prednisolone treatment in mdx mice. *Signifies different from Placebo.
Table 1.
EDL muscle masses of mdx mice that were treated with a placebo or prednisolone for 8 wks.
| Placebo (n=8) | Prednisolone (n=15) | P-value | |
|---|---|---|---|
| EDL wet mass (mg) | 17.6 ± 1.0 | 14.9 ± 3 | 0.005 |
| EDL dry mass (mg) | 3.5 ± 0.1 | 3.3 ± 0.1 | 0.143 |
| EDL dry:wet mass (%) | 20.3 ± 0.8 | 22.4 ± 0.5 | 0.029 |
To determine if a change in fiber size contributed to the prednisolone-induced loss of EDL muscle mass, fibers from this muscle were assessed histologically (Figure 3A & 3B). EDL muscles from prednisolone-treated mdx mice had an overall mean fiber CSA that was 21% lower than those from placebo-treated mice (Figure 3C; P=0.040). The frequency distribution of fiber size also showed that EDL muscles from prednisolone-treated mdx mice had a shift toward smaller fibers (Figure 3D). Small fibers were categorized as those being less than 500 μm2, and large fibers were greater than 3000 μm2. Mice that received prednisolone had twice as many small fibers (12% vs. 6%; P<0.001) and 38% fewer large fibers (8% vs. 13%; P<0.001) compared to placebo-treated mice. Centralized nuclei are used as a marker of muscle fiber degeneration-regeneration history, and these centrally-nucleated fibers are prominent in mdx mice and accumulate with age 35. The number of centrally-nucleated fibers in the EDL was 20% lower in prednisolone-treated mice (69 ± 2 vs. 55 ± 4%; P=0.027). Collectively, these data indicate that prednisolone treatment of mdx mice leads to smaller muscle mass and myofibers and improved histological appearance.
Figure 3.
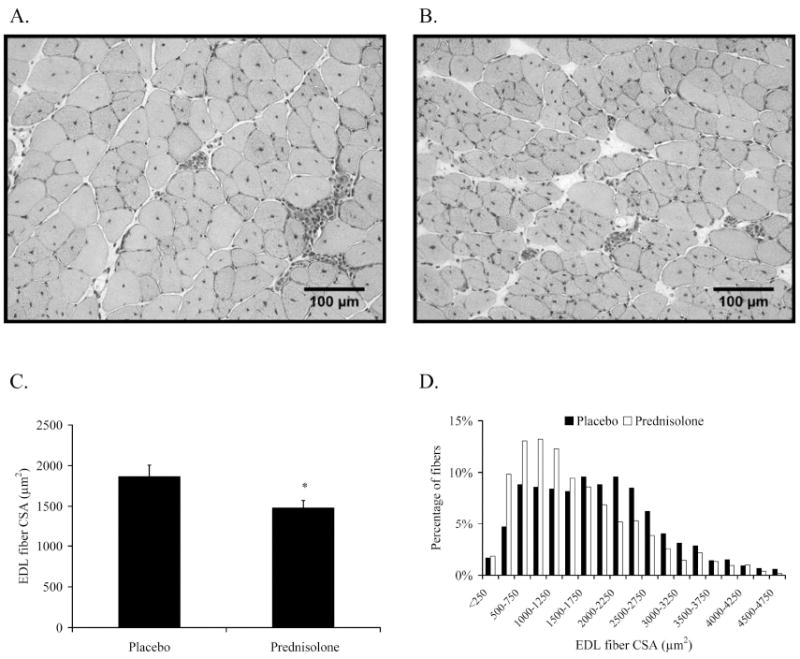
Prednisolone affected fiber CSA of the EDL muscle of mdx mice. A. Representative H&E image of an EDL muscle from a placebo-treated mdx mouse. B. Representative H&E image of an EDL muscle from a prednisolone-treated mdx mouse. C. Mean CSAs of fibers from EDL muscles. D. Frequency distributions of fiber CSAs from EDL muscles. A sample size of n=4-5 per treatment group were assessed. *Signifies different from Placebo.
Since prednisolone improves strength in boys with DMD, we sought to determine if prednisolone also improves muscle contractile function in mdx mice. Contractile properties of the EDL muscle were measured in vitro. As shown in Table 2, there was no improvement in peak twitch force, Po, maximal rate of force development or relaxation, passive stiffness, or active stiffness with prednisolone treatment. However, EDL muscle specific force was 26% greater in prednisolone-treated mice due to the muscle’s smaller cross-sectional area (P=0.021). To further probe these effects of prednisolone, a second study was undertaken which assessed size and functional parameters of muscles in addition to the EDL.
Table 2.
Contractility of EDL muscles from mdx mice that were treated with placebo or prednisolone for 8 wks.
| Placebo (n=8) | Prednisolone (n=15) | P-value | |
|---|---|---|---|
| Pt (mN) | 69.2 ± 3.0 | 76.3 ± 2.3 | 0.078 |
| Po (mN) | 291 ± 19 | 311 ± 12 | 0.340 |
| Specific Po (N/cm2) | 9.93 ± 0.92 | 12.49 ± 0.57 | 0.021 |
| +dP/dt (N/s) | 8.04 ± 0.37 | 8.85 ± 0.25 | 0.077 |
| -dP/dt (N/s) | -13.3 ± 1.3 | -14.8 ± 0.8 | 0.293 |
| Passive stiffness (N/m) | 16.6 ± 1.1 | 15.9 ± 0.7 | 0.549 |
| Active stiffness (N/m) | 410 ± 19 | 440 ± 10 | 0.137 |
Pt=peak twitch force. Po=maximal isometric tetanic force. +dP/dt=maximal rate of tetanic force development. −dP/dt=maximal rate of relaxation.
Study 2: The effects of prednisolone on muscle size and in vivo strength
Similar to Study 1, prednisolone-treated mice weighed less than placebo-treated mice after 1, 2, 3, 5, and 6 wks of treatment (data not shown). Because side-effects of prednisolone include hyperphagia and hyperactivity which can impact body mass, food intake and cage activities were assessed. Food intake did not differ between the treatment groups (4.1 ± 0.1 vs. 4.1 ± 0.2 g/mouse/d; P=0.636). Ambulatory cage activity was also similar between placebo and prednisolone-treated mice (0.42 ± 0.04 vs. 0.45 ± 0.05 km/d; P=0.617).
Muscle mass was also reduced by prednisolone treatment, as in Study 1. EDL wet mass was 13% lower (16.5 ± 0.5 vs. 14.4 ± 0.6 mg; P=0.014), and TA wet mass was 9% lower (75.9 ± 1.7 vs. 68.8 ± 2.1 mg; P=0.020) in prednisolone-treated mice. EDL and TA fiber CSA were also 26% (1,475 ± 54 vs. 1,084 ± 92; P=0.004) and 21% lower (2,114 ± 77 vs. 1,764 ± 124; P=0.032), respectively, in prednisolone-treated mice, confirming the results of the Study 1. Prednisolone slowed the accumulation of centrally-nucleated fibers in both EDL and TA muscles during the 8-wk treatment period (Figure 4). Prednisolone-treated mice had 14% and 15% fewer centrally-nucleated fibers in the EDL (P=0.038) and TA muscles (P=0.002), respectively, than placebo-treated mice. Circulating CK was also assayed as an indicator of muscle damage. There were no differences between placebo and prednisolone-treated mice before treatment (10,577 ± 1,929 vs. 10,624 ± 1,219) after 2 wks (10,508 ± 3,147 vs. 9,539 ± 2,099), 4 wks (10,440 ± 2,436 vs. 10,749 ± 2,115) or 6 wks (8,773 ± 1,480 vs. 8,956 ± 1,951 U/L) of treatment (P=0.956).
Figure 4.
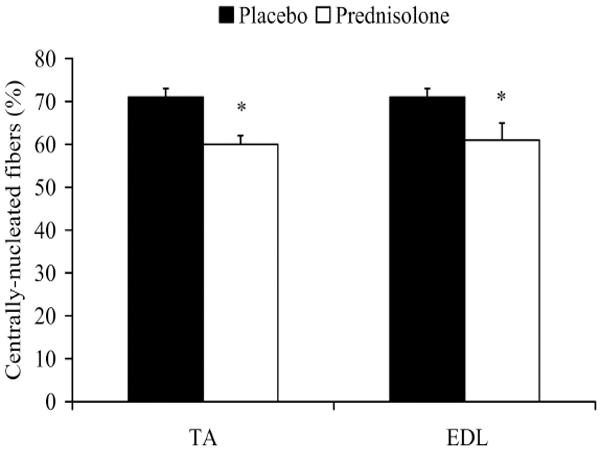
Percentage of centrally-nucleated fibers with prednisolone treatment in mdx mice. A sample size of n=6 per group was used for the EDL and n=8 for the TA. *Signifies different from Placebo.
In study 2, in vivo torque of the anterior crural muscles was measured every 2 wks to determine if prednisolone could either improve or offset strength loss due to disease progression. There were no differences between placebo- and prednisolone-treated mice in raw torque before treatment (1.6 ± 0.1 vs. 1.5 ± 0.1 N*mm), after 2 wks (2.1 ± 0.1 vs. 1.8 ± 0.1 N*mm), after 4 wks (2.1 ± 0.2 vs. 1.8 ± 0.1 N*mm), or after 6 wks (2.0 ± 0.2 vs. 1.9 ± 0.1 N*mm; P=0.651). Considering the results of both studies, prednisolone consistently decreased muscle mass, fiber size, and the frequency of centrally-nucleated fibers without changing overall maximal strength.
Study 1 and Study 2: The effect of prednisolone on the induction of eccentric injury
With the lack of dystrophin, mdx mice are extremely susceptible to eccentric contraction-induced injury 37. We determined if prednisolone protects hindlimb skeletal muscles from eccentric injury via two different preparations. First, injury was induced in isolated EDL muscles by 5 high-force, eccentric contractions in mice that had been previously treated with prednisolone or placebo for 8 wk. There was no difference between prednisolone- and placebo-treated mice in maximal isometric force before (P=0.340) or after the eccentric injury protocol (Figure 5A; P=0.186). There was also no difference in force generated on the first (P=0.353) or last eccentric contraction (P=0.244), between prednisolone- and placebo-treated mice. LDH activity of the Krebs buffer in which the EDL muscle was incubating was assayed both before and after the eccentric injury, and the change in activity was calculated. An increase in LDH activity would indicate more damage to the fibers, and hence, more LDH leaking out of the muscle and into the bath. Both treatment groups demonstrated an increase in LDH activity following injury, but prednisolone did not alter the increase relative to placebo (38 ± 9% vs. 36 ± 3%; P=0.805).
Figure 5.
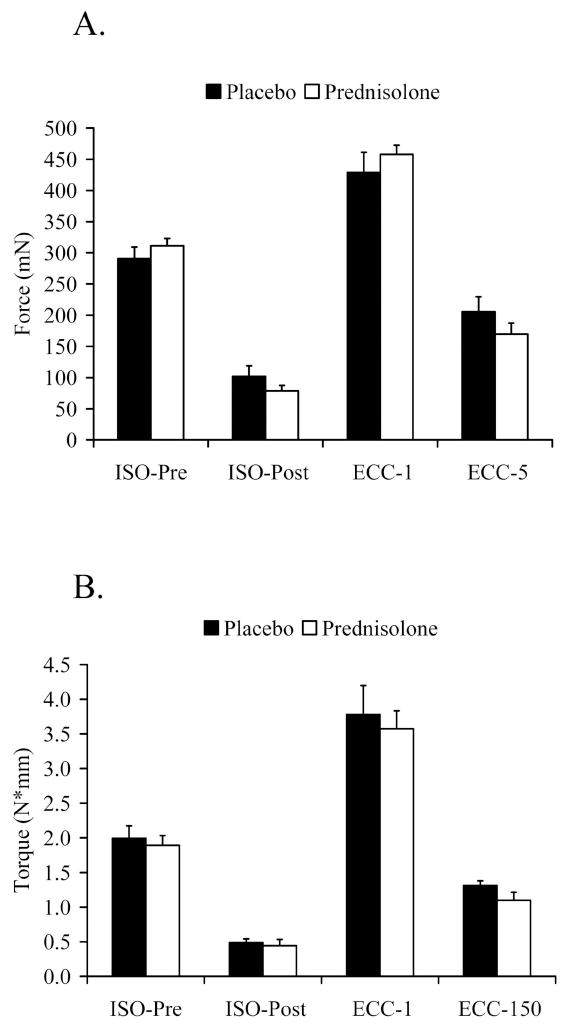
Prednisolone treatment in mdx mice did not affect susceptibility to eccentric-contraction induced injury in vitro (Study 1) or in vivo (Study 2). A. In vitro force of EDL muscles before and after injurious eccentric contractions. B. In vivo torque of the anterior crural muscles before and after the eccentric contraction injury protocol. Data are means ± SE. ECC=Eccentric. ISO=Isometric. Isometric-post measurements were made 2-3 min following the final eccentric contraction.
Second, an in vivo injury to the anterior crural muscle group was induced by 150 eccentric contractions following 6 wks of prednisolone or placebo treatment (Figure 5B). Similar to the in vitro findings, there was no difference in maximal isometric torque before (P=0.663) or after (P=0.865) the eccentric injury protocol between mice that were receiving prednisolone and placebo. Eccentric torque after the first contraction (P=0.686) and the 150th contraction (P=0.141) were also similar between groups. These data show that prednisolone does not protect skeletal muscle against injury induced by eccentric contractions, whether the contractions are evoked by directly opening sodium channels (in vitro) or through the nerve (in vivo).
Study 2: The effect of prednisolone on recovery following injury
After the in vivo eccentric contraction protocol, mice were allowed 2 wks to recover. At that time in vivo isometric torque was reassessed, and EDL muscles were isolated and further assessed in vitro. Prednisolone failed to improve in vivo isometric torque (Table 3). All mdx mice recovered to 108% of their pre-injury isometric torque during the 2-wk recovery period. In addition, multiple parameters of in vitro EDL contractility were not any better with prednisolone treatment compared to placebo treatment 2 wks following eccentric injury. Serum CK levels also remained similar between placebo- and prednisolone-treated mice 2 wk following injury (6,486 ± 1,417 vs. 6,893 ± 1,322 U/l; P=0.837). There was no difference in the frequency of centrally-nucleated fibers between placebo and prednisolone-treated mice in the TA muscle (63 ± 2% vs. 58 ± 3%; P=0.198) or the EDL muscle (62 ± 3 vs. 57 ± 3%; P=0.258). These data indicate that prednisolone does not confer a benefit to muscle function in mdx mice following a physiological muscle injury.
Table 3.
In vivo and in vitro muscle contractility following recovery of eccentric injury in mdx mice that were treated with a placebo or prednisolone for 8 wks.
| Placebo (n=8) | Prednisolone (n=8) | P-value | |
|---|---|---|---|
| Isometric torque (N*mm) | 1.97 ± 0.23 | 2.22 ± 0.15 | 0.390 |
| Isometric torque (N*mm/kg BW) | 60.4 ± 6.5 | 70.4 ± 5.5 | 0.261 |
| Isometric torque (N*mm/g muscle) | 25.5 ± 2.2 | 28.9 ± 1.7 | 0.247 |
| Pt (mN) | 81.7 ± 3.2 | 76.3 ± 5.2 | 0.387 |
| Po (mN) | 357 ± 15 | 349 ± 19 | 0.765 |
| Specific Po (N/cm2) | 14.0 ± 0.8 | 14.9 ± 0.5 | 0.319 |
| +dP/dt (N/s) | 9.20 ± 0.25 | 8.55 ± 0.61 | 0.344 |
| -dP/dt (N/s) | -18.3 ± 1.1 | -18.0 ± 1.5 | 0.868 |
| Passive stiffness (N/m) | 11.4 ± 0.5 | 11.5 ± 0.3 | 0.888 |
| Active stiffness (N/m) | 431 ± 16 | 447 ± 22 | 0.575 |
Eccentric injury was induced in vivo with 150 eccentric contractions (Study 2). Two wks later, torque of the anterior crural muscles was measured in vivo and then EDL contractility was immediately assessed in vitro.
DISCUSSION
To our knowledge, this is the first report of muscle contractile function properties in mdx mice following prednisolone treatment. We found that prednisolone treatment in mdx mice from 5 to 13 wks of age increased specific force of the EDL muscle and improved the histological appearance of the muscle by decreasing the percentage of fibers that express centralized nuclei. The increase in specific force was mostly due to the effect of prednisolone on decreasing skeletal muscle mass and muscle fiber cross-sectional area. Prednisolone failed to increase EDL maximal isometric tetanic or twitch force or maximal isometric torque of the anterior crural muscles. Prednisolone also failed to confer a functional benefit to muscles of mdx mice over time or following muscle injury and recovery. In summary, these data demonstrate that 8 wks of prednisolone treatment only improves specific force in dystrophic mouse muscle, but it does not improve muscle contractile function after acute injury or following recovery.
Several randomized control trials have been conducted to investigate the effects of prednisolone on strength in boys with DMD, and the results of those studies show very consistent and significant improvement in strength 3, 17, 29, 38. On the contrary, animal studies have found conflicting evidence for improvement 10, 13, 15, 16, 18, 22, 36, 49. These contradictory results may be due to the timing or regimen of prednisolone treatment, the imprecise methods used for measuring muscle function, or perhaps the mild dystrophy of the mdx mouse. In the current study, we selected doses of prednisolone very similar to what has been used previously in mdx mice to improve muscle performance 10, 13, 16, 18, 22, which is also close to what individuals with DMD receive. In fact, higher doses of prednisolone given to mdx mice actually decrease voluntary whole-body strength 16. One limitation of the dosing regimen is that the relative dose (in mg/kg BW) decreases slightly over time as the mice grow, since the release of the hormone is constant. The treatment time we selected coincides with the most severe period of disease in the mdx mouse which would seem to have the best possibility of being beneficial. Therefore, we surmise that prednisolone dose or timing is not the explanation for the contradictory findings. As for methodology, we used two methods to comprehensively determine muscle contractile function capacity. First, in vitro contractility of the EDL muscle after 8 wks of prednisolone treatment failed to show improvement in several contractile parameters, including maximal isometric force. In our second study, prednisolone did not increase maximal isometric torque measured via in vivo percutaneous electrical stimulation at multiple time points. These data show that 8-wk of prednisolone treatment does not improve overall muscle contractile function in the mdx mouse. Thus, it may be that the beneficial effects of prednisolone are not realized in the mdx mouse because of its mild dystrophy. Using a more progressive disease model, like mdx/utrophin or mdx/MyoD knockout mice, may provide more insight into prednisolone’s mechanism of improved strength in individuals with DMD.
It has been well-characterized that muscles of mdx mice are extremely susceptible to acute injury induced by eccentric contractions 37, 47. We found that prednisolone-treated mice displayed fewer fibers that exhibit centralized nuclei, indicative of a previously damaged fiber. This result supports the findings of others in DMD patients and mdx mice 19, 36 suggesting that prednisolone therapy prevents the accumulation of damaged fibers. To test if prednisolone is protective, Yang et al. 49 investigated the effect of methylprednisolone on force following contraction-induced injury of the diaphragm. In both 1- and 3-mo-old mdx mice, treated and untreated mice demonstrated equivalent decrements in force of the diaphragm during an eccentric contraction injury protocol. In the current study, we found similar results using two methods of contraction-induced injury of hindlimb skeletal muscles. Five eccentric contractions of isolated EDL muscles produced similar force drops between treated and untreated mice. Comparable results were also found when the anterior crural muscles were injured by 150 eccentric contractions in vivo. In addition, LDH released into the bath and CK in the circulation were not affected by prednisolone, again indicating a lack of protection by prednisolone. Together these support previous results that show prednisolone does not protect dystrophic skeletal muscle from injury. Since fewer centrally-nucleated fibers were found in prednisolone-treated mice in the absence of injury, it could be speculated that prednisolone protects muscle fibers from daily, minor injury. Both of our eccentric injury protocols produced approximately a 75% drop in force and such an injurious protocol could overwhelm a subtle effect by prednisolone. Therefore, it is possible that prednisolone may be able to prevent minor injury to the muscle, but it is not protective during eccentric contractions that generate high force and major injury. This possibility should be explored further.
There is some evidence that prednisolone can improve skeletal muscle regeneration by promoting satellite cell proliferation. For example, myoblast and myotube density and the number of proliferating myoblasts are increased with prednisolone 30, 34, 40. We extended the current literature on the effect of prednisolone on regeneration by using contractile function as our primary marker of muscle recovery. Two wks following injury, muscle contractile function, serum CK, and centrally-nucleated fibers were similar between the treatment groups. These data show that prednisolone does not improve muscle function following injury in dystrophic skeletal muscle. These results confirm previous work which found that prednisolone did not affect the number of regenerating fibers or proliferating myonuclei 4 days following crush injury in mdx mice 1. One explanation may be that mdx mice appear to have normal or improved regenerative capabilities following injury compared to wild-type mice 4, 25, 28, 42. If prednisolone does indeed improve muscle regeneration in a more severe dystrophic phenotype, this effect may be masked, since mdx mice already are capable of enhanced regeneration.
One of the most consistent results of prednisolone we found was its catabolic effect on body and muscle size. Few reports have documented changes in muscle mass due to prednisolone treatment in dystrophic muscle, and results from studies are conflicting. In mdx mice there are reports of little or no change in muscle mass due to prednisolone 1, 12, and in boys with DMD prednisone causes an increase in mass 17. Muscle fiber size has also been shown to not change 48 and increase 1 with prednisolone treatment. In our study, we found that TA and EDL muscle mass, mean fiber cross-sectional area, and the variability in fiber size decreased with prednisolone treatment. While muscle and fiber atrophy is typically seen as detrimental, young mdx mice have larger muscles than wild-type mice. 7 We surmise that prednisolone is driving muscle mass back toward normal, and thus this could be interpreted as being beneficial. Despite these changes in size, muscle strength remained equal to placebo-treated mice. In fact, when EDL force was normalized to muscle CSA, prednisolone-treated muscles were stronger. Overall maximum force is typically not depressed when comparing muscles from mdx and wild-type mice. Mdx mice are weaker than wild-type mice only when their strength is normalized to muscle size 7. These data indicate that the catabolic effects of prednisolone on dystrophic mouse muscle may again be considered beneficial, since muscle mass is returning normal without compromising muscle function.
One could speculate that the decrease in muscle mass and fiber cross-sectional area from prednisolone could be anti-inflammatory due to prevention of water accumulation in the muscle. We found that most of the muscle mass loss with prednisolone treatment was due to water, because the dry mass of the EDL was similar between treatment groups. It is possible that the infiltration of inflammatory mediators drives water retention in the extracellular matrix and muscle fibers in mdx mice. However, when muscle water content was decreased with prednisolone, possibly through an anti-inflammatory mechanism, muscle contractility was maintained. In other words, inflammation is not leading to muscle weakness, because controlling inflammation through glucocorticoid treatment is not increasing muscle strength. Further data is needed to correlate changes in muscle size, water content, and muscle contractility with both muscle and systemic inflammatory markers.
In summary, 8 wks of prednisolone treatment to mdx mice improved specific force of the EDL muscle, but it did not improve other components of contractile function. This was mostly attributed to the effect of prednisolone on lowering muscle size, mean fiber cross-sectional area, and fiber variability. While prednisolone did result in fewer centrally-nucleated fibers, this did not translate into increased maximal contractile function. Prednisolone also failed to prevent force loss during eccentric injury or improve function during the 2-wk recovery process. Prednisolone administered to boys with DMD results in rapid and significant strength gains, but this same mechanism of action is not apparent in the mdx mouse.
Acknowledgments
The authors would like to thank Gordon Warren for assistance in setting up the in vivo torque apparatus. The authors would like to thank Steven Nelson, Sarah Greising, and James McKeehen for technical assistance.
ABBREVIATIONS
- CK
Creatine Kinase
- DMD
Duchenne Muscular Dystrophy
- EDL
Extensor Digitorum Longus
- LDH
Lactate Dehydrogenase
- OCT
Optimum Cutting Temperature
- TA
Tibialis Anterior
References
- 1.Anderson JE, McIntosh LM, Poettcker R. Deflazacort but not prednisone improves both muscle repair and fiber growth in diaphragm and limb muscle in vivo in the mdx dystrophic mouse. Muscle Nerve. 1996;19:1576–85. doi: 10.1002/(SICI)1097-4598(199612)19:12<1576::AID-MUS7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 2.Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 1998;94:557–72. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 3.Beenakker EA, Fock JM, Van Tol MJ, Maurits NM, Koopman HM, Brouwer OF, et al. Intermittent prednisone therapy in Duchenne muscular dystrophy: a randomized controlled trial. Arch Neurol. 2005;62:128–32. doi: 10.1001/archneur.62.1.128. [DOI] [PubMed] [Google Scholar]
- 4.Boer JM, de Meijer EJ, Mank EM, van Ommen GB, den Dunnen JT. Expression profiling in stably regenerating skeletal muscle of dystrophin-deficient mdx mice. Neuromuscul Disord. 2002;12(Suppl 1):S118–24. doi: 10.1016/s0960-8966(02)00092-5. [DOI] [PubMed] [Google Scholar]
- 5.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burrow KL, Coovert DD, Klein CJ, Bulman DE, Kissel JT, Rammohan KW, et al. Dystrophin expression and somatic reversion in prednisone-treated and untreated Duchenne dystrophy. CIDD Study Group Neurology. 1991;41:661–6. doi: 10.1212/wnl.41.5.661. [DOI] [PubMed] [Google Scholar]
- 7.Coulton GR, Curtin NA, Morgan JE, Partridge TA. The mdx mouse skeletal muscle myopathy: II. Contractile properties. Neuropathol Appl Neurobiol. 1988;14:299–314. doi: 10.1111/j.1365-2990.1988.tb00890.x. [DOI] [PubMed] [Google Scholar]
- 8.Courdier-Fruh I, Barman L, Briguet A, Meier T. Glucocorticoid-mediated regulation of utrophin levels in human muscle fibers. Neuromuscul Disord. 2002;12(Suppl 1):S95–104. doi: 10.1016/s0960-8966(02)00089-5. [DOI] [PubMed] [Google Scholar]
- 9.Croxtall JD, van Hal PT, Choudhury Q, Gilroy DW, Flower RJ. Different glucocorticoids vary in their genomic and non-genomic mechanism of action in A549 cells. Br J Pharmacol. 2002;135:511–9. doi: 10.1038/sj.bjp.0704474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Luca A, Pierno S, Liantonio A, Cetrone M, Camerino C, Fraysse B, et al. Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J Pharmacol Exp Ther. 2003;304:453–63. doi: 10.1124/jpet.102.041343. [DOI] [PubMed] [Google Scholar]
- 11.Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- 12.Fisher I, Abraham D, Bouri K, Hoffman EP, Muntoni F, Morgan J. Prednisolone-induced changes in dystrophic skeletal muscle. Faseb J. 2005;19:834–6. doi: 10.1096/fj.04-2511fje. [DOI] [PubMed] [Google Scholar]
- 13.Golumbek PT, Keeling RM, Connolly AM. Strength and corticosteroid responsiveness of mdx mice is unchanged by RAG2 gene knockout. Neuromuscul Disord. 2007;17:376–84. doi: 10.1016/j.nmd.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Gordon T, Stein RB. Comparison of force and stiffness in normal and dystrophic mouse muscles. Muscle Nerve. 1988;11:819–27. doi: 10.1002/mus.880110804. [DOI] [PubMed] [Google Scholar]
- 15.Granchelli JA, Avosso DL, Hudecki MS, Pollina C. Cromolyn increases strength in exercised mdx mice. Res Commun Mol Pathol Pharmacol. 1996;91:287–96. [PubMed] [Google Scholar]
- 16.Granchelli JA, Pollina C, Hudecki MS. Pre-clinical screening of drugs using the mdx mouse. Neuromuscul Disord. 2000;10:235–9. doi: 10.1016/s0960-8966(99)00126-1. [DOI] [PubMed] [Google Scholar]
- 17.Griggs RC, Moxley RT, 3rd, Mendell JR, Fenichel GM, Brooke MH, Pestronk A, et al. Duchenne dystrophy: randomized, controlled trial of prednisone (18 months) and azathioprine (12 months) Neurology. 1993;43:520–7. doi: 10.1212/wnl.43.3_part_1.520. [DOI] [PubMed] [Google Scholar]
- 18.Hudecki MS, Pollina CM, Granchelli JA, Daly MK, Byrnes T, Wang JC, et al. Strength and endurance in the therapeutic evaluation of prednisolone-treated MDX mice. Res Commun Chem Pathol Pharmacol. 1993;79:45–60. [PubMed] [Google Scholar]
- 19.Hussein MR, Hamed SA, Mostafa MG, Abu-Dief EE, Kamel NF, Kandil MR. The effects of glucocorticoid therapy on the inflammatory and dendritic cells in muscular dystrophies. Int J Exp Pathol. 2006;87:451–61. doi: 10.1111/j.1365-2613.2006.00470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ingalls CP, Warren GL, Lowe DA, Boorstein DB, Armstrong RB. Differential effects of anesthetics on in vivo skeletal muscle contractile function in the mouse. J Appl Physiol. 1996;80:332–40. doi: 10.1152/jappl.1996.80.1.332. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs SC, Bootsma AL, Willems PW, Bar PR, Wokke JH. Prednisone can protect against exercise-induced muscle damage. J Neurol. 1996;243:410–6. doi: 10.1007/BF00869001. [DOI] [PubMed] [Google Scholar]
- 22.Keeling RM, Golumbek PT, Streif EM, Connolly AM. Weekly oral prednisolone improves survival and strength in male mdx mice. Muscle Nerve. 2007;35:43–8. doi: 10.1002/mus.20646. [DOI] [PubMed] [Google Scholar]
- 23.Landisch RM, Kosir AM, Nelson SA, Baltgalvis KA, Lowe DA. Adaptive and nonadaptive responses to voluntary wheel running by mdx mice. Muscle Nerve. 2008;38:1290–3. doi: 10.1002/mus.21141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, Habelitz S, et al. Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. J Bone Miner Res. 2006;21:466–76. doi: 10.1359/JBMR.051103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Louboutin JP, Fichter-Gagnepain V, Pastoret C, Thaon E, Noireaud J, Sebille A, et al. Morphological and functional study of extensor digitorum longus muscle regeneration after iterative crush lesions in mdx mouse. Neuromuscul Disord. 1995;5:489–500. doi: 10.1016/0960-8966(95)00006-9. [DOI] [PubMed] [Google Scholar]
- 26.Lowe DA, Warren GL, Hayes DA, Farmer MA, Armstrong RB. Eccentric contraction-induced injury of mouse soleus muscle: effect of varying [Ca2+]o. J Appl Physiol. 1994;76:1445–53. doi: 10.1152/jappl.1994.76.4.1445. [DOI] [PubMed] [Google Scholar]
- 27.Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008:CD003725. doi: 10.1002/14651858.CD003725.pub3. [DOI] [PubMed] [Google Scholar]
- 28.Matecki S, Guibinga GH, Petrof BJ. Regenerative capacity of the dystrophic (mdx) diaphragm after induced injury. Am J Physiol Regul Integr Comp Physiol. 2004;287:R961–8. doi: 10.1152/ajpregu.00146.2004. [DOI] [PubMed] [Google Scholar]
- 29.Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320:1592–7. doi: 10.1056/NEJM198906153202405. [DOI] [PubMed] [Google Scholar]
- 30.Metzinger L, Passaquin AC, Warter JM, Poindron P. alpha-Methylprednisolone promotes skeletal myogenesis in dystrophin-deficient and control mouse cultures. Neurosci Lett. 1993;155:171–4. doi: 10.1016/0304-3940(93)90700-u. [DOI] [PubMed] [Google Scholar]
- 31.Moran AL, Nelson SA, Landisch RM, Warren GL, Lowe DA. Estradiol replacement reverses ovariectomy-induced muscle contractile and myosin dysfunction in mature female mice. J Appl Physiol. 2007;102:1387–93. doi: 10.1152/japplphysiol.01305.2006. [DOI] [PubMed] [Google Scholar]
- 32.Moran AL, Warren GL, Lowe DA. Soleus and EDL muscle contractility across the lifespan of female C57BL/6 mice. Exp Gerontol. 2005;40:966–75. doi: 10.1016/j.exger.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Pasquini F, Guerin C, Blake D, Davies K, Karpati G, Holland P. The effect of glucocorticoids on the accumulation of utrophin by cultured normal and dystrophic human skeletal muscle satellite cells. Neuromuscul Disord. 1995;5:105–14. doi: 10.1016/0960-8966(94)00042-8. [DOI] [PubMed] [Google Scholar]
- 34.Passaquin AC, Metzinger L, Leger JJ, Warter JM, Poindron P. Prednisolone enhances myogenesis and dystrophin-related protein in skeletal muscle cell cultures from mdx mouse. J Neurosci Res. 1993;35:363–72. doi: 10.1002/jnr.490350403. [DOI] [PubMed] [Google Scholar]
- 35.Pastoret C, Sebille A. Age-related differences in regeneration of dystrophic (mdx) and normal muscle in the mouse. Muscle Nerve. 1995;18:1147–54. doi: 10.1002/mus.880181011. [DOI] [PubMed] [Google Scholar]
- 36.Payne ET, Yasuda N, Bourgeois JM, Devries MC, Rodriguez MC, Yousuf J, et al. Nutritional therapy improves function and complements corticosteroid intervention in mdx mice. Muscle Nerve. 2006;33:66–77. doi: 10.1002/mus.20436. [DOI] [PubMed] [Google Scholar]
- 37.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90:3710–4. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahman MM, Hannan MA, Mondol BA, Bhoumick NB, Haque A. Prednisolone in Duchenne muscular dystrophy. Bangladesh Med Res Counc Bull. 2001;27:38–42. [PubMed] [Google Scholar]
- 39.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–80. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 40.Sklar RM, Brown RH., Jr Methylprednisolone increases dystrophin levels by inhibiting myotube death during myogenesis of normal human muscle in vitro. J Neurol Sci. 1991;101:73–81. doi: 10.1016/0022-510x(91)90019-4. [DOI] [PubMed] [Google Scholar]
- 41.Stein RB, Gordon T. Nonlinear stiffness--force relationships in whole mammalian skeletal muscles. Can J Physiol Pharmacol. 1986;64:1236–44. doi: 10.1139/y86-209. [DOI] [PubMed] [Google Scholar]
- 42.Turk R, Sterrenburg E, de Meijer EJ, van Ommen GJ, den Dunnen JT, t Hoen PA. Muscle regeneration in dystrophin-deficient mdx mice studied by gene expression profiling. BMC Genomics. 2005;6:98. doi: 10.1186/1471-2164-6-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warren GL, Hayes DA, Lowe DA, Prior BM, Armstrong RB. Materials fatigue initiates eccentric contraction-induced injury in rat soleus muscle. J Physiol. 1993;464:477–89. doi: 10.1113/jphysiol.1993.sp019646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warren GL, Hayes DA, Lowe DA, Williams JH, Armstrong RB. Eccentric contraction-induced injury in normal and hindlimb-suspended mouse soleus and EDL muscles. J Appl Physiol. 1994;77:1421–30. doi: 10.1152/jappl.1994.77.3.1421. [DOI] [PubMed] [Google Scholar]
- 45.Wehling-Henricks M, Lee JJ, Tidball JG. Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscul Disord. 2004;14:483–90. doi: 10.1016/j.nmd.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone J Clin Invest. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weller B, Karpati G, Carpenter S. Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J Neurol Sci. 1990;100:9–13. doi: 10.1016/0022-510x(90)90005-8. [DOI] [PubMed] [Google Scholar]
- 48.Weller B, Massa R, Karpati G, Carpenter S. Glucocorticoids and immunosuppressants do not change the prevalence of necrosis and regeneration in mdx skeletal muscles. Muscle Nerve. 1991;14:771–4. doi: 10.1002/mus.880140812. [DOI] [PubMed] [Google Scholar]
- 49.Yang L, Luo J, Petrof BJ. Corticosteroid therapy does not alter the threshold for contraction-induced injury in dystrophic (mdx) mouse diaphragm. Muscle Nerve. 1998;21:394–7. doi: 10.1002/(sici)1097-4598(199803)21:3<394::aid-mus14>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]