

Abstract
Apoptosis or programmed cell death is an important outcome of cell fate and is influenced by several factors. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is a member of the EGF family of growth factors and is synthesized as a membrane-associated precursor molecule (proHB-EGF). Under stressful conditions proHB-EGF is proteolytically cleaved and released as a soluble ligand (sHB-EGF) that activates the EGF receptor. We show that antibody against CD9, a membrane tetraspanin, induces apoptosis in mouse embryonic stem cells through the activation of specific EGF receptor residues (Y-1148 and Y-1173), caspase-3 and MAPK signalling. HB-EGF and the p38 inhibitor PD169316 act in a pro-survival manner by perturbing phosphorylation of EGFR Y-1173, suggesting its importance in inducing apoptosis. Caspase-3 activation was attenuated in the presence of HB-EGF and PD169316. Furthermore, HB-EGF and PD169316 prevent p38 phosphorylation while promoting the phosphorylation of the pro-survival SAPK/JNK and ERK. These results suggest a role for CD9 as an endogenous suppressor of apoptosis, an effect that is mimicked by HB-EGF and PD169316.
Keywords: apoptosis, CD9, EGFR, HB-EGF, PD169316
Apoptosis of embryonic stem (ES) cells is a critical component of stem-cell fate choice and maintenance of the chromosomal integrity of ES cell lines (1). Apoptosis is regulated by complex interacting signals and is greatly influenced by growth factors (2–4). Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is a 21–27 kD member of the epidermal growth factor (EGF) family and promotes cell survival in normal tissue and inhibits apoptosis caused by ischaemia reperfusion injury. Ectodomain shedding of proHB-EGF is an important mechanism to regulate the biological activities of many tyrosine kinase membrane proteins (2, 4, 5). However, proHB-EGF itself is also biologically active and forms complexes with both CD9 integrin and EGF receptor (EGFR) (2, 3). The influence of HB-EGF on apoptosis has been linked to its heterobimolecular associations with integrin, EGFR and CD9 (3, 6). CD9, a membrane bound tetraspanin, is associated with HB-EGF in several cell types. In monkey kidney cells HB-EGF acts as the receptor to diphtheria toxin and exposure to toxin leads to the activation of the mitogen activated protein kinase (MAPK) pathway (2, 5). Activation of the MAPK pathway can promote cell survival and inhibit apoptosis and typically can occur through HB-EGF binding to EGFR. Loss of CD9/HB-EGF association lowers the affinity of HB-EGF for diphtheria toxin and perturbs EGFR signalling, suggesting that HB-EGF/EGFR signalling depends on CD9/HB-EGF interactions (2, 5).
The anti-human CD9 monoclonal antibody (ALB6) can inhibit cell proliferation, reduce cell viability and induce MAPK signalling changes specific to apoptosis, suggesting a CD9-dependent apoptotic pathway (7). The anti-CD9 antibody ALB6 activates c-Jun NH2-terminal kinase/stress-activated protein kinase (JNK/SAPK) and p38 MAPK within 5–15 min, and induces caspase-3 activation within 24–48 h (7). PD169316, a MAPK inhibitor, prevents p38 phosphorylation and apoptosis in neuroblastoma cells (8). Of note, ALB6 induces specific tyrosine phosphorylation of the p46 Shc isoform, and over expression of dominant-negative p46 Shc completely suppresses ALB6-induced activation of JNK/SAPK, p38 MAPK and caspase-3, resulting in the inhibition of apoptotic cell death (7). Similarly, in mouse embryonic stem cells (mESCs) a blocking antibody to CD9 (KMC8) induces apoptosis (9).
In this study, using the mESCs R1 and D3 as our models, we show that the MAPK pathway contributes to CD9-dependent apoptosis. Furthermore, we demonstrate the ability of soluble HB-EGF and PD169316 (p38 inhibitor) to prevent CD9 dependent apoptosis through the activation of ERK and JNK and the suppression of p38 MAPK.
MATERIALS AND METHODS
Reagents and Antibodies
Anti-CD9 KMC8 and isotype control were purchased from Pharmingen (San Diego, CA). The antibodies against ERK 1/2, phosphorylated ERK 1/2, SAPK/JNK, phosphorylated SAPK/JNK, p38 MAPK, phosphorylated p38 MAPK, SRC, phosphorylated SRC, PLCγ, c-CBL, GRB, cleaved caspase-3, phosphorylated EGFR (Tyr 845, 1068 and 1173) and PY-100 were purchased from Cell Signaling Technology (Beverly, MA). EGFR (Tyr 1086) antibody was from Sigma-Aldrich (St Louis, MO). EGFR (Tyr 1148) antibody, PD169316 and recombinant HB-EGF were purchased from Calbiochem (Darmstadt, Germany). Recombinant mouse EGF was purchased from Cell Sciences (Canton, MA). DAPI was obtained from KPL (Gaithersburg, MD).
ES Cell Culture
Mouse ES cell lines R1 and D3 (provided by Dr S. Dalton, The University of Georgia, Athens, GA, USA) were maintained on gelatin-coated dishes in complete media containing 1000 U/ml recombinant mouse Leukemia Inhibitory Factor (LIF-ESGRO; Chemicon International, Temecula, CA), 20% fetal calf serum, Knockout serum replacement (Gibco, BRL, Grand Island, NY), 2 mM l-glutamine, 100 U/ml penicillin/streptomycin, (VWR International, Bridgeport, New Jersey), 2 mM sodium pyruvate (VWR International, Bridgeport, NJ), 0.2 mM 2-mercaptoethanol (Invitrogen, Carlsbad, CA), and DMEM (VWR International, Bridgeport, NJ). Initial plating density was 2 × 104 cells/cm2.
Immunoprecipitation and Western Blot Analysis
Cells were lysed in buffer containing 0.5 μM 3-[(3-cholamidopropyl)-dimethylammonio]-1- propanesulfonate (CHAPS) (Calbiochem, San Diego, CA), 100 mM Tris (pH 7.2) (Sigma-Aldrich), phosphatase inhibitor cocktail set II (Calbiochem, San Diego, CA), and protease inhibitor cocktail tablets-complete (Roche, Penzberg, Germany). Immunoprecipitations were performed with 50 μg cell lysate for 1 h followed by the addition of protein-G-Sepharose for 1 h at 4°C. Mini protean II system from Bio-Rad (Hercules, CA) was used for western blotting. Proteins were separated on pre-cast Tris–HCl gels (4–15% gradient, Bio-Rad, Hercules, CA) at 30 mA (two gels) for 1 h, then transferred to nitrocellulose membranes (0.4 μm) (Bio-Rad, Hercules, CA) at 250 mA for 1 h. Membranes were then blocked in 5% bovine serum albumin (BSA) (Pierce, Rockford, IL) in PBS. Primary antibody was incubated with the membrane in 1% BSA/PBS (Pierce) at room temperature for 1 h. Membranes were washed for 30 min in wash buffer (Bio-Rad, Hercules, CA). HRP-conjugated secondary antibody was added to the membrane in 1% BSA/PBS (Pierce) at room temperature for 1 h. After a 30-min wash, membranes were incubated in ECL substrate for 1 min (Pierce). Filters were developed on Ektar Kodak film using an AGFA developer (Gevaert, NV).
Immunofluorescence Microscopy
Mouse ES cells were cultured on gelatin-coated chamber slides. After PBS washes, cells were fixed with 2% paraformaldehyde (PFA) at room temperature for 15 min. Cells were washed twice with PBS and treated with 0.5% Triton X/PBS for 5 min, washed, then blocked with 2% PBS for 1 h at room temperature. Cells were incubated with primary antibody overnight. After two washes with PBS, cells were incubated with the appropriate fluorescein isothiocyanate-conjugated antibody (Jackson Immunoresearch Laboratories, West Grove, PA) and mounted with Vectashield (Vector Labs, Burlingame, CA). Propidium iodide (PI) (Calbiochem, Gibbstown, NJ) was added (final 10 μg/ml) directly to the culture medium. After incubating for 30 min at room temperature, staining was observed using a fluorescent microscope (Nikon, Melville, NY).
RNAi Transfection Preparation
Twenty-four hours before transfection cells were plated on 1.75 × 105 inactivated mouse embryonic fibroblast (MEF) cells in 1 ml of MEF growth medium per well (six-well dish). Each transfection sample was prepared using Stealth RNAi-Lipofectamine 2000 as the following: Samples were diluted to 50 pmol of Stealth™ RNAi (i.e. 2.5 µl of 20 µM Stealth RNAi) in 100 µl of Opti-MEM® I Reduced Serum Medium. Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was gently mixed before use, then diluted 2 µl in 100 µl of Opti-MEM® I Reduced Serum Medium, mixed gently and incubated for 15 min at room temperature. After a 15-min incubation, the diluted Stealth RNAi and the diluted Lipofectamine 2000 (total volume ∼205 µl) were combined and mixed gently and incubated for 15 min at room temperature.
During the 15 min incubation (Step 2c), mESCs were prepared for plating. After selecting a flask of low passage (p10), healthy mES cells maintained on an inactivated MEF feeder layer were dissociated by trypsinization, counted and replated on inactivated MEF feeder cells to a final density of 2 × 105 cells per well in 1 ml of complete medium. Approximately 205 µl of Stealth RNAi-Lipofectamine 2000 complex was added to each well containing cells and complete medium. After mixing gently by rocking the plate back and forth cells were incubated at 37°C in a humidified CO2 incubator for 16–24 h and assayed for gene knockdown.
Transcript Analysis by Quantitative Real-Time PCR (Q-PCR)
RNA was prepared using Qiagen RNeasy Mini Kits. Chromosomal DNA was removed using RNase-free DNase (Qiagen). cDNA was prepared using the Superscript First Strand Synthesis System (Invitrogen) using 2 µg total RNA. Target mRNAs were assayed using TaqMan Gene Expression Assays (Applied Biosystems). PCR reactions were performed on an Applied Biosystems ABI 7700 Sequence Detector.
RESULTS
Anti-CD9 Induces Apoptosis and Decreases Viability in mESCs
In murine ES cells plated at a concentration of 2 × 103 cells/ml, anti-CD9 KMC8 is reported to induce apoptosis (9). Cells appeared to be dead within 24 h of culture after treatment with KMC8 (25 μg/ml). We assayed for cleaved caspase-3, a hallmark of apoptosis (10), after treating mESCs with KMC8. KMC8 treatment induces activation of caspase-3. Apoptosis by KMC8 was observed in both R1 and D3 mES cells (Fig. 1). In this setting, the presence of HB-EGF reduced activation of caspase-3 by 2.5-fold, whereas, EGF was not as effective in preventing apoptosis (Fig. 1 and Table 1). HB-EGF treatment (without KMC8) does not induce apoptosis (Fig. 1 and Table 1). Furthermore, PI staining confirmed a decrease in cell viability after treatment with KMC8 (Table 1). Non-specific rat IgG2a antibody did not affect the colony formation or survival of ES cells. These results show that apoptosis is specific to the use of KMC8 and CD9-dependent.
Fig. 1.

Anti-CD9 antibody treatment causes cleavage of caspase-3, which is blunted by simultaneous treatment with HBE-GF or EGF (20×). After 24 h of treatment, cells were fixed and stained for cleaved caspase-3 (green). Nuclei are labelled with DAPI. HBE-GF independently does not promote apoptosis (p = <0.05), nor does matching isotype control antibody. Data from these treatments are analysed in Table 1.
Table 1.
Cleaved Caspase-3 and PI staining as a function of anti-CD9, HB-EGF and EGF treatment of mouse embryonic stem cells
| Anti-CD9 | Anti-CD9 (%) | Anti-CD9 + HB-EGF | HB-EGF + EGF | EGF | Control | |
|---|---|---|---|---|---|---|
| Cleaved caspase-3 | 80 ± 3.6 | 31 ± 2.1 | 42 ± 1.0 | 6 ± 0.3 | 7 ± 0.5 | 5 ± 0.8 |
| PI | 78 ± 4.2 | 31 ± 4.0 | 50 ± 2.2 | 11 ± 1.7 | 15 ± 1.7 | 11 ± 3.4 |
Data are presented as the percentage of cells which are positive for cleaved caspase-3 and PI. Comparisons between groups were determined using Tukey multiple comparison tests, using cell counts from n = 3 independent experiments (p < 0.05).
HB-EGF and PD169316 Perturb Anti-CD9-Stimulated Phosphorylation of EGFR Tyrosine-1147 and -1173
Antibodies against phosphorylated EGFR were used to determine if anti-CD9 treatment induced EGFR phosphorylation and activated the MAPK pathway. MAPK has been shown to be involved with cell survival and apoptosis (7). Treatment of cells with KMC8 induced tyrosine-1148 and -1173 phosphorylation (Fig. 2), which are known to activate MAPK signalling. In the presence of anti-CD9, both HB-EGF and PD169316 suppressed phosphorylation of EGFR tyrosine-1173, while tyrosine-1148 phosphorylation was unaffected (Fig. 3). These results indicate that apoptosis induced by KMC8 requires tyrosine-1173 phosphorylation. Protein phosphorylation induced by anti-CD9 is specific and does not induce phosphorylation of tyrosine-845, -1068 and -1086 in the EGFR (Fig. 2). As a result, we did not see phosphorylation of the down-stream proteins associated with their phosphorylation, such as phospholipase C gamma (PLCγ) or Casitas b-lineage lymphoma (c-Cbl) (Fig. 4B). However, phosphorylation of growth factor receptor-binding protein 2 (GRB2) was observed (Fig. 4B), which can occur as a result of tyrosine-1173 and -1148 phosphorylation on the EGFR (11). These results show KMC8 apoptosis has involves specific EGFR tyrosine residues. HB-EGF and PD169316 are able to perturb their involvement.
Fig. 2.

Identification of EGFR phosphorylation sites involved in anti-CD9-induced apoptosis (20×). Twenty-four hrs of anti-CD9 antibody treatment was performed, cells were fixed and stained for activated EGFR Y845, 1068, 1086, 1148 and 1173 (green). Nuclei are labelled with DAPI. Anti-CD9 antibody treatment induced activation of EGFR Y1148 and Y1173. Matching isotype control antibody did not activate any of the EGFR residues of interest (negative control). Separately treated cells with EGF or HB-EGF produced activated EGFR (positive control).
Fig. 3.

Phosphorylation of EGFR is suppressed by HB-EGF and p38 MAPK inhibitor (20×). After 24 h of anti-CD9 antibody treatment and fixation cells were stained for activated EGFR 1148 and 1173 (green). Nuclei are labelled with DAPI. Anti-CD9 antibody treatment induced phosphorylation of EGFR Y1148 and Y1173. HB-EGF and p38 inhibitor suppressed the activation of Y1173 while EGF did not as significantly. Matching isotype control antibody did not activate any of the EGFR residues of interest (negative control). Separately treated cells with EGF or HB-EGF produced activated EGFR (positive control).
Fig. 4.

Effects of anti-CD9 antibody on SAPK/JNK and ERK pathway activation. (A) After 5 min of antibody treatment, cells were lysed, then later probed for reactivity to SAPK/JNK and phosphorylated-SAPK/JNK antibodies. Anti-CD9 induces significant phosphorylation of SAPK/JNK and ERK, while HB-EGF promotes a greater amount of SAPK/JNK and ERK phosphorylation. The p38 inhibitor produced less phosphorylation while anti-CD9 + EGF treatment produced similar results as the p38 inhibitor (not shown). (B) Phosphorylation of EGFR-related proteins PLC and CBL did not occur.
CD9 RNAi Increases Activation of Caspase-3
To determine if CD9 is required for cell survival, we inhibited its expression through RNAi transfection (Fig. 5A). Three commercially available RNAi (Invitrogen) caused profound suppression of CD9 expression in mESCs (Figs 5B and C). Additionally an increase in caspase-3 activation was observed, which coincides with the silencing of CD9 transcription. The amount of activated caspase-3 in RNAi transfected cells was not as great as staurosporin treated cells, which represents a positive control. These results show that expression of CD9 is essential for mESC survival in vitro.
Fig. 5.
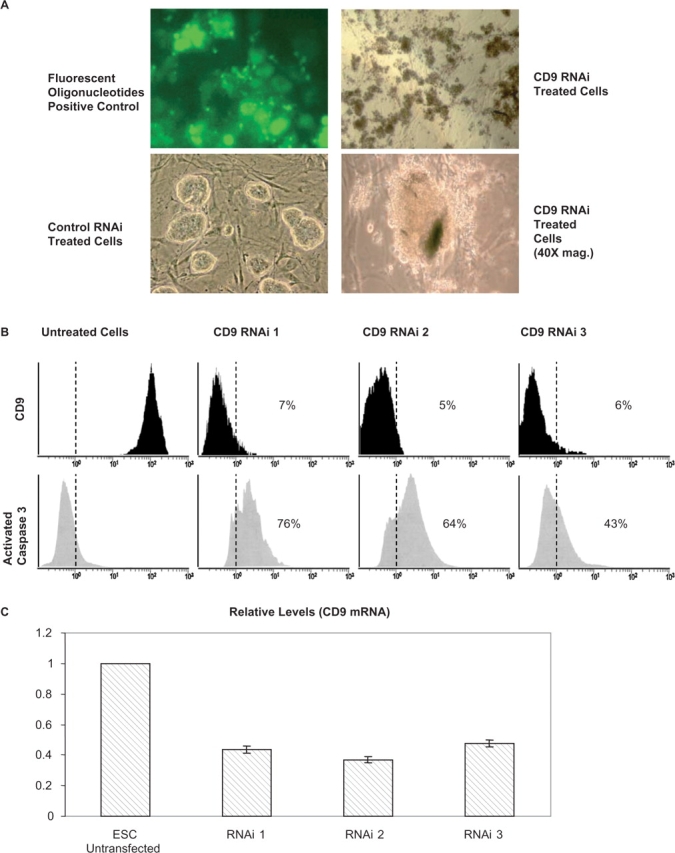
Caspase-3 activation correlates with CD9 expression. (A) Cells were transfected with a commercially available RNAi. Fluorescently labelled oligonucleotides were used to confirm transfection (20×). Untreated cells were used to display normalized cells (20×). Treated cells display a loss of integrity (40×). (B) Flow cytometry analysis shows how caspase-3 activation increased while the presence of CD9 on the cell surface diminished. (C) Transcript profiling of mESCs by Q-PCR for CD9 after RNAi transfection. Transcript levels were determined in triplicate and shown as ±standard error of the mean (SEM) after being normalized against GAPDH. Levels of transcripts in mESCs were normalized against GAPDH and assigned a value of one.
HB-EGF and PD169316 Prevent Multiple Species Phosphorylation of p38
Within 5 min of anti-CD9 treatment, phosphorylation of JNK and ERK (p44/p42) were observed and were significantly reduced when cells were treated with PD169316, whereas treatment with HB-EGF did not reduce their phosphorylation (Fig. 4A). However, multiple species of phosphorylated p38 MAPK were completely suppressed by both HB-EGF and PD169316 (Fig. 6). These results suggest MAPK signalling, specifically p38 phosphorylation, is a requirement for anti-CD9-induced cell death (Fig. 6).
Fig. 6.

Effects of anti-CD9 antibody, HB-EGF and p38 MAPK inhibitor on p38 MAPK pathway. Cells were treated with anti-CD9 antibody, anti-CD9 Ab + HB-EGF, Anti-CD9Ab + p38 inhibitor and control IgG2a. Cells were lysed with CHAPS buffer, scrapped and quantitated. Immunoprecipitations were performed using anti-pan phospho-p38 antibody. Samples were run on a 4–15% gradient gel, transferred to nitrocellulose membranes and anti-pan phospho-p38 antibody primary immunoglobulins were added overnight. HRP-anti-rabbit antibody was used as secondary. Membranes were developed via ECL.
DISCUSSION
Our goal in this study was to determine the role of signalling pathways, specifically the MAPK pathway, on apoptosis in mESCs induced through CD9 ligation. In addition, we explored the anti-apoptotic effect of HB-EGF and PD169316 on MAPK signalling. The MAPK pathway is a core component of both pro-apoptotic and pro-survival signalling (7, 12, 13), but its involvement in CD9 dependent apoptosis of (non-transformed) mESCs has not been explored. Additionally, the effect of HB-EGF and PD169316 on MAPK signalling in mouse stem-cell survival is also unknown.
Several studies suggest mechanisms by which anti-CD9 reduces mouse stem-cell survival, specifically through the interference with cell adhesion (3). However, few studies report associations between CD9 and apoptosis (9). We found KMC8 capable of inducing apoptosis in sparsely plated mESCs (2 × 103/cm2) (2) however, anti-CD9 was ineffective against murine ES cells plated at a higher density (2 × 104/cm2) (data not shown). CD9 associates with HB-EGF and is of known importance in cell survival under stressful conditions (4). It is possible that in higher density plated stem cells CD9 may support apoptosis resistance as juxtacrine orientated (RAJO) proHB-EGF. Soluble HB-EGF perturbs anti-CD9 induced apoptosis at lower plating density, possibly through similar mechanisms as proHB-EGF.
In cancer cell lines, anti-CD9 has proven to be the best method for studying the CD9-mediated signalling pathway because a natural ligand for CD9 has yet to be discovered (7, 9). An important characteristic regarding the molecular mechanisms involved with anti-CD9 induced apoptosis is the involvement of at least two pathways, caspase and the MAPK pathway (7). Caspase activation is a hallmark of apoptosis and is activated intrinsically and extrinsically. Caspase-3 activation is essential for apoptosis because of its involvement in proteolytic cleavage of several proteins (7, 10). Our results clearly show that anti-CD9-induced apoptosis recruits caspase-3, while HB-EGF and PD169316 block anti-CD9 antibody induced apoptosis. The MAPK pathway, which consists of JNK, ERK (p44/p42) and p38, is a core component involved in the terminal stages of signalling cascades that are related to both pro-apoptotic and pro-survival signalling. ERK has cell survival properties that are initiated by such stimuli as hypoxia and growth factor withdrawal, while JNK and p38 have distinct apoptotic properties which are initiated through a range of cellular stresses such as UV-light and osmotic shock. The up-regulation of JNK and p38 in addition to the down regulation of ERK has been associated with apoptosis (8, 13). In contrast, JNK expression has also been associated with pro-survival (13). Stem-cell differentiation and cell fate might also be determined by the balance between expression of JNK, ERK and p38. Our data shows that within 5 min of addition of KMC8, there was phosphorylation of p46 JNK, p42 ERK and multiple isoforms of p38 MAPK. The addition of KMC8 and HB-EGF perturbed the phosphorylation of all isoforms of p38 MAPK, while increasing activation of p46 JNK and p42 ERK proving consistent with a setting in which the changes in MAPK signalling pathway is involved with CD9 antibody apoptosis. Furthermore, we found that PD169316 poorly increased activation of JNK and ERK, but was equally as effective as HB-EGF in suppressing p38 MAPK phosphorylation. These results suggests that PD169316 is an effective inhibitor against phosphorylation of JNK and ERK as well as p38 MAPK. It has not been reported as to which isoform of p38 MAPK is essential for apoptosis.
We have shown that KMC8 causes phosphorylation of the EGFR within 5 min after treatment. Specifically EGFR Y-1148 and Y-1173 phosphorylation is known to promote the activation of the MAPK pathway. Cells treated with HB-EGF and PD 169316 do not undergo tyrosine phosphorylation or apoptosis, while those that were treated with EGF does not perturb cell death or change phosphorylation. These results suggest that this is a growth factor-specific phenomenon.
Several groups have shown CD9 to be essential for cellular adhesion, gamete fusion and migration (14–17). Our data confirm that expression of CD9 is required for mESC survival. RNAi studies show a correlation between the silencing of CD9 and an increase in caspase-3 activation, suggesting CD9 in some manner acts as a suppressor of cellular stress, whereas its absence favors apoptosis through caspase-3 activation. In terms of mechanisms there are likely to be several factors leading to caspase-3 activation, but these remain to be identified in future studies. We expect that uncovering more of the mechanistic changes associated with CD9 antibody-induced apoptosis using human stem cells as a model will lead to potential targets for cancer therapeutics through the indication of apoptosis.
FUNDING
This work was supported by The National Science Foundation (Science Behind Our Foods), National Institutes of Health [NIH 2T32 NS 007480-06] and Emory University School of Medicine Department of Anesthesiology.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENTS
Special thanks to Drs Richard D. Cummings (Emory University), Allan I. Levey (Emory University), Gregory Buck (Texas A&M University-Corpus Christi), Steve Stice (The University of Georgia), Steve Nickerson (The University of Georgia) and Mark Froetschel (The University of Georgia). IANAFS.
Glossary
Abbreviations:
- CD9
cluster of differentiation 9
- EGF
epidermal growth factor
- EGFR
EGF receptor
- ES
embryonic stem
- HB-EGF
heparin-binding epidermal growth factor-like growth factor
- JNK/SAPK
c-Jun NH2-terminal kinase/stress-activated protein kinase
- MAPK
mitogen-activated protein kinase
- mESCs
mouse embryonic stem cells
- RAJO
resistance as juxtacrine orientated
REFERENCES
- 1.Hamazaki T, Iiboshi Y, Oka M, Papst PJ, Meacham AM, Zon LI, Terada N. Hepatic maturation in differentiating embryonic stem cells in vitro. FEBS Lett. 2001;497:15–19. doi: 10.1016/s0014-5793(01)02423-1. [DOI] [PubMed] [Google Scholar]
- 2.Cha JH, Brooke JS, Ivey KN, Eidels L. Cell surface monkey CD9 antigen is a coreceptor that increases diphtheria toxin sensitivity and diphtheria toxin receptor affinity. J. Biol. Chem. 2000;275:6901–6907. doi: 10.1074/jbc.275.10.6901. [DOI] [PubMed] [Google Scholar]
- 3.Ono M, Handa K, Withers DA, Hakomori S. Motility inhibition and apoptosis are induced by metastasis-suppressing gene product CD82 and its analogue CD9, with concurrent glycosylation. Cancer Res. 1999;59:2335–2339. [PubMed] [Google Scholar]
- 4.Iwamoto R, Mekada E. Heparin-binding EGF-like growth factor: a juxtacrine growth factor. Cytokine Growth Factor Rev. 2000;4:335–344. doi: 10.1016/s1359-6101(00)00013-7. [DOI] [PubMed] [Google Scholar]
- 5.Iwamoto R, Higashiyama S, Mitamura T, Taniguchi N, Klagsbrun M, Mekada E. Heparin-binding EGF-like growth factor, which acts as the diphtheria toxin receptor, forms a complex with membrane protein DRAP27/CD9, which up-regulates functional receptors and diphtheria toxin sensitivity. EMBO J. 1994;13:2322–2330. doi: 10.1002/j.1460-2075.1994.tb06516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J. 1997;6:428–442. [PubMed] [Google Scholar]
- 7.Murayama Y, Miyagawa J, Oritani K, Yoshida H, Yamamoto K, Kishida O, Miyazaki T, Tsutsui S, Kiyohara T, Miyazaki Y, Higashiyama S, Matsuzawa Y, Shinomura Y. CD9-mediated activation of the p46 Shc isoform leads to apoptosis in cancer cells. J. Cell. Sci. 2004;117:3379–3388. doi: 10.1242/jcs.01201. [DOI] [PubMed] [Google Scholar]
- 8.Thellunga S, Villaa V, Corsaroa A, Arenaa S, Millob E, Damonteb G, Benattib U, Tagliavinic F, Floriod T, Schettinia G. p38 MAP kinase mediates the cell death induced by PrP106–126 in the SH-SY5Y neuroblastoma cells. Neurobiology of Disease. 2002;9:69–81. doi: 10.1006/nbdi.2001.0461. [DOI] [PubMed] [Google Scholar]
- 9.Oka M, Tagoku K, Russell TL, Nakano Y, Hamazaki T, Meyer EM, Yokota T, Terada N. CD9 is associated with leukemia inhibitory factor-mediated maintenance of embryonic stem cells. Mol. Biol. Cell. 2002;13:1274–1281. doi: 10.1091/mbc.02-01-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lakhani SA, Masud A, Kuida K, Porter Jr GA, Booth CJ, Mehal WZ, Inayat I, Flavell RA. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–480. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- 12.Hackel PO, Zwick E, Prenzel N, Ullrich A. Epidermal growth factor receptors: critical mediators of multiple receptor pathways. Curr. Opin. Cell. Biol. 1999;2:184–189. doi: 10.1016/s0955-0674(99)80024-6. [DOI] [PubMed] [Google Scholar]
- 13.Ortiz MA, Lopez-Hernandez FJ, Bayon Y, Pfahl M, Piedrafita FJ. Retinoid-related molecules induce cytochrome c release and apoptosis through activation of c-Jun NH(2)-terminal kinase/p38 mitogen-activated protein kinases. Cancer Res. 2001;61:8504–8512. [PubMed] [Google Scholar]
- 14.Seigneuret M, Delaguillaumie A, Lagaudriere-Gesbert C, Conjeaud H. Structure of the tetraspanin main extracellular domain. A partially conserved fold with a structurally variable domain insertion. J. Biol. Chem. 2001;276:40055–40064. doi: 10.1074/jbc.M105557200. [DOI] [PubMed] [Google Scholar]
- 15.Stipp CS, Kolesnikova TV, Hemler ME. Functional domains in tetraspanin proteins. Trends Biochem. Sci. 2003;28:106–112. doi: 10.1016/S0968-0004(02)00014-2. [DOI] [PubMed] [Google Scholar]
- 16.Wright MD, Tomlinson MG. The ins and outs of the transmembrane 4 superfamily. Immunol. Today. 1994;12:588–94. doi: 10.1016/0167-5699(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 17.Berditchevski F, Zutter MM, Hemler ME. Characterization of novel complexes on the cell surface between integrins and proteins with 4 transmembrane domains (TM4 proteins) Mol. Biol. Cell. 1996;2:193–207. doi: 10.1091/mbc.7.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]