

Abstract
CD95 (also called Fas and APO-1) is a prototypical death receptor that regulates tissue homeostasis mainly in the immune system through induction of apoptosis 1-3. During cancer progression CD95 is frequently downregulated or cells are rendered apoptosis resistant 4,5 raising the possibility that loss of CD95 is part of a mechanism for tumour evasion. However, complete loss of CD95 is rarely seen in human cancers 4 and many cancer cells express large quantities of CD95 and are highly sensitive to CD95 mediated apoptosis in vitro. Furthermore, cancer patients frequently have elevated levels of the physiological ligand for CD95, CD95L 6. These data raise the intriguing possibility that CD95 could actually promote the growth of tumours through its nonapoptotic activities 7. Here we show that cancer cells in general, regardless of their CD95 apoptosis sensitivity, depend on constitutive activity of CD95, stimulated by cancer-produced CD95L, for optimal growth. Consistently, loss of CD95 in mouse models of ovarian cancer and liver cancer reduces cancer incidence as well as the size of the tumours. The tumorigenic activity of CD95 is mediated by a pathway involving JNK and c-Jun. These results demonstrate that CD95 plays a growth promoting role during tumorigenesis and suggest that efforts to inhibit its activity rather than to enhance its activation should be considered during cancer therapy.
To test the function of endogenous CD95 in tumour cells, expression of CD95 was reduced in various human cancer cell lines using CD95 specific shRNA lentiviruses (Supplementary Fig. 1). Infection of the CD95 high expressing ovarian cancer cell line HeyA8, which in vitro is sensitive to CD95 mediated apoptosis, with a lentiviral shRNA against CD95 (R#6) substantially reduced CD95 protein and surface expression resulting in loss of CD95 apoptosis sensitivity (Fig. 1a). Abrogation of CD95 expression also resulted in substantial growth reduction. This was confirmed with another CD95 targeting shRNA (R#4) (Supplementary Fig. 2) and in another ovarian cancer cell line, SKOV3ip1, which expresses very low amounts of CD95 and is completely resistant to CD95L induced apoptosis (Fig. 1b and data not shown). Reconstitution of the SKOV3ip1 CD95 knock down cells with an siRNA resistant version of CD95 to endogenous levels restored the growth of SKOV3ip1 cells (Supplementary Fig. 3). A growth inhibiting effect of reducing CD95 expression was also found in cell lines derived from colon (HCT116), renal (CAKI-1), breast (MCF7), and liver (HepG2) cancer (Fig. 1c-f). Knocking down CD95 in MCF7 cells with the R#6 virus resulted, 6 days after infection, in growth arrest (data not shown). However, 3 weeks after infection cells expressing intermediate CD95 levels grew out, albeit with reduced growth as compared to vector control infected cells (Fig. 1e). Neither MCF7 nor CAKI-1 cells benefited from overexpressing CD95 (Supplementary Fig. 4) suggesting that cancer cells, regardless of the absolute level of CD95 expression, maintain expression of CD95 at a level sufficient to promote optimal growth. This is consistent with our previous report that in 22 tumour cell lines stimulation of CD95 did not result in increased proliferation 6. However, incubating cells with the neutralising CD95L monoclonal antibody (mAb), NOK-1, reduced cell growth (Supplementary Fig. 5a) suggesting that the small amount of CD95L produced by tumour cells (Supplementary Fig. 5b) contributes to their growth. To directly test this assumption we used lentivirus based shRNAs specific for CD95L which knocked down CD95L in a murine cell line expressing full length human CD95L (Supplementary Fig. 5c). We monitored growth of HepG2 cells infected with each of three independent CD95L specific shRNA viruses all of which caused reduction of CD95L expression (Fig. 1g). Paralleling the efficiency of the knock down the viruses caused different degrees of growth inhibition ranging from a reduction in growth (L#2) to a complete loss of growth (L#1) (Fig. 1h). Knocking down CD95L using an siRNA SmartPool (Supplementary Fig. 5c) also resulted in profound growth inhibition of HepG2 cells (Supplementary Fig. 5d). Finally, knocking down CD95L also reduced growth of all other cancer cell lines suggesting that CD95L is essential for the growth of many tumour cells (Fig. 1i). The knock down of CD95 in HepG2 only caused a moderate reduction in growth, possibly because this knock down may not have been efficient enough to cause a more pronounced effect. Given the small amount of CD95L expressed in tumour cells, reducing its expression is more likely to cause severe effects by falling below a threshold of minimal expression. This interpretation is consistent with a recent analysis that determined that the threshold for CD95 to signal apoptosis versus nonapoptotic signalling is 1000 times higher 8. Our data suggest that almost undetectable amounts of CD95 and CD95L are sufficient and in some cases required to promote growth of tumour cells.
Figure 1. Reducing CD95 or CD95L expression inhibits cell proliferation of cancer cells.
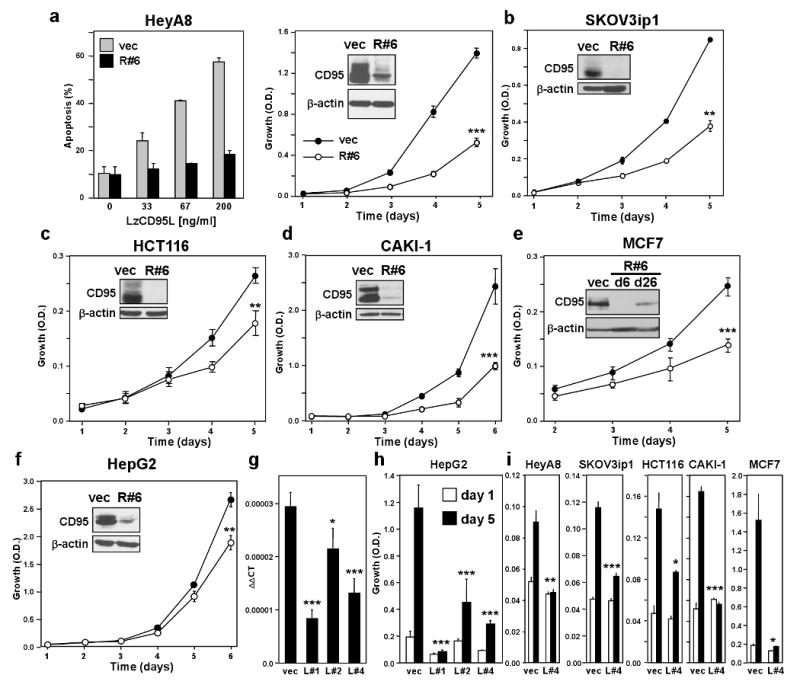
a-f, Growth of different cell lines infected with the CD95 specific shRNA lentivirus CD95shRNA#6 (R#6). Inserts show the total CD95 expression levels of cells expressing scrambled control (vec) or R#6 as determined by western blot analysis. A similar effect was also found with a CD95 expressing variant of the neuroblastoma cell line NB4 (not shown). Apoptosis sensitivity of HeyA8 vec and R#6 cells by LzCD95 ligand treatment was determined by quantifying DNA fragmentation (a). g, HepG2 cells were stably infected with three different CD95L specific shRNA lentiviruses and CD95L mRNA was quantified using real time PCR. h, Growth of cells in g over 5 days. i, Growth of different cell lines infected with the L#4 virus. Proliferation of cells was examined by SRB assay (a-f and h and i). * p<0.05, ** p<0.01, *** p<0.001. Values in graphs in a to i represent mean -/+ s.d. from three independent experiments.
CD95 is highly expressed in epithelial ovarian cancer but these cancer cells acquire resistance to CD95 mediated apoptosis 9. Large quantities of CD95L are found in patient ascites 10,11 and patient serum 12. As a first step toward understanding the role of CD95 in ovarian cancer in vivo we employed an intraperitoneal xenograft model of metastatic ovarian cancer 13. Nude mice injected with SKOV3ip1 cells infected with either the R#6 (Fig. 2a) or the R#4 (Fig. 2b) virus had about a 50% reduction in tumour mass, number of metastases and ascites formation. To test whether in vivo CD95 dependent tumour growth was driven by CD95L, SKOV3ip1 cells were grown for one week in nude mice and then treated with either a neutralising anti-CD95L mAb specific for mouse CD95L (MFL3) or for human CD95L (NOK-1). Tumour growth was attenuated only in mice injected with the human CD95L specific antibody (Fig. 2d) suggesting that it is predominantly the CD95L produced by the human cancer cells (see insert in Fig. 2d), rather than CD95L produced by the microenvironment in the mouse host, that drives CD95-mediated tumour growth.
Figure 2. Loss of CD95 expression inhibits ovarian cancer in vivo.
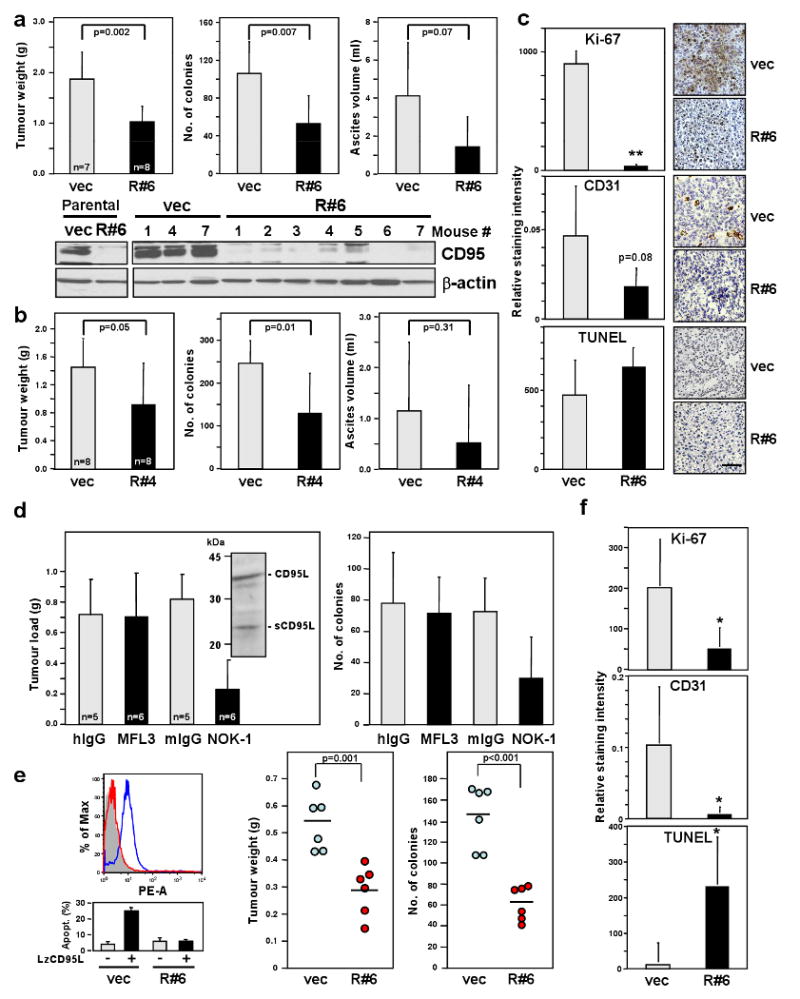
a Tumour weight, number of tumour colonies and ascites from mice injected with SKOV3ip1 vec or R#6 cells. Lysates of cells and tumour tissues were examined for CD95 level by western blot analysis. b, Same parameters as in (a) were measured from mice injected with SKOV3ip1 vec or R#4 cells. c, Histology and immunohistochemistry staining for Ki-67, TUNEL and CD31 of SKOV3ip1 vec and R#6 tumours. Scale bar = 100 μm. ** p-value <0.001. d, Tumour load and number of tumour colonies of mice treated with neutralising mAb for murine CD95L (MFL3), human CD95L (NOK-1) or corresponding isotype control mAbs were measured. Inset, Western blot analysis of SKOV3ip1 cell lysate for CD95L. e, Surface CD95 staining (upper left) and apoptosis sensitivity by LzCD95L (100 ng/ml) treatment (lower left) of MONTY-1 vec and R#6 cells. Weight and number of colonies of tumours formed by MONTY-1 vec and R#6 cells are shown (right). f, The staining intensity for Ki-67, TUNEL and CD31 of tumours from MONTY-1 vec and R#6 cells were quantified. * p-value <0.05. Values in graphs in a to e and f represent mean -/+ s.d. from three independent experiments. The horizontal bars in right part part of e represent the mean of 6 animals.
To test whether the growth promoting role of CD95 could also be found in actual ovarian cancer we used a primary ovarian cancer cell line (MONTY-1 14) at an early passage number. CD95 was knocked down in MONTY-1 cells (again resulting in reduced proliferation (Supplementary Fig. 6)) after which these cells were injected into nude mice. Similar to findings with SKOV3ip1 cells, MONTY-1 cells with reduced CD95 expression were completely resistant to CD95 mediated apoptosis and showed reduced intra-abdominal tumour load (Fig. 2e). Also, similar to findings with SKOV3ip1 cells (Fig. 2c) this result could, at least in part, be explained by a reduced proliferation rate (reduced Ki-67 staining) and reduced vascularization (reduced CD31 staining). The tumours showed an increase in TUNEL staining suggesting that the inhibition of the growth promoting CD95 receptor caused cancer cells to die (Fig. 2f).
Similar to other subtypes of epithelial ovarian cancers 9, the endometrioid subtype expresses CD95 (Fig. 3a) suggesting that in ovarian cancer CD95 could promote tumorigenesis. To test this we used a genetic model of epithelial endometrioid ovarian cancer based on the expression of activated mutant K-ras and deletion of Pten 15. For these studies we generated K-ras/Pten mutant mice carrying loxP sites in the CD95 gene 16 (Supplementary Fig. 7). Cancer formation was initiated by injecting Adenovirus (AdV) Cre into the right ovarian bursa of the mice. Eight weeks after injection 9 out of 12 mice expressing wt CD95 had formed an ovarian cancer in the injected ovary with no cancer in the uninjected control ovary while only one of the 11 mice with knocked out CD95 had an early cancer (Fig. 3b-d). While wt mice started to die 8-9 weeks after AdV Cre injection in this model, only one of 4 mice lacking CD95 in the ovaries had an early cancer after 14 weeks. IHC confirmed that CD95 was highly expressed in the cancerous ovaries of wt mice and efficiently knocked out in the AdV-Cre injected ovaries of mice carrying the floxed CD95 alleles (Fig. 3d). PCR analysis to detect deleted pten confirmed that the Cre recombinase was equally active in cells of either genotype (Supplementary Fig. 7c). In summary, the data suggest that CD95 plays a role in the tumorigenesis of ovarian cancer.
Figure 3. Deletion of CD95 leads to reduction in tumour formation in a spontaneous model of endometrioid ovarian cancer.
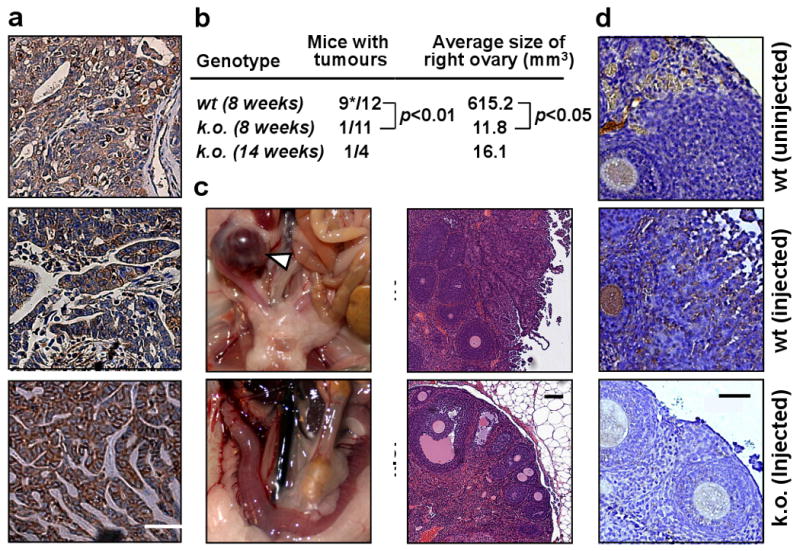
a, Staining for CD95 in three primary endometrioid ovarian cancers. Scale bar = 50 μm. b, Number of mice that formed visible tumours either 8 or 14 weeks after injection of AdV Cre into the right ovarian bursa. * one mouse died from ovarian cancer 42 days after AdV Cre injection. c, Representative image and histology of right ovaries from wt and k.o. mice 8 weeks after injection of AdV Cre. Arrow head indicates ovarian tumour. Scale bar = 100 μm. d, CD95 staining of ovary from untreated wt, AdV Cre treated wt or AdV Cre treated k.o. mice. Scale bar = 50 μm. wt, LSL-K-rasG12D/+PtenloxP/loxPCD95wt/wt; k.o., K-rasG12D/+PtenloxP/loxPCD95loxP/loxP.
Liver is the tissue with the highest constitutive CD95 expression. It was previously shown that mice expressing an apoptosis signalling deficient mutant of CD95 have a defect in liver regeneration after partial hepatectomy (PH) 17 raising the possibility that CD95 is involved in liver regeneration. To determine if it is CD95 expressed on hepatocytes that drives their proliferation, we generated hepatocyte-specific CD95 knock out mice by crossing CD95 loxP mice with mice expressing the Cre recombinase under control of the liver specific albumin promoter (Alb-Cre mice) (Supplementary Fig. 8). These mutant mice were then subjected to 2/3 PH. Proliferation of hepatocytes was severely impaired 48 hrs after PH in the absence of CD95 on hepatocytes (Fig. 4a,b) confirming that CD95 expression on hepatocytes is required for their proliferation. This raised a possibility that CD95 might also promote liver cancer.
Figure 4. Deletion of CD95 in the liver leads to a decrease in tumour formation caused by reduced ability of hepatocytes to proliferate and to activate JNK.
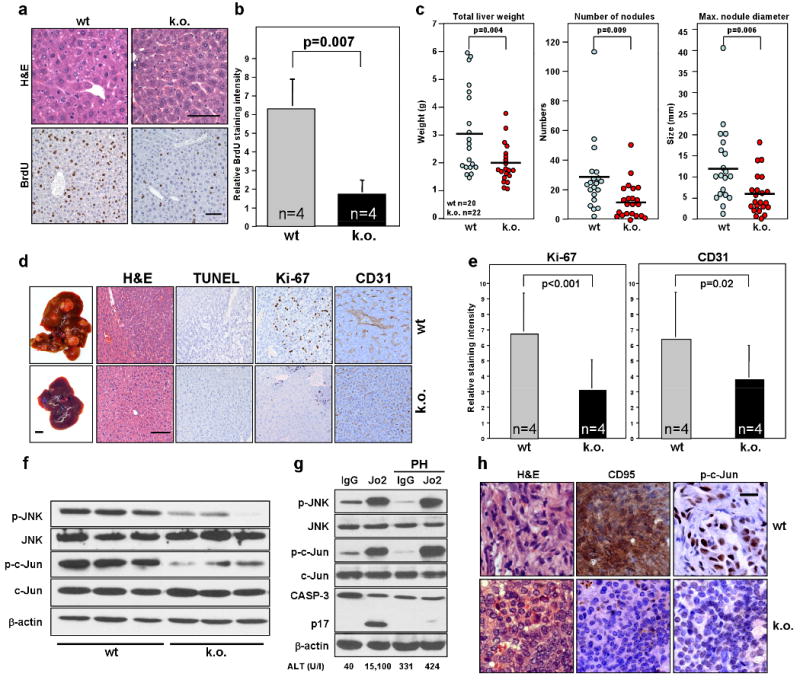
a, H&E and BrdU staining of livers from wild-type (wt) and liver specific CD95 k.o. mice 48 hrs after partial hepatectomy. Scale bars = 100 μm. b, Quantification of relative BrdU staining intensity of the mice in (a). c, wt and liver specific CD95 k.o. mice were injected with a single dose of DEN i.p. to induce liver tumour formation. 8 months later, all mice were sacrificed and parameters of total liver weight, number of liver surface nodules and maximum nodule diameter were recorded. d, Intact livers (scale bar = 1 cm), H&E staining and immunohistochemistry of TUNEL, Ki-67 and CD31 from mice in (c). Scale bar = 100 μm. e, Quantification of Ki-67 and CD31 staining for liver samples in (d). There was no detectable TUNEL staining. f, Western blot for phospho-JNK and phospho-c-Jun in three untreated wt and three CD95 k.o. mouse livers. g, wt mice with or without PH were injected i.p with 10 μg of murine CD95-specific agonistic antibody, Jo2 or isotype matched control mAb. After 6 hours phospho-JNK, p-c-Jun levels and cleavage of caspase-3 in livers were measured by western blot analysis. Concentration of the liver enzyme ALT in the serum of injected mice is given. h, Immunohistochemistry staining of ovarian tumours from wt (LSL-K-rasG12D/+PtenloxP/loxPCD95wt/wt) or k.o. (K-rasG12D/+PtenloxP/loxPCD95loxP/loxP) mice 8 weeks after injection of Adv Cre for CD95 and phospho-c-Jun. Scale bar = 20 μm. Values in graphs in b and e represent mean -/+ s.d. from three independent experiments. The horizontal bars in c represent the mean.
Prolonged damage to the liver accompanied by compensatory hepatocyte proliferation can promote hepatocarcinogenesis 18 which can be recapitulated in a mouse model of diethylnitrosamine (DEN) induced hepatocellular carcinoma (HCC). As compared to wt mice, mice lacking expression of CD95 in the liver showed a clear reduction of HCC 8 months following a single injection of DEN (Fig. 4c). These data suggest that CD95 contributes to hepatocarcinogenesis. Ki-67 staining in the tumours of CD95 deficient livers was reduced (Fig. 4d,e), consistent with a contribution of CD95 to the growth of liver cancer. Because it was previously shown that expression of Cre recombinase can affect the apoptosis sensitivity of certain tissues 19, we confirmed our data in Cre expressing mice of a different genotype (Supplementary Fig. 9). DEN has been described to induce liver damage through a mechanism that involves ROS 18. Consistently, reduction of HCC in mice lacking CD95 expression in the liver was not due to a decrease in DEN induced damage because injection of DEN into 15 day old or adult wt or CD95 k.o. mice did not induce significantly increased apoptosis and caused a similar activation of JNK (Supplementary Fig. 10). However, the proliferative response (Ki-67 staining) of hepatocytes in DEN injected mice was strongly reduced in CD95 deficient livers (Supplementary Fig. 10a). In summary, our data support a role of CD95 in liver regeneration that translates into an increase in hepatocarcinogenesis in a chemically induced model of liver cancer.
JNK1 deficient mice have a reduced ability to recover from PH and are less susceptible to DEN induced liver cancer 20,21. Recently this link between JNK1 activation and HCC was confirmed in human HCC by two independent studies. HCC patients showed an increased activation of JNK1 in the cancer cells when compared to adjacent noncancerous tissue 20,22. Consistent with a function of JNK in proliferation of tumour cells we found that treatment of tumour cell lines with the JNK inhibitor SP600125 completely inhibited their growth (Supplementary Fig. 11a and Supplementary Results). Strikingly, hepatocytes from CD95 deficient mice showed reduced phosphorylation of JNK and the JNK substrate c-Jun (Fig. 4f) suggesting that loss of CD95 reduced the basal activity of JNK resulting in loss of proliferative capacity. To test whether stimulation of CD95 in the liver causes activation of JNK we injected wt mice with the agonistic anti-CD95 mAb, Jo2. Injection of Jo2 caused a massive increase in the phosphorylation of JNK and c-Jun both in mice that possessed an intact liver (these mice died when injected with Jo2) and in mice subjected to PH (known to protect mice from liver death 17, as evidenced by a lack of caspase-3 cleavage, TUNEL staining and increase in liver enzyme ALT) (Fig. 4g and Supplementary Fig. 12a). A clear increase in intracellular p-c-Jun staining of hepatocytes (Supplementary Fig. 12b) indicated that JNK was activated in hepatocytes in response to CD95 stimulation in mice protected from apoptosis by PH. The link between CD95 and the JNK pathway was also found in endometrioid tumours from K-ras/Pten mutant mice. Tumour tissue taken from cancerous ovaries expressing CD95 stained strongly for both CD95 and nuclear p-c-Jun, whereas tumour tissue from mice deficient for CD95 expression in the ovaries not only lacked expression of CD95, but was also devoid of p-c-Jun staining (Fig. 4h).
In aggregate the data suggest that cancer cells and the regenerating liver require a basal activity of CD95 to activate JNK resulting in phosphorylation of c-Jun and driving proliferation. To identify further downstream effectors in this pathway we isolated RNA from HeyA8 cells with knocked down CD95 using two independent shRNAs, and from HepG2 cells with knocked down CD95, and from CD95 deficient livers and compared gene expression profiles to the corresponding parental/wt cells. Interestingly, among all three systems, there were only two genes that were downregulated to a greater extent than CD95 itself—early growth 1 (Egr1) and the AP1 component c-Fos (Supplementary Fig. 13a-c). Both genes are downstream of JNK and are essential growth promoting transcription factors 23,24. Consistent with their function, both Egr1 and c-Fos were strongly upregulated in the liver of Jo2-injected, partial hepatectomised mice (Supplementary Fig. 13d) and their expression was strongly reduced in CD95 deficient ovarian cancer (Supplementary Fig. 13e).
Our data suggest that the CD95/CD95L system, rather than acting tumour suppressive, drives cancer growth joining the ranks of the TNFR1 and TNFα in stimulating tumour growth. In line with our results, studies suggest that CD95 activates neuronal stem cells 25 and acts as a tumour promoter for glioblastoma by activating src kinases 26. In addition it was recently shown that mice that only express soluble CD95L suffer from large histiocytic sarcomas in the liver 27 likely due to a lack of apoptosis induction and a tumorigenic activity of CD95L. Our data suggest that CD95 exerts a growth promoting activity mainly through a pathway involving JNK, c-Jun, Erg1 and c-Fos, and that tumour cells are stimulated by their own CD95L. The data provide an explanation for the long standing mystery of the role of CD95 expression in many tissues and in the majority of human cancers without signs of apoptosis in vivo, and suggest that efforts to inhibit rather than to promote CD95 activity should be considered during cancer therapy.
Methods Summary
Endometrioid ovarian cancer induction
The method of ovarian cancer induction using LSL-K-rasG12D/+PtenloxP/loxP and LSL-K-rasG12D/+PtenloxP/loxPCD95loxP/loxP female mice was described previously 28. Briefly, mice were sedated, the right ovary was exposed and the ovarian bursa was injected with AdCre (2.5 × 107 plaque-forming units) (University of Iowa Gene Transfer Vector Core). The left ovary was not injected and served as an internal control. Mice were evaluated weekly for palpable tumour and all were sacrificed 8 or 14 weeks after the injection of the virus. At the time of sacrifice the primary tumour was excised, weighed, measured and the number of metastatic nodules and volume of ascites were recorded. All tissue was fixed in 10% formalin, embedded in paraffin, and stained with haematoxylin and eosin.
Partial hepatectomy
2/3 partial hepatectomy was performed as previously described 29,30. In brief, mice were anaesthetised with ketamine (100 mg/kg) and xylazine (10 mg/kg) i.p., the liver was exposed through a midline incision, and the right and left lobes were sequentially ligated with a silk 4-0 suture, and resected. BrdU solution (50 mg/kg in PBS) was injected i.p. 2 hrs prior to analysis. DNA synthesis was measured by immunohistochemical staining of paraffin liver sections with anti-BrdU antibody. The percentage of BrdU positive hepatocyte nuclei among total hepatocyte nuclei was calculated using the Cellular Image Analysis System (ACIS, Clarient, San Juan Capistrano, CA).
Liver cancer induction
15 day old CD95loxP/loxP (wt) and CD95loxP/loxPAlbumin-Cre (CD95 k.o.) male mice were injected with a single dose of DEN (25 μg/g body weight, i.p.). Eight months later, all mice were sacrificed and livers were excised. Parameters of total liver weight, number of liver surface nodules and maximum nodule diameters were recorded.
Full Methods and associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Material
Acknowledgments
We are grateful to Drs. Alexander Chervonsky and Daniela Dinulescu for providing the CD95 loxP/loxP mice and the K-rasG12D/+Ptenfl/fl mice, respectively, and to Dr. Syed Ahmed for help with one experiment. We are grateful to Terry Li for performing the IHC and to Dr. You-Jia Hua for analysing the gene array data. This work was funded by grants CA112240 from the NCI and CCFA 1661 from the Crohn's and Colitis Foundation of America.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions L.C. and S.M.P performed the experiments; A.T. performed the PH; A.H., K.S., C.F. and I.R. performed some experiments; J.T. performed pathology analyses; Y.X.F. and E.L., supervised some experiments; M.E.P designed experiments and supervised the project.
Author Information Reprints and permission information is available at www.nature.com/reprints.
References
- 1.Nagata S. Fas ligand-induced apoptosis. Annu Rev Genet. 1999;33:29–55. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
- 2.Krammer PH. CD95's deadly mission in the immune system. Nature. 2000;407:789–95. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- 3.Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–92. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peter ME, Legembre P, Barnhart BC. Does CD95 have tumor promoting activities? Biochim Biophys Acta. 2005;1755:25–36. doi: 10.1016/j.bbcan.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23:2950–66. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- 6.Barnhart BC, et al. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. Embo J. 2004;23:3175–85. doi: 10.1038/sj.emboj.7600325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peter ME, et al. The CD95 receptor: apoptosis revisited. Cell. 2007;129:447–50. doi: 10.1016/j.cell.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 8.Lavrik IN, et al. Analysis of CD95 threshold signaling: triggering of CD95 (FAS/APO-1) at low concentrations primarily results in survival signaling. J Biol Chem. 2007;282:13664–71. doi: 10.1074/jbc.M700434200. [DOI] [PubMed] [Google Scholar]
- 9.Baldwin RL, Tran H, Karlan BY. Primary ovarian cancer cultures are resistant to Fas-mediated apoptosis. Gynecol Oncol. 1999;74:265–71. doi: 10.1006/gyno.1999.5448. [DOI] [PubMed] [Google Scholar]
- 10.Abrahams VM, et al. Epithelial ovarian cancer cells secrete functional Fas ligand. Cancer Res. 2003;63:5573–81. [PubMed] [Google Scholar]
- 11.Rabinowich H, et al. Lymphocyte apoptosis induced by Fas ligand- expressing ovarian carcinoma cells. Implications for altered expression of T cell receptor in tumor-associated lymphocytes. J Clin Invest. 1998;101:2579–88. doi: 10.1172/JCI1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor DD, Lyons KS, Gercel-Taylor C. Shed membrane fragment-associated markers for endometrial and ovarian cancers. Gynecol Oncol. 2002;84:443–8. doi: 10.1006/gyno.2001.6551. [DOI] [PubMed] [Google Scholar]
- 13.Sawada K, et al. C-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res. 2007;67:1670–1680. doi: 10.1158/0008-5472.CAN-06-1147. [DOI] [PubMed] [Google Scholar]
- 14.Kaur S, et al. {beta}3-integrin expression on tumor cells inhibits tumor progression, reduces metastasis, and is associated with a favorable prognosis in patients with ovarian cancer. Am J Pathol. 2009;175:2184–96. doi: 10.2353/ajpath.2009.090028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dinulescu DM, et al. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 16.Stranges PB, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–41. doi: 10.1016/j.immuni.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desbarats J, Newell MK. Fas engagement accelerates liver regeneration after partial hepatectomy. Nat Med. 2000;6:920–3. doi: 10.1038/78688. [DOI] [PubMed] [Google Scholar]
- 18.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–90. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. 2007;8:665–8. doi: 10.1038/ni0707-665. [DOI] [PubMed] [Google Scholar]
- 20.Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest. 2008;118:3943–53. doi: 10.1172/JCI37156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakurai T, Maeda S, Chang L, Karin M. Loss of hepatic NF-kappa B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation. Proc Natl Acad Sci U S A. 2006;103:10544–51. doi: 10.1073/pnas.0603499103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang Q, et al. Sustained JNK1 activation is associated with altered histone H3 methylations in human liver cancer. J Hepatol. 2009;50:323–33. doi: 10.1016/j.jhep.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim CP, Jain N, Cao X. Stress-induced immediate-early gene, egr-1, involves activation of p38/JNK1. Oncogene. 1998;16:2915–26. doi: 10.1038/sj.onc.1201834. [DOI] [PubMed] [Google Scholar]
- 24.Cavigelli M, Dolfi F, Claret FX, Karin M. Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J. 1995;14:5957–64. doi: 10.1002/j.1460-2075.1995.tb00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corsini NS, et al. The death receptor CD95 activates adult neural stem cells for working memory formation and brain repair. Cell Stem Cell. 2009;5:178–90. doi: 10.1016/j.stem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Kleber S, et al. Yes and PI3K Bind CD95 to Signal Invasion of Glioblastoma. Cancer Cell. 2008;13:235–48. doi: 10.1016/j.ccr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 27.La OR, et al. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature. 2009;461:659–63. doi: 10.1038/nature08402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romero IL, et al. Effects of oral contraceptives or a gonadotropin-releasing hormone agonist on ovarian carcinogenesis in genetically engineered mice. Cancer Prev Res (Phila Pa) 2009;2:792–9. doi: 10.1158/1940-6207.CAPR-08-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greene AK, Puder M. Partial hepatectomy in the mouse: technique and perioperative management. J Invest Surg. 2003;16:99–102. [PubMed] [Google Scholar]
- 30.Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc. 2008;3:1167–70. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.