

Abstract
The G protein-coupled receptor GPR30 (GPER) binds 17β-estradiol (E2), yet differs from classical estrogen receptors (ERα and ERβ). GPR30 can mediate E2-induced non-genomic signaling, but its role in ERα-positive breast cancer remains unclear. Gene expression microarray data from 5 cohorts comprising 1,250 breast carcinomas showed an association between increased GPR30 expression and ERα-positive status. We therefore examined GPR30 in estrogenic activities in ER-positive MCF-7 breast cancer cells using G-1 and diethylstilbestrol, ligands which selectively activate GPR30 and ER, respectively, and small interfering RNAs (siRNAs). In expression studies, E2 and diethylstilbestrol but not G-1 down-regulated both ER and GPR30, indicating this was ER mediated. In Ca2+ mobilization studies, GPR30 but not ERα mediated E2-induced Ca2+ responses, since E2, 4-hydroxytamoxifen (also activates GPR30) and G-1, but not DES, elicited cytosolic Ca2+ increases not only in MCF-7 cells, but also in ER-negative SKBr3 cells. Additionally, in MCF-7 cells, GPR30 depletion blocked E2- and G-1-induced Ca2+ mobilization, but ERα depletion did not. Interestingly, GPR30-coupled Ca2+ responses were sustained and inositol triphosphate receptor-mediated in ER-positive MCF-7 cells, but transitory and ryanodine receptor-mediated in ER-negative SKBr3 cells. Proliferation studies involving GPR30 depletion indicated that GPR30's role was to promote SKBr3 cell growth, but reduce MCF-7 cell growth. Supporting this, G-1 profoundly inhibited MCF-7 cell growth, potentially via p53 and p21 induction. Further, flow cytometry showed that G-1 blocked MCF-7 cell cycle progression at the G(1)-phase. Thus, GPR30 antagonizes growth of ERα-positive breast cancer, and may represent a new target to combat this disease.
Keywords: GPR30, estrogen receptor α, G-1, calcium mobilization, MCF-7 cells, SKBr3 cells
Introduction
The G protein-coupled receptor GPR30 (also termed GPER for G protein-coupled estrogen receptor 1) is a seven transmembrane-domain protein identified as a novel 17β-estradiol (E2)-binding protein structurally distinct from the classical estrogen receptors α and β (ERα and ERβ). GPR30 can mediate rapid E2-induced non-genomic signaling events including stimulation of adenylyl cyclase, and via transactivation of epidermal growth factor receptors, induces mobilization of intracellular calcium (Ca2+) stores and activation of mitogen-activated protein kinase (MAPK) and phosphatidyl inositol 3-kinase (PI3K) signaling pathways (1, 2). GPR30 also exhibits prognostic utility in endometrial (3) ovarian (4), and breast cancer (5, 6), and can modulate growth of hormonally responsive cancer cells (7-11). Therefore, GPR30 likely plays important roles in modulating estrogen responsiveness, and in the development and/or progression of hormonally responsive cancers. Moreover, GPR30 represents a promising new target for drug discovery in hormonally responsive disease.
In addition to E2, the selective estrogen receptor modulator (SERM) tamoxifen (TAM), one of its active metabolites 4-hydroxytamoxifen (4OHT), and the complete antiestrogen fulvestrant, all activate GPR30 (12-14). GPR30 does not significantly bind the non-steroidal full ER agonist diethylstilbestrol (DES) (12). Drug discovery efforts have yielded two GPR30-selective high affinity ligands, an agonist termed G-1 (15), and recently, an antagonist termed G-15 (16). G-1 and G-15 do not bind ERs at concentrations up to 10-6 M (15, 16). Moreover, G-1 specificity for GPR30 was illustrated by showing it does not significantly bind 25 other G-protein coupled receptors (17).
GPR30 has been shown to mediate E2's proliferative effects in thyroid (7), endometrial (8, 18), ovarian (9) and ER-negative SKBr3 breast cancer cell lines (9, 10), since GPR30 depletion, using antisense oligonucleotides or RNA interference (RNAi) methodologies, abrogated E2-stimulated growth in these cells. However, GPR30-mediated growth effects differ in ER-positive MCF-7 breast cancer cells. Ahola et al. (11) reported that transient overexpression of GPR30 in MCF-7 cells inhibited bromodeoxyuridine incorporation, an indicator of proliferation.
We investigated GPR30 largely in ER-positive MCF-7 with some comparisons to ER-negative SKBr3 breast cancer cells. First, a statistical association was sought between GPR30 and ERα-positive status in publicly available breast carcinoma microarray data sets. Next, the contribution of ERα and GPR30 in several E2-responsive activities, including regulation of GPR30 expression, intracellular Ca2+ mobilization, cellular growth, and cell cycle progression, was studied using receptor-specific ligands and small interfering RNA (siRNA) methodology.
Materials and Methods
Breast Cancer Microarray Data Mining
GPR30 mRNA levels and ER status were extracted from gene expression microarrays comprising 1,250 breast carcinomas across five distinct cohorts. The first cohort or the NKI cohort (n = 295; Netherlands Cancer Institute, Amsterdam) was derived from van de Vijver et al. [(19); www.rii.com/publications/2002/nejm.html]. NKI data were obtained using 2-color 60-polymer oligonucleotide arrays. cRNA from one tumor was competitively hybridized against a pooled reference cRNA from all tumors. Expression values correspond to normalized log2 ratio intensity units. ER-positive status was supplied with the microarray data (19). Pearson's correlation coefficients were computed between GPR30 and all other genes using the R software package (www.R-project.org). Cohorts 2 through 5 were obtained from GEO (Gene Expression Omnibus) (20). These cohorts are termed the Uppsala cohort (GSE3494/GSE4922/GSE6532; samples collected in Uppsala County, Sweden), the Stockholm cohort (GSE1456; samples collected at the Karolinska Hospital in Stockholm, Sweden), the EMC cohort (GSE2034/GSE5327; samples collected at the Erasmus Medical Center, Rotterdam, Netherlands), and the TRANSBIG cohort (GSE7390; samples collected by the translational research network managed by the Breast International Group). The Uppsala and EMC cohorts contained samples processed at the same institution that span multiple GEO accession numbers. These 4 cohorts utilized Affymetrix microarray technology (Santa Clara, CA). Where available, raw data (in the form of CEL files) were downloaded, otherwise MAS5.0 normalized data were downloaded (CEL files were available for all studies except GSE2034 and GSE5327). All data pre-processing and MAS5.0 normalization were performed using R software, and the justMAS function in the simpleAffy library from Bioconductor (no background correction, target intensity of 600) (21). After normalization, gene expression data were extracted for the GPR30 probe 210640_s_at. ER status was provided via Supplementary Information in GEO.
Compounds and Cell Lines
E2, DES and 2-aminoethyldiphenylborinate (2APB) were from (Sigma-Aldrich, St. Louis, MO). G-1 and fulvestrant (FUL; ICI 182,780, Faslodex) were from Tocris (Ellisville, MO). Xestospongin C (XeC) and ryanodine (Ry) were from Calbiochem, EMD Biosciences (La Jolla, CA). All agents were added to culture medium at 1:10,000 to 1:1,000 (v/v). Fura-2 AM and all cell culture reagents were from Invitrogen (Carlsbad, CA). MCF-7:WS8 human mammary carcinoma cells were used in all experiments indicating MCF-7 cells; they were clonally selected for sensitivity to E2-stimulated growth (22). SKBr3 cells were purchased from ATCC (Manassas, VA). Both cell lines were maintained in estrogenized media [RPMI-1640 media plus 10% fetal bovine serum (FBS)]. MCF-7 cells were switched to estrogen-free media (phenol red-free RPMI 1640 media plus 10% charcoal-stripped FBS) for 2 days before all experiments, except where noted.
Real-time quantitative polymerase chain reaction (qPCR) assays
qPCR was conducted as previously described (23). Target mRNA levels were normalized to PUM1 [pumilio homolog 1 (Drosophila)] mRNA levels (24). See Supplemental Materials for primer sequences. Data were analyzed by comparison to a serial-dilution series of MCF-7 cell cDNA. Values in each group were averaged from four biological replicates (unless otherwise indicated), and each biological replicate was averaged from four technical replicates.
siRNA Transfection
MCF-7 cells that had been maintained in estrogenized media were transfected with siRNAs for 6 h in serum-free Opti-MEM (Invitrogen) using Dharmafect 1 (Dharmacon RNAi Technologies, Lafayette, CO), followed by overnight recovery in estrogenized media. The transfection was carried out a second time, and then the cells were immediately switched to estrogen-free media and again allowed overnight recovery. siRNAs were transfected at 200 nM final concentration, except in Ca2+ experiments in which siRNAs were transfected at 100 nM and co-transfected with siGLO Green, a 6-FAM-labeled inactive double-stranded RNA. See Supplementary Materials for ERα and GPR30 siRNA sequences (Dharmacon RNAi Technologies).
Ca2+ Imaging
Cytoplasmic Ca2+concentrations were measured using Fura-2 AM and microscopy as previously described (25). Flat SKBr3 cells were imaged since they were the major morphologic cell type, whereas rounded SKBr3 cells were not imaged. All compounds were administered at 1 m.
Cellular Proliferation
Cell growth was assessed using Hoechst 33258 (Invitrogen) and compared to a standard curve of serial-diluted calf thymus DNA as previously described (26, 27).
Cell Cycle Analyses
Cell cycle distribution was determined by propidium iodide staining and using a fluorescence-activated cell sorter (Becton Dickinson, San Jose, CA) as previously described (27). Data were analyzed using FloJo 7.2.5 for Windows (Tree Star, Ashland, OR).
Immunoblot Analyses
Immunoblotting was carried out using 40 μg protein per lane as previously described (23). Membranes were probed using antibodies against ERα (AER 611; Lab Vision), p53 (DO-1; Calbiochem), p21 (F-5; Santa Cruz), cyclin D1 (DCS-6; Santa Cruz), cyclin B1 (D-11; Santa Cruz), and β-actin (AC-15; Sigma-Aldrich). Membranes were visualized using the Odyssey Infrared Imaging System (Li-Cor Biosciences; Lincoln, NE).
Statistical Analyses
Mann Whitney tests were performed using Prism 4.03 for Windows (GraphPad Software, San Diego, CA). All other statistical analyses were performed using Excel 2003 for Windows (Microsoft, Redmond, WA). Error is represented by SEMs in Ca2+ mobilization experiments and by SDs in all other experiments. Also in the Ca2+ experiments, P-values reflect 1-way ANOVA comparing the maximum Ca2+ concentration vs. the concentration at the treatment start time, unless otherwise stated.
Results
Increased GPR30 mRNA expression associates with ERα-positive status in 1,250 breast carcinomas
Evidence of a relationship between GPR30 and ERα expression was sought by mining publicly available and well-annotated gene expression microarray data sets across 5 independent cohorts comprising 1,250 breast carcinomas. In the NKI cohort (n = 254), data were collected using 2-color oligonucleotide microarrays (Fig. 1A). According to the non-parametric Mann-Whitney rank sum test, GPR30 mRNA levels were significantly higher in ERα-positive versus ERα-negative tumors (P < 0.0001). The upper range of GPR30 expression was 7.7-fold higher on a linear scale in the ERα-positive compared to ERα-negative carcinomas. Also, GPR30 and ERα mRNA levels correlated as continuous variables (Pearson's coefficient ρ = 0.30, adjusted P < 0.0001). The other 4 cohorts utilized 1-color Affymetrix oligonucleotide microarrays (Fig. 1B). In each of these 4 cohorts, GPR30 mRNA levels were significantly higher in the ERα-positive compared to the ER-negative breast cancers (Uppsala cohort, n = 244, P = 0.040; Stockholm cohort, n = 159, P = 0.0091; EMC cohort, n = 344, P = 0.0050; TRANSBIG cohort, n = 198, P = 0.0024).
Figure 1. GPR30 mRNA expression shows an association with ERα-positive status in human breast carcinomas.

(A) GPR30 mRNA levels in the NKI cohort derived from 2-color arrays. Expression values are normalized log2 ratio intensity units corresponding to a single tumor cRNA hybridized against a pooled reference cRNA from all tumors. B) GPR30 mRNA levels in the Uppsala, Stockholm, EMC, and TRANSBIG cohorts all derived from 1-color arrays. Expression values are MAS5.0 normalized intensity units. A-B, Sample sizes of ERα-positive (ER+) and ER-negative (ER-) cancers are shown, and bars indicate the 75th, 50th (median), and 25th percentiles. Significance was assessed using the non-parametric Mann-Whitney rank test.
E2 down-regulates GPR30 mRNA expression via ER and not GPR30
GPR30 regulation in response to E2 was investigated. MCF-7 cells were treated with E2 or without (control, vehicle only) over a 96 h time course, followed by determination of ERα and GPR30 mRNA levels by qPCR. As expected, E2 steadily down-regulated ERα mRNA levels by 59% over 96 h (Fig. 2A). E2 also down-regulated GPR30 but with faster kinetics than with ERα (Fig. 2B); GPR30 mRNA levels were decreased by 37 % at 2 h (P = 0.0013), and by 79 % at 24 h (P < 0.0001). Afterwards, GPR30 mRNA levels rebounded. Additionally, GPR30 mRNA expression decreased in a concentration-dependent manner from 10-12 M E2 to 10-10 M E2 (Fig. 2C). The GPR30-specific agonist G-1 did not alter GPR30 mRNA expression, but the ER-specific agonist DES did repress GPR30 expression relative to control treatment by 54 % P = 0.0009), which was very similar to E2's effect (Fig. 2D). FUL completely blocked E2 and DES effects. Therefore E2 likely acted via ER and not GPR30 to down-regulate GPR30 mRNA expression.
Figure 2. E2 represses ERα and GPR30 mRNA levels via ER and not GPR30 in MCF-7 cells.

E2 regulation of (A) ERα and (B) GPR30 mRNA levels across a time course. MCF-7 cells were treated with 10-9 M E2 or with the vehicle ethanol alone for 2, 6, 12, 24, 48, 72, and 96 h. (C) GPR30 mRNA levels in response to 24 h treatment with a serial dilution series of E2. (D) ERα and GPR30 mRNA levels in response to 48 h treatment with ER and GPR30 ligands as determined by qPCR. Each data point represents the average of 6 (A-B), or 4 (C-D) biological replicates.
GPR30 and not ERα mediates E2-induced Ca2+ mobilization responses
To begin to delineate whether endogenous ERα and/or GPR30 mediates E2-induced Ca2+ responses in breast cancer cells, changes in intracellular Ca2+ concentrations [Ca2+]i were measured in ER-positive MCF-7 and ER-negative SKBr3 breast cancer cells at the single cell level using Fura-2AM (Fig. 3). In ER-positive MCF-7 cells (Fig. 3A), E2 induced [Ca2+]i by 112 ± 1.6 nM (n = 47 cells, P = 0.0063), G-1 by 511 ± 3.4 nM (n = 58 cells, P = 0.0007), and 4OHT by 234 ± 3.4 nM (n = 31 cells, P = 0.0017), while DES did not significantly increase the [Ca2+]i (change = 41 ± 0.8 nM, n = 23 cells, P = 0.66). In ER-negative SKBr3 cells (Fig. 3B), the rank order of ligand-induced cytosolic Ca2+ increases was the same as in MCF-7 cells, but the magnitude of the increases was much greater and the responses were transitory instead of sustained. In ER-negative SKBR3 cells, E2 induced oscillating increases in [Ca2+]i with an average maximum of 294 ± 1.6 nM (n = 36 cells, P = 0.0037). G-1 and 4OHT induced transitory increases in [Ca2+]i of 1,517 nM ± 10.3 nM (n = 79 cells, P = 0.0001) and of 558 nM ± 2.7 nM (n = 37 cells, P = 0.0013), respectively, while DES did not ([Ca2+]i change = 51 ± 1.3 nM, n = 21 cells, P = 0.26). Therefore, since G-1 and two ER ligands that also bind GPR30, E2 and 4OHT, but not ER-selective DES, elicited Ca2+ responses in both ER-positive MCF-7 cells and ER-negative SKBr3 cells, they likely did so via GPR30.
Figure 3. ER ligands that also activate GPR30 induce Ca2+ mobilization responses in both ER-positive MCF-7 and ER-negative SKBr3 cells.

Ligand-induced Ca2+ responses (A-B), and blockade of G-1-induced responses using Ca2+ channel inhibitors (C-D) in MCF-7 and SKBr3 cells. Cells were loaded with Fura-2AM and intracellular Ca2+ concentrations [Ca2+]i were determined in individual cells vs. time using fluorescence microscopy. Cells were perfused with all ligands at 10-6 M starting at 1 m. 2APB was used at 10-4 M, xestospongin C (XeC) at 10-5 M, and ryanodine (Ry) at 10-5 M. SKBr3 cells with flat, not rounded, morphology were imaged. G-1-induced Ca2+ traces in (A, B) were redrawn in (C, D), respectively.
Two of the major Ca2+ channels, inositol triphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) (28), were tested for whether they mediated G-1-induced Ca2+ mobilization. The pharmacological probes 2APB and XeC, both of which inhibit IP3Rs, and Ry, which at high concentrations blocks RyRs, were employed. Cells were pretreated for 30 m before inducing Ca2+ responses with G-1. In MCF-7 cells, both 2APB and XeC blocked G-1-induced [Ca2+]i increases by 78 % (both 2APB + G-1 vs. G-1 alone and XeC + G-1 vs. G-1 alone, P = 0.0018), but Ry did not. In contrast in SKBR3 cells, Ry blocked G-1-induced [Ca2+]i increases by 80 % (Ry + G-1 vs. G-1 alone, P = 0.0006), whereas XeC did not. Also, 2APB allowed G-1 to almost fully induce [Ca2+]i increases, although the response was significantly lower by 17 % versus G-1 alone (P = 0.0094), but this was likely due to blockade of store operated Ca2+ entry (SOCE), another activity of 2APB. Therefore, GPR30 was coupled to IP3Rs in ER-positive MCF-7 cells, but to RyRs in ER-negative SKBr3 cells.
To confirm that GPR30 and not ERα mediates Ca2+ mobilization in response to E2 in MCF-7 cells, cells were transfected with siRNAs targeting these receptors (characterization of siRNAs in Supplementary Materials), and a non-targeting siRNA pool as a control. First, the GPR30 siRNA was validated by showing it led to an almost complete blockade of G-1 induced Ca2+ responses ([Ca2+]i increase = 58 ± 1.3 nM, n = 19 cells, P = 0.62) (Fig. 4A). Next, E2-induced Ca2+ mobilization responses were investigated (Fig. 4B). In non-targeting siRNA-transfected cells, E2 induced an [Ca2+]i increase of 159 ± 1.6 nM (n = 14 cells, P = 0.0075). However, in ERα siRNA-transfected cells, E2 caused almost a 2-fold further rise in [Ca2+]i (314 ± 3.2 nM, n = 17 cells; P = 0.00058). In GPR30 siRNA-transfected cells, the E2-induced Ca2+ response was blocked ([Ca2+]i increase = 52 ± 0.8 nM, n = 16 cells, P = 0.35). ERα and GPR30 expression were depleted in the appropriate siRNA-transfected cells (Fig. 4C). However, GPR30 mRNA levels were increased by 73% in ERα-depleted cells, a finding consistent with the prior observation that E2 and DES repressed GPR30 expression (Fig. 2B-D). Hence, the 2-fold potentiation of E2-induced [Ca2+]i in ERα-depleted cells likely reflected, at least in part, the increased GPR30 expression.
Figure 4. GPR30 and not ERα mediates E2-induced Ca2+ mobilization in MCF-7 cells.
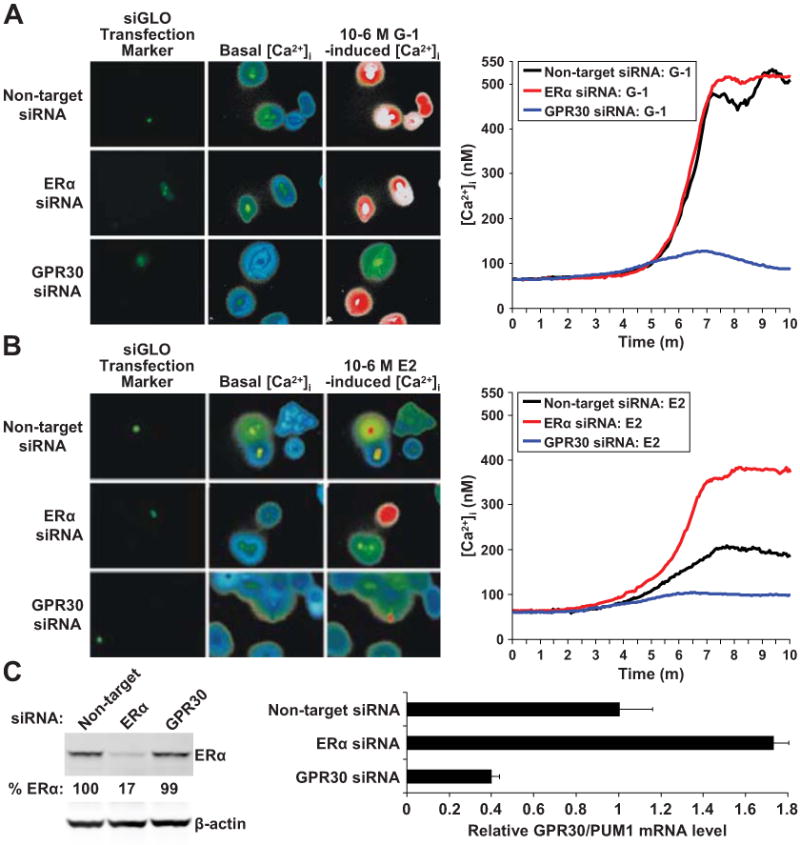
(A) G-1-induced and (B) E2-induced Ca2+ responses. Cells were transfected with non-targeting pool, ERα, and GPR30 siRNAs. Transfected cells were labeled using siGLO Green and appear green. Ca2+ imaging was performed 48 h following the transfection as in Fig. 3. Low levels of basal [Ca2+]i are visualized as blue and then green, while higher levels of [Ca2+]i are seen as red and then white. (C) ERα protein levels were measured by immunobloting and GPR30 mRNA levels by qPCR in siRNA-transfected cells 48 h following transfection.
GPR30 functions to promote growth of ER-negative SKBr3 cells, but to inhibit growth of ER-positive MCF-7 cells
GPR30's role in cellular proliferation was examined by transfecting cells with non-targeting and GPR30 siRNAs, and then measuring cellular DNA mass after 5 days of growth. First, SKBr3 cells were evaluated (Fig. 5A). The number of cells seeded in each group was similar as indicated by a lack of difference in DNA masses at day 0. After 5 days of growth, DNA mass was 45 % lower in GPR30 compared to non-targeting siRNA-transfected cells (P < 0.0001). Thus, GPR30's function was to promote growth of SKBr3 cells, in accordance with other reports (9, 10). Next, MCF-7 cells were similarly evaluated (Fig. 5B). Again equivalent numbers of non-targeting and GPR30 siRNA-transfected cells were seeded as indicated by DNA masses at day 0. After 5 days, GPR30 depletion did not affect basal growth (control treatment). However, GPR30 depletion did potentiate E2-stimulated growth by 2.1-fold (non-targeting vs. GPR30 siRNA-transfected cells, P < 0.0001). Analysis of progesterone receptor (PgR) and TFF1 (pS2) mRNA levels by qPCR indicated no significant differences in their induction by E2 between non-targeting and GPR30 siRNA-transfected cells (data not shown). Therefore, in contrast to SKBr3 cells, GPR30's function in MCF-7 cells was to inhibit growth.
Figure 5. GPR30 promotes growth of ER-negative SKBr3 but inhibits growth of ER-positive MCF-7 cells.

Proliferation of (A) SKBr3 and (B) MCF-7 cells transfected with non-targeting pool and GPR30 siRNAs. Cells were transfected and seeded at 15,000 cells per well in 24-well dishes. Media was replenished the day after seeding on day 0, and every other day thereafter. Cells were collected on days 0 and 5. SKBr3 cells were cultivated in their passage media, and MCF-7 cells in estrogen-free media supplemented with 10-9 M E2 (E2) or without E2 (Control, C). Proliferation was assessed as cellular DNA mass (μg/well) using 24 replicate wells. GPR30 mRNA levels were determined by qPCR 48 h following the transfection in both cell lines, and in MCF-7 cells, after 24 h of 10-9 M E2 or control treatment. (C-D) Proliferation of MCF-7 cells over 6 days treated with (C) a serial dilution series of E2, or with (D) 10-9 M DES, in the absence and presence of 10-6 M G-1. Twelve replicate wells were used per group.
GPR30's role in MCF-7 cell proliferation was further evaluated by examining effects of G-1 on E2 (Fig. 5C) and DES-stimulated (Fig. 5D) growth over 6 days. G-1 blocked E2's concentration-dependent growth stimulatory response (all E2 treatment groups vs. paired E2 + G-1 treatment groups, P = 0.0001, 1-way ANOVA); in particular, G-1 inhibited 10-10 M E2-stimulated growth by 77 % relative to E2 alone (P < 0.0001, T test). G-1 also blocked DES-stimulated growth by 72 % (DES vs. DES + G-1, P < 0.0001). Additionally, in both the E2 and DES experiments, G-1 inhibited basal (control treatment) growth by 32% (P < 0.0001) and 47 % (P < 0.0001), respectively. Therefore, G-1-activated GPR30 blocked growth of ER-positive breast cancer cells, but did so independently of ligand-activated ER.
G-1-activated GPR30 blocks cell cycle progression at G(1)-phase
The effect of G-1 on cell cycle progression was investigated. MCF-7 cells were synchronized by estrogen withdrawal, and then treated with E2 and G-1 for 24 h, followed by propidium iodide staining and flow cytometric analysis (Fig. 6A). Treatment with G-1 alone significantly decreased the proportion of S-phase cells from 19.8 % (control) to 14.7 % (G-1; P < 0.0001). Importantly, the addition of G-1 to E2 led to retention of an additional 11.6 % of the cells in G(1)-phase (42.7 % in E2 vs. 54.4 % in E2 + G-1-treated cells, P < 0.0001), and prevented 13.2 % of cells from entering S-phase (37.7 % in E2 vs. 24.5 % in E2 + G-1-treated cells, P < 0.0001). Therefore, G-1 blocked E2-stimulated cells from cell cycle progression at the G(1)-phase.
Figure 6. G-1 inhibits cell cycle progression in E2-stimulated MCF-7 cells by producing a block at G(1)-phase.
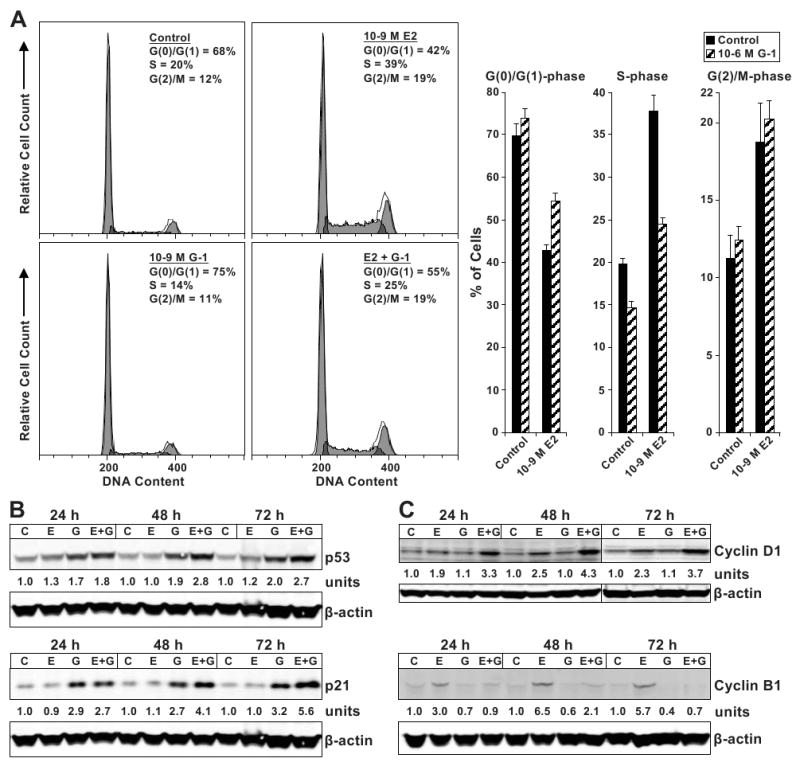
(A) Cell cycle distribution as determined by propidium iodide staining of DNA content and flow cytometry. Cells were synchronized by 3 days cultivation in estrogen-free media, and then treated as indicated for 24 h. Thirty-thousand cells per sample and 3 replicates per group were collected. Representative histograms are shown. Immunoblot analyses of (B) p53 and p21, and of (C) cyclins D1 and B1 protein levels. MCF-7 cells were control (C), 10-9 M E2 (E) and 10-6 M G-1 (G) -treated as indicated. Quantitated protein levels normalized to β-actin are indicated.
The G-1-induced cell cycle block was further investigated by measuring protein expression of the tumor suppressor p53, the cyclin dependent kinase inhibitor (CDK-I) p21 (Fig. 6B), the G(1)-phase-specific cyclin D1, and the G(2)/M-phase-specific cyclin B1 (Fig. 6C). MCF-7 cells were treated with E2 and G-1, and then collected at 24 h, 48 h, and 72 h for immunoblot analysis. Both p53 and p21 proteins were up-regulated in G-1 and E2 + G-1-treated cells across all time points compared to control-treated cells (Fig. 6B). As expected, E2 up-regulated both cyclins D1 and B1 across the time course compared to control treatment, whereas G-1 alone did not (Fig. 6C). However, the addition of G-1 to E2 potentiated the up-regulation of cyclin D1 while nearly completely preventing cyclin B1 accumulation compared to E2 alone across the time points. Since cyclin D1 is induced during G(1)-phase and degraded in S-phase (29, 30), whereas cyclin B1 accumulates during G(2)-phase and degrades upon M-phase entry (31), these data are consistent with G-1 blocking cell cycle progression in G(1)-phase before cyclin D1 degradation occurred and before cylin B1 accumulated.
Discussion
Filardo et al. (5) and Kuo et al. (6) have previously shown in breast carcinomas a positive association between GPR30 and ERα expression by IHC and qPCR, respectively. We confirmed and extended this finding by examining gene expression microarray data sets of 5 independent patient cohorts comprising 1,250 breast cancers. In all cohorts, high GPR30 mRNA levels showed an association with ERα-positivity (Fig. 1). It is unknown why high GPR30 levels would be selected for in ERα-positive breast carcinomas given GPR30 attenuates growth of ER-positive breast cancer, but some GRP30-dependent functions may be necessary for tumorigenesis and cell survival, such as activation of adenylyl cyclase, PI3K, and MAPK (1, 2). Additional roles of GPR30 may be needed for disease progression, such as in cell migration (15, 32), which may then promote metastasis (5).
The interplay between GPR30 and ERα was further investigated using MCF-7 breast cancer cells. E2 repressed GPR30 expression in a time- and concentration-dependent manner (Fig. 2B-C). Also, DES but not G-1 down-regulated GPR30, therefore ER mediated this effect (Fig. 2D). The inverse functional relationship between ERα and GPR30 was also demonstrated by depleting ERα, which led to de-repression of GPR30 mRNA expression and consequently potentiated E2-induced Ca2+ mobilization responses (Fig.4). E2 is known to down-regulate ERα expression in MCF-7 cells as a negative feedback-regulatory loop to prevent over-responsiveness (26). Likewise, GPR30 may also be negatively-regulated by E2 via ER to prevent excessive GPR30-dependent activity, such as aberrantly high [Ca2+]i. Interestingly, the maximum increases in [Ca2+]i. were much larger in SKBr3 cells than in MCF-7 cells (Fig. 3B vs. 3A). It is possible that this was due to the lack of ERs in SKBr3 cells which translated into a lack of negative-feedback regulation.
GPR30 depletion decreased growth of SKBr3 cells (Fig. 5A), but potentiated E2-stimulated growth in MCF-7 cells (Fig. 5B), indicating that GPR30 functions to promote SKBr3 but inhibit MCF-7 cellular proliferation. Also in MCF-7 cells, G-1 profoundly inhibited E2-stimulated (Fig. 5C) and DES-stimulated growth (Fig. 5D), as well as decreased the percentage of cells entering S-phase (Fig. 6A). However, these G-1 effects occurred in both the presence and absence of E2. These findings in MCF-7 cells complement those of Ahola et al. (11) who reported that transient GPR30 overexpression decreased the percentage of proliferating MCF-7 cells independent of E2. Indeed, GPR30 likely does not directly regulate ER transcriptional activity since there were no significant differences in E2-induced mRNA expression of well established ER target genes PgR and TFF1 between GPR30 siRNA and non-targeting siRNA-transfected cells (data not shown).
Rather, we propose GPR30 antagonizes growth of MCF-7 cells by inducing sustained increases in cytosolic Ca2+ concentrations (Fig. 3A and 4), in contrast to transitory increases in SKBr3 cells where GPR30 promotes growth. Aberrant sustained increases in intracellular Ca2+ levels can lead to inhibition of proliferation, and induce apoptosis (33). For example, the plasma membrane Ca2+-ATPase (PMCA) pumps Ca2+ across the plasma membrane out of the cell to lower cytosolic Ca2+ levels after Ca2+ increases. Partial inhibition of PMCA in MCF-7 cells causes a moderate increase in intracellular Ca2+ levels which leads to inhibition of proliferation by altering cell cycle kinetics (34). Additionally, the mechanism of action of numerous antitumor agents involves increases in [Ca2+]i (35).
It is possible that differences in GPR30-coupled Ca2+ signaling which mediate sustained vs. transitory responses associate with ER status. In support of this hypothesis, GPR30 was coupled to differing Ca2+ channels: to IP3Rs in ER-positive MCF-7 cells but to RyRs in ER-negative SKBr3 cells. Alternatively, sustained vs. transitory Ca2+ responses could have been due to potential alterations in factors which participate in lowering cytosolic Ca2+, such as plasma membrane or sarco/endoplasmic reticulum Ca2+-ATPase pumps. We intend to explore these possibilities involving differing Ca2+ responses in future studies.
As shown by propidium iodide staining and flow cytometry, G-1-induced a cell cycle block at the G(1)-phase (Fig. 6A). Consisitent with a G(1)-phase arrest, G-1 increased accumulation of the tumor suppressor p53, the CDK-I p21, and the G(1)-phase-specific cyclin D1, but prevented E2-induced accumulation of the G(2)/M-phase-specific cyclin B1 (Fig. 6B-C). Ca2+ signaling has been shown to induce p53 via activation of CREB (cAMP response element binding protein) (28). In MCF-7 cells, p53 induction by E2 has been shown to be Ca2+ and calmodulin kinase IV dependent (36). Also, aberrant Ca2+ mobilization in response to anticancer/cytotoxic agents correlates with p53 induction (35). Thus, G-1 could lead to p53 induction via Ca2+ mobilization in MCF-7 cells. Then, p53 could induce p21 via a p53 response element to mediate arrest in G(1)-phase of the cell cycle (28).
As a SERM, tamoxifen acts as an antiestrogen in ER-positive breast cancer, but as an estrogen in the endometrium and bone (37). Similarly, G-1 inhibits growth of ER-positive MCF-7 breast cancer cells, but promotes growth of the endometrium (16) and plays an important role in promoting bone growth in vivo (38, 39). Thus, G-1's tissue specific proliferative effects may parallel those of tamoxifen. It is interesting to speculate that 4OHT-induced Ca2+ mobilization (Fig. 3A-B) may be involved in some of tamoxifen's tissue-specific effects.
Taken together, GPR30 inhibits growth of ERα-positive breast cancer. Our studies also indicate that pharmacologic activation of GPR30 shows promise in combating ER-positive breast cancer. G-1 would also probably be well-tolerated since it, like E2, exerts beneficial effects against an animal model of multiple sclerosis, but without E2-associated side effects (17, 40). Thus, G-1 may represent the first in a new class of therapeutically relevant agents for use alone or in conjunction with conventional antihormonal therapeutics in breast cancer.
Supplementary Material
Acknowledgments
Grant support: Department of Defense BC050277 and the Weg and Genuardis funds (V.C. Jordan). NIH grants P30CA006927 (Fox Chase Cancer Center), HL090804 and HL090804-01A2S109 (E. Brailoiu), CA127731 (E.R. Prossnitz, T.I. Oprea, J.B. Arterburn), CA116662 (E.R. Prossnitz), U54MH084690 and CA118100 (University of New Mexico Cancer Center), and NS18710 and HL51314 (N.J. Dun).
References
- 1.Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol. 2008;70:165–90. doi: 10.1146/annurev.physiol.70.113006.100518. [DOI] [PubMed] [Google Scholar]
- 2.Prossnitz ER, Barton M. Signaling, physiological functions and clinical relevance of the G protein-coupled estrogen receptor GPER. Prostaglandins Other Lipid Mediat. 2009;89:89–97. doi: 10.1016/j.prostaglandins.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith HO, Leslie KK, Singh M, et al. GPR30: a novel indicator of poor survival for endometrial carcinoma. Am J Obstet Gynecol. 2007;196:386 e1–9. doi: 10.1016/j.ajog.2007.01.004. discussion e9-11. [DOI] [PubMed] [Google Scholar]
- 4.Smith HO, Arias-Pulido H, Kuo DY, et al. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol. 2009;114:465–71. doi: 10.1016/j.ygyno.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filardo EJ, Graeber CT, Quinn JA, et al. Distribution of GPR30, a seven membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin Cancer Res. 2006;12:6359–66. doi: 10.1158/1078-0432.CCR-06-0860. [DOI] [PubMed] [Google Scholar]
- 6.Kuo WH, Chang LY, Liu DL, et al. The interactions between GPR30 and the major biomarkers in infiltrating ductal carcinoma of the breast in an Asian population. Taiwan J Obstet Gynecol. 2007;46:135–45. doi: 10.1016/S1028-4559(07)60007-2. [DOI] [PubMed] [Google Scholar]
- 7.Vivacqua A, Bonofiglio D, Albanito L, et al. 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the G protein-coupled receptor GPR30. Mol Pharmacol. 2006;70:1414–23. doi: 10.1124/mol.106.026344. [DOI] [PubMed] [Google Scholar]
- 8.Vivacqua A, Bonofiglio D, Recchia AG, et al. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17{beta}-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol. 2006;20:631–46. doi: 10.1210/me.2005-0280. [DOI] [PubMed] [Google Scholar]
- 9.Albanito L, Madeo A, Lappano R, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17{beta}-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67:1859–66. doi: 10.1158/0008-5472.CAN-06-2909. [DOI] [PubMed] [Google Scholar]
- 10.Albanito L, Sisci D, Aquila S, et al. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology. 2008;149:3799–808. doi: 10.1210/en.2008-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahola TM, Manninen T, Alkio N, Ylikomi T. G protein-coupled receptor 30 is critical for a progestin-induced growth inhibition in MCF-7 breast cancer cells. Endocrinology. 2002;143:3376–84. doi: 10.1210/en.2001-211445. [DOI] [PubMed] [Google Scholar]
- 12.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–32. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 13.Filardo EJ, Quinn JA, Bland KI, Frackelton AR., Jr Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via transactivation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–60. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 14.Filardo EJ, Quinn JA, Frackelton AR, Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16:70–84. doi: 10.1210/mend.16.1.0758. [DOI] [PubMed] [Google Scholar]
- 15.Bologa CG, Revankar CM, Young SM, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2:207–12. doi: 10.1038/nchembio775. [DOI] [PubMed] [Google Scholar]
- 16.Dennis MK, Burai R, Ramesh C, et al. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;5:421–7. doi: 10.1038/nchembio.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blasko E, Haskell CA, Leung S, et al. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol. 2009;214:67–77. doi: 10.1016/j.jneuroim.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin BC, Suzawa M, Blind RD, et al. Stimulating the GPR30 estrogen receptor with a novel tamoxifen analogue activates SF-1 and promotes endometrial cell proliferation. Cancer Res. 2009;69:5415–23. doi: 10.1158/0008-5472.CAN-08-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 20.Barrett T, Suzek TO, Troup DB, et al. NCBI GEO: mining millions of expression profiles--database and tools. Nucleic Acids Res. 2005;33:D562–6. doi: 10.1093/nar/gki022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang SY, Wolf DM, Yingling JM, Chang C, Jordan VC. An estrogen receptor positive MCF-7 clone that is resistant to antiestrogens and estradiol. Mol Cell Endocrinol. 1992;90:77–86. doi: 10.1016/0303-7207(92)90104-e. [DOI] [PubMed] [Google Scholar]
- 23.Ariazi EA, Kraus RJ, Farrell ML, Jordan VC, Mertz JE. Estrogen-related receptor alpha1 transcriptional activities are regulated in part via the ErbB2/HER2 signaling pathway. Mol Cancer Res. 2007;5:71–85. doi: 10.1158/1541-7786.MCR-06-0227. [DOI] [PubMed] [Google Scholar]
- 24.Lyng MB, Laenkholm AV, Pallisgaard N, Ditzel HJ. Identification of genes for normalization of real-time RT-PCR data in breast carcinomas. BMC Cancer. 2008;8:20. doi: 10.1186/1471-2407-8-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brailoiu E, Churamani D, Cai X, et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J Cell Biol. 2009;186:201–9. doi: 10.1083/jcb.200904073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res. 1996;5623:21–30. [PubMed] [Google Scholar]
- 27.Liu H, Lee ES, Gajdos C, et al. Apoptotic action of 17{beta}-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. J Natl Cancer Inst. 2003;95:1586–97. doi: 10.1093/jnci/djg080. [DOI] [PubMed] [Google Scholar]
- 28.Lipskaia L, Lompre AM. Alteration in temporal kinetics of Ca2+ signaling and control of growth and proliferation. Biol Cell. 2004;96:55–68. doi: 10.1016/j.biolcel.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 29.Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer. 2003;10:179–86. doi: 10.1677/erc.0.0100179. [DOI] [PubMed] [Google Scholar]
- 30.Guo Y, Yang K, Harwalkar J, et al. Phosphorylation of cyclin D1 at Thr 286 during S phase leads to its proteasomal degradation and allows efficient DNA synthesis. Oncogene. 2005;24:2599–612. doi: 10.1038/sj.onc.1208326. [DOI] [PubMed] [Google Scholar]
- 31.Pines J, Hunter T. Isolation of a human cyclin cDNA: evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell. 1989;58:833–46. doi: 10.1016/0092-8674(89)90936-7. [DOI] [PubMed] [Google Scholar]
- 32.Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signaling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009;28:523–32. doi: 10.1038/emboj.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+transfer in the control of apoptosis. Oncogene. 2008;27:6407–18. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee WJ, Robinson JA, Holman NA, McCall MN, Roberts-Thomson SJ, Monteith GR. Antisense-mediated Inhibition of the plasma membrane calcium-ATPase suppresses proliferation of MCF-7 cells. J Biol Chem. 2005;280:27076–84. doi: 10.1074/jbc.M414142200. [DOI] [PubMed] [Google Scholar]
- 35.Olofsson MH, Havelka AM, Brnjic S, Shoshan MC, Linder S. Charting calcium-regulated apoptosis pathways using chemical biology: role of calmodulin kinase II. BMC Chem Biol. 2008;8:2. doi: 10.1186/1472-6769-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qin C, Nguyen T, Stewart J, Samudio I, Burghardt R, Safe S. Estrogen up-regulation of p53 gene expression in MCF-7 breast cancer cells is mediated by calmodulin kinase IV-dependent activation of a nuclear factor kappaB/CCAAT-binding transcription factor-1 complex. Mol Endocrinol. 2002;16:1793–809. doi: 10.1210/me.2002-0006. [DOI] [PubMed] [Google Scholar]
- 37.Ariazi EA, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. In: Ottow E, Weinmann H, editors. Nuclear Receptors as Drug Targets. Mörlenbach: Wiley-VCH; 2008. pp. 127–99. [Google Scholar]
- 38.Martensson UE, Salehi SA, Windahl S, et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. 2009;150:687–98. doi: 10.1210/en.2008-0623. [DOI] [PubMed] [Google Scholar]
- 39.Chagin AS, Savendahl L. GPR30 estrogen receptor expression in the growth plate declines as puberty progresses. J Clin Endocrinol Metab. 2007;92:4873–7. doi: 10.1210/jc.2007-0814. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Dehghani B, Li Y, et al. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J Immunol. 2009;182:3294–303. doi: 10.4049/jimmunol.0803205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.