

Summary
Based on the finding that tissue factor belongs to a group of genes upregulated in endothelial cells by VEGF, but not by EGF, we investigated signals selectively triggered by VEGF. Whereas the transcription factor early growth response (EGR)-1,which has previously been shown by us to be essentially involved in tissue factor gene regulation, was similarly induced by both factors, one major difference between VEGF and EGF signaling was the activation of the Ca++-mediated calcineurin/nuclear factor of activated T cells (NFAT) pathway by VEGF. Consistent with the importance of this pathway for tissue factor induction, treatment of endothelial cells with the Ca++ chelator BAPTA-AM, as well as the calcineurin inhibitor cyclosporin A, partially inhibited VEGF-induced tissue factor upregulation. Furthermore, tissue factor reporter gene assays revealed a synergistic cooperation of NFAT and EGR-1 in the induction of the TF promoter, and a physical interaction between the two factors was indicated by co-immunoprecipitation assays. Another gene upregulated by VEGF predominantly via NFAT, which is not induced by EGF, is the DSCR-1 gene. The calcineurin inhibitor DSCR-1 seems to be induced by VEGF in a negative feed-back loop to limit NFAT activation. When we tested adenoviral overexpression of DSCR-1, VEGF-mediated induction of tissue factor mRNA was reduced, and complete suppression could be achieved by a combination of viruses expressing DSCR-1 and NAB2, a corepressor of EGR-1. These findings support that both, NFAT and EGR-1, are required for tissue factor upregulation in response to VEGF.
Keywords: EGR-1, NFAT, VEGF, endothelial cells, tissue factor, DSCR-1
Introduction
Among the endothelial-specific receptors the vascular endothelial growth factor (VEGF) receptor family, in particular VEGFR-2 or Flk-1, is the major initiator of physiological and pathological angiogenesis (1). Additional endothelial cell-specific tyrosine kinase receptors of the Tie and ephrin receptor families control later stages of angiogenesis and vessel stabilization (2, 3). Aside from these receptors, which are almost exclusively expressed on endothelial cells, additional growth factor receptors such as EGF and TGF-β receptors are found in endothelial cells (3, 4). These are generally expressed on many different cell types and are presumably involved in the regulation of survival, growth and differentiation in concert with the cell type specific receptors.
We have previously found that the tissue factor (TF) gene is among the genes strongly upregulated by VEGF and have investigated its regulation by VEGF (5-7). TF is a distant member of the cytokine receptor family and functions as a high-affinity receptor for factor VII/VIIa initiating the blood coagulation cascade (8, 9). Aside from its important role in hemostasis (10-12), TF has been proposed to play a role in vessel formation. It is found to be expressed on tumor endothelium (13) and TF knockout mice die at gestation day 9.5 due to hemostatic problems indicating a possible failure in vasculogenesis (14). Additional data provide support for a signaling role of TF in the initiation of physiologic as well as pathologic angiogenesis through indirect factor VII-mediated activation of the PAR-2 receptor and direct TF cytoplasmic tail signaling (15-17).
The pathways leading to transcriptional upregulation of TF by inflammatory cytokines have been intensively investigated by us and others and are mainly dependent on activation of the nuclear factor kappa B (NF-κB) (10, 18, 19). Consistently, the NF-κB responsive element within the TF promoter is essential for inflammatory upregulation and overexpression of the natural inhibitor of NF-κB (IκB) and almost completely abrogates inflammatory TF upregulation (20). In contrast, TF upregulation during VEGF stimulation of endothelial cells does not appear to involve NF-κB. It is dependent in part on the transcription factor early growth response-1 (EGR-1), which is rapidly induced in response to VEGF treatment. The respective EGR-1 binding sites in the TF promoter have been previously shown by us to be essential for VEGF-mediated TF reporter gene transcription (6, 7).
Another strongly VEGF-upregulated gene which has turned out to be important for the control of gene expression has been recently identified by differential gene expression profiling (21, 22; B. Schweighofer and E. Hofer, unpublished data). This gene known as DSCR-1, calcipressin or MCIP1, was originally defined as one of the genes in the Down Syndrome Critical Region on human chromosome 21 (23, 24), and the protein encoded by this gene was found to bind to and inhibit the enzymatic activity of the serine/threonine phosphatase calcineurin (25). It was reported that VEGF can trigger DSCR-1 gene upregulation via multiple NEAT binding sites in its promoter (26).
As part of an effort to define characteristic signaling patterns induced by VEGF via VEGFR-2/Flk-l important for gene regulation and distinct from other tyrosine kinase receptors, we have compared the capacity of VEGF and EGF to induce signaling pathways and genes in endothelial cells. We show that in contrast to VEGF, EGF is not capable of inducing signals in endothelial cells sufficient for upregulation of TF. We define the Ca++-dependent activation of the transcription factor nuclear factor of activated T cells (NEAT) as a pathway important for TF induction, which is activated selectively by VEGF but not by EGF. We further provide evidence that NEAT functionally and physically cooperates with EGR-1 in TF gene regulation. In addition, we show that the DSCR-1 gene, which similar to TF is selectively upregulated by VEGF, acts as a negative regulatory factor together with the EGR-specific corepressor NAB2 (5) in limiting VEGF-mediated TF gene regulation.
Materials and methods
Cell culture and materials
Human umbilical vein endothelial cells (HUVEC) were cultured at 37°C and 5% CO2 in medium 199 (Invitrogen, Carlsbad, CA, USA) supplemented with 20% SCS (HyClone, Logan, UT, USA), 1 U/ml heparin, 50 μg/ml ECGS (Technoclone, Vienna, Austria), 2 mM glutamine, 100 U/ml penicillin and 1 mg/ml streptomycin (7). Cells were used for experiments up to passage number 5. Short-starved cells were obtained by starving with 1 % serum for 5 hours (h). Two hundred ninety-three HEK cells were grown in MEM alpha medium (Invitrogen) supplemented with 10% FCS (HyClone), 2 mM glutamine, 100 U/ml penicillin and 1 mg/ml streptomycin.
Recombinant human VEGF165 was obtained from PromoCell (Heidelberg, Germany) or Peprotech (London, England). Phorbol 12-myristate 13-acetate (PMA), citrated plasma and calcium ionophore A23187 were purchased from Sigma Chemicals (St. Louis, MO, USA). Cyclosporin A (CsA) was kindly provided by the Novartis Research Institute (Vienna, Austria). Polyclonal rabbit anti-EGR-1 and anti-GFP antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and New England Biolabs Inc. (Beverly, MA, USA), respectively. Peroxidase-conjugated donkey anti-rabbit immunoglobulin G (IgG) and sheep anti-mouse IgG were purchased from GE Healthcare Bio-Sciences (Little Chalfont, UK), Immobilon-P transfer membranes from Millipore (Bedford, MA, USA) and LumiGLO chemiluminescent reagent from New England BioLabs.
Recombinant adenoviral constructs and infection
For the construction of a recombinant adenovirus expressing N-terminal Flag-tagged DSCR1 splice form 1, a human full-length DSCR1 cDNA for splice form 1 (clone IRALp962A2315Q2, RZPD – Heidelberg, Germany) was sub-cloned into the vector pACCMVpLpASR+ (5). For this purpose a 637-bp fragment was amplified by PCR and inserted between the BamHI and HindIII sites of the polylinker. The forward primer providing a BamHI site and encoding an N-terminal Flag-tag (27) was 5′-CGGGATCCATGGACTACAAGGACGACGATGACCAAGGAGGAGGTGGACCTGCAGGAC-3′, the reverse primer including a HindIII site was 5′-CCCAAGCTTTCAGCTGAGGTGGATCGGCGT-3′. The NAB2-expressing adenovirus and the control adenovirus without insert have been described previously (5). Production of high-titer virus stocks was performed as reported in reference (5). For infection HUVEC grown in six-well plates for two days were incubated with the recombinant adenoviruses for 24-48 h at a multiplicity of infection (MOI) of 100.
Tissue factor activity assay
Cells were seeded in six-well plates at 70% confluency and grown overnight. Following treatment with growth factors or other reagents, cells were scraped from the plates and analyzed for TF activity as described (7). Briefly, after induction for 6 h with VEGF or EGF cells were washed twice and then scraped in 1 ml clotting buffer (12 mM sodium acetate, 7 mM diethylbarbitate and 130 mM sodium chloride; pH 7.4). 100 μl of resus pended cells were mixed with 100 μl of citrated plasma and clotting times were measured after recalcification with 100 μl 20 mM CaCl2 at 37°C. TF equivalents were determined by using a standard curve established with rabbit brain thromboplastin.
RNA isolation and real time RT-PCR
After incubation with the indicated stimuli, cells were treated with RNAlater (Ambion, Austin, TX, USA), washed once with DEPC-treated water and processed for RNA isolation. RNA was extracted from cultured endothelial cells with Trizol (Invitrogen). Two μg of total RNA were reverse transcribed into cDNA (SuperscriptTM II RT, Invitrogen) using oligo-dT primers and real-time PCR was used to monitor gene expression using a Light Cycler instrument (Roche Diagnostics GmbH, Mannheim, Germany) according to established procedures (28, 29). As internal standard the amount of β2-microglobulin mRNA was used for normalization. The oligonucleotide primers used were as follows:
TF-forward: 5′-CCGAACAGTTAACCGGAAGA-3′, TF-reverse: 5′-TCAGTGGGGAGTTCTCCTTC-3′; EGR-1-forward: 5′-AGCCCTACGAGCACCTGAC-3′, EGR-1-reverse: 5′-TGGGTTGGTCATGCTCACTA-3′; DSCRl_E4-forward: 5′-TTAGCTCCCTGATTGCCTGT-3′; DSCRl_E4-reverse: 5 ′-GGAGAAGGGGTTGCTGAAGT-3′; DSCR1-forward: 5 ′-GCCAAATTTGAGTCCCTCTTT-3′; DSCR1-reverse: 5 ′-GGTCGCATCTTCCACTTGTT-3′; β2-microglobulin-forward: 5′-GATGAGTATGCCTGCCGTGTG-3′; β2-microglobulin-reverse: 5′-CAATCCAAATGCGGCATCT-3′.
Western blot analysis
After the various treatments, cells were washed twice with ice-cold phosphate-buffered saline (PBS), lysed in 100 μl of Laemmli buffer, scraped and heated for 5 minutes (min) at 95°C. Total cell lysates were subjected to SDS-PAGE and the separated proteins transferred to Immobilon-P membranes according to described procedures (30). For staining, the membranes were blocked for 30 min with PBS containing 0.1% Tween-20 and 3% skim milk and incubated with the primary antibody diluted in blocking buffer for 1 hat room temperature (RT). Then the membranes were washed three times for 5 min with PBS containing 0.1% Tween-20 and incubated with peroxidase-conjugated secondary antibody for 1 h at RT After a washing step, the membranes were incubated for 1 min with ECL reagent (GE Healthcare Bio-Sciences) and exposed to X-ray films as required. For reprobing with an additional antibody, the membranes were washed twice in PBS, stripped for 30 min at 55°C with stripping buffer (62.5 mM Tris-HCL, pH 6.8, 2% SDS, 100 mM 2-mercaptoethanol) and washed three times for 5 min with PBS at RT. The membrane was stored wet wrapped in SaranWrap at 4°C after each immunodetection.
Co-immunoprecipitation
Two hundred ninety-three HEK cells were grown in six-well plates to about 70% confluency, and calcium phosphate transfection was performed with the expression constructs for EGR-1-myc, GFP-NFAT or a combination of both plasmids. The construct for the expression of human EGR-1 was obtained in the pcDNA 3.1/myc vector, which provides for the addition of a C-terminal myc-tag (Invitrogen, Carlsbad, CA, USA), by ligating a PCR product encompassing the coding region of the human EGR-1 gene including the single intron into the vector (31). The expression vector for a GFP-NFAT fusion protein (N-terminal pRSGFP-C1-NFATc) was kindly provided by L. Gerace (32). Twenty-four h post-transfection the cells were treated for 1 h with PMA (100 nM) or A23187 (2.5 mM). Then the cells were lysed for 20 min in 500 μl IP-lysis buffer (50 mM HEPES pH 7.3, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) containing protease inhibitors (PMSF, TPCL and TLCK) and the lysate centri fuged for 10 min at 15,000 rpm. For immunoprecipitation 2.5 μg rabbit polyclonal anti-myc antibody and 50 μl washed protein A Sepharose beads were added to 400 μl cleared cell lysate. Immunoadsorption to the protein A Sepharose beads was performed o/n at 4°C. The beads were then washed five times in NP40 IP-buffer (50 mM Tris pH7.5, 150 mM NaCl, 1 mM EDTA and 0.25% NP-40), the bound proteins solubilized in Laemmli buffer and analyzed by Western blotting.
Transient transfections
Reporter gene constructs containing a fragment of the TF promoter (−330 to +118 bp) cloned into the luciferase expression vector pUBT-luc were previously described (18). Transient transfections of HUVEC were performed using the Lipofectamine Plus™ reagent (Invitrogen). Twenty-four h prior to transfection, HUVEC were seeded in six-well tissue culture plates to reach 70% confluency the next morning. Cells were incubated with transfection mixture containing a total of 1.5 μg DNA of the various cotransfected plasmids (including a CMV-β-gal or a TK promoter driven renilla-luciferase construct as internal control), 6 μl Plus™ reagent and 3 μl Lipofectamine in a total volume of 1 ml medium 199 per well for 2 h. Then cells were washed and incubated in M199 medium and 20% serum for 20-30 h before the treatment of the cells with growth factors or other reagents. Cells were lysed and luciferase values determined from triplicate wells and normalized to β-galactosidase produced from cotransfected expression plasmids. Several identical experiments were performed with plasmids from different DNA preparations to exclude an influence of DNA quality. In some cases the dual-luciferase® reporter assay system was used. Cells were then lysed for 20 min in ice-cold passive lysis buffer and measurement of firefly- and renilla-luciferase activity was performed on a luminoscan. Reagents and components of the dual-luciferase reporter assay system were purchased from Promega (San Luis Obispo, CA, USA).
Immunofluorescence studies
Endothelial cells were seeded on LabTek 8-well tissue culture chamber slides (NUNC, Wiesbaden, Germany) coated with fibronectin. The cells were rinsed briefly in PBS and fixed in 3% paraformaldehyde or 3.7% formaldehyde/2% sucrose. Permeabilization was achieved by addition of PBS containing 0.25% Triton X-100. EGR-1 specific rabbit antibody were diluted in PBS/0.5% BSA and incubated for 2 h at RT The cells were washed three times in PBS/0.5% BSA. Secondary FITC-conjugated anti-rabbit antibodies (Accurate Scientific, Westbury NY, USA) were used for visualization. Cover slips were mounted in FA mounting fluid (BD Diagnostics, Franklin Lakes, NJ, USA) and results analyzed on a Nikon Diaphot TMD microscope. Images were taken by a CCD camera (Kappa GmbH, Gleichen, Germany).
Statistical analysis
To show significance of the data obtained p-values were calculated by Student’s t-test. Statistical significance was assumed if the two-tailed p-value was <0.05.
Results
VEGF and EGF differ in their capacity to upregulate TF in endothelial cells
In an attempt to define signaling pathways characteristic for VEGF/VEGFR-2 we compared VEGF and EGF in their ability to upregulate TF expression in endothelial cells. When HUVEC were activated with VEGF a strong increase in TF mRNA, and protein expression was observed, whereas treatment with EGF failed to induce significant TF expression (Fig. 1A, B). According to our previous data, EGR-1 is decisively involved in the VEGF-mediated upregulation of TF, since deletion of the EGR-1 sites from the TF promoter abrogates VEGF-mediated induction of TF reporter genes (6, 7) and an adenovirus overexpressing the specific corepressor of EGR-1, NAB-2, strongly inhibited the VEGF effect on TF mRNA (5).
Figure 1. Tissue factor and EGR-1 upregulation by VEGF as compared to EGF.

A) TF mRNA levels in HUVEC in response to VEGF or EGF. HUVEC were treated with VEGF (100 ng/ml) or EGF (10 ng/ml) for 1 and 2 hours, respectively, and the amount of TF mRNA was determined by real-time RT-PCR as described in Materials and methods. The mean values ± SD obtained from three independent experiments are shown. B) TF activity following treatment of HUVEC with VEGF or EGF. HUVEC were harvested in clotting buffer after 6 h stimulation with VEGF (100 ng/ml) or EGF (10 ng/ml). Aliquots of endothelial cell lysates were used in one stage clotting assays to determine tissue factor activity. The mean values ± SD obtained from three independent experiments are shown. C) EGR-1 mRNA levels in HUVEC in response to VEGF (100 ng/ml) or EGF (10 ng/ml) as determined by real-time RT-PCR. D) Induction of EGR-1 by VEGF or EGF. HUVEC were stimulated for the indicated time periods with VEGF (100 ng/ml) or EGF (10 ng/ml). The cells were lysed in SDS-sample buffer. Equal amounts of the whole-cell lysates were loaded on SDS-PAGE and blotted with an anti-EGR-l antibody. A representative experiment of five performed with similar results is shown.
However, when we next tested whether the inability of EGF to induce TF induction would be reflected in a diminished capacity of EGF to trigger EGR-1 production, we observed that EGF was at least as efficient as VEGF in the induction of EGR-1 mRNA and protein (Fig. 1C, D). It appeared therefore that another pathway selectively induced by VEGF would be important in addition to EGR-1 to activate the TF gene. One of the candidate pathways, which is critically involved in the regulation of TF by inflammatory mediators, the NF-κB pathway (18, 20), was excluded since according to our previous data VEGF did not cause significant activation of this pathway (6, 7) and overexpression of IκB, the natural inhibitor of NF-κB, had no strong inhibitory effects on TF upregulation in response to VEGF (data not shown). IκBα overexpression by adenoviruses had been previously shown to largely abrogate TF induction by TNF-α (20).
The role of Ca++ signaling and NFAT activation for VEGF-mediated TF upregulation
Based on previous data that NFAT can bind to and activate the TF promoter (33), we investigated the quantitative contribution of Ca++ signaling and NFAT activation to TF upregulation by VEGF. For this purpose we used the calcineurin inhibitor cyclosporin A (CsA). The obtained data show that VEGF-mediated upregulation of TF mRNA and protein expression was inhibited by CsA to an extent of about 50% and 40% supporting a significant contribution of this pathway to TF upregulation (Fig. 2A, B). To confirm that Ca++ signals can upregulate TF, we further tested the induction of TF by the Ca++ ionophore A23187 and its inhibition by CsA. TF mRNA and protein was strongly induced by the Ca++ ionophore and CsA inhibited this induction by about 80 and 50% (Fig. 2A, B). Usually ionophore induction reached only levels of about 30–50% of the VEGF-mediated induction (data not shown). This and the stronger inhibition of the A23187 effect by CsA when compared to the inhibition of the VEGF induction suggests that, in the case of VEGF, pathways in addition to calcineurin/NFAT have an important role in the induction. This is further supported by the finding that when the Ca++ chelator BAPTA-AM is used to inhibit Ca++ signals, the effects of the Ca++ ionophore are almost completely inhibited whereas the VEGF effects again are inhibited only partially by about 30% (Fig. 2C).
Figure 2. Inhibition of VEGF- and A23187 ionophore-mediated tissue factor upregulation by CsA and BAPTA-AM.

A) HUVEC were seeded in six-well plates and grown to confluency. Cells were preincubated with CsA (I μg/ml) for 30 min and then induced with VEGF (100 ng/ml) or A23187 (2.S μM) for 2 h. Tissue factor mRNA levels were determined by real time RT-PCR as described Materials and methods. Data are displayed as % of the VEGF- or A23187-induced mRNA levels. The mean values ± SD obtained from three independent experiments are shown. B) HUVEC were seeded in six-well plates and grown to confluency. Pretreatment with CsA (I μg /ml) was performed 30 min before the cells were stimulated with VEGF (100 ng/ml) or A23187 (2.5 μM) for 6 h. Aliquots of endothelial cell lysates were used in an one stage clotting assay to determine tissue factor activity. Data are displayed as % of the VEGF- or A23187-induced mRNA levels. The mean values ± SD obtained from three independent experiments are shown. C) HUVEC were preincubated with BAPTA-AM (10 μM) for 30 min and then induced with VEGF (100 ng/ml) or A23187 (2.5 μM) for 6 h. Cells were harvested in clotting buffer, and tissue factor activities were determined. A representative experiment of three performed with similar results is shown.
To test whether NFAT induction might be a substantial difference between VEGF and EGF signaling and would be significantly blocked by CsA in endothelial cells, we transfected HUVEC with an expression vector for an NEAT-GFP fusion protein (32). One day after transfection the cells were stimulated with VEGF, A23187 or EGF in the presence or absence of CsA. Then the cytoplasmic-nuclear translocation of NEAT was analyzed by immunofluorescence. The obtained data showed that upon stimulation with VEGF and ionophore, NFAT translocated rapidly to the nucleus within 30 min, and this translocation could be largely inhibited by preincubation of the cells with CsA (Fig. 3A). In contrast, no stimulation of translocation could be observed for EGF, which obviously is not competent to induce Ca++ signals and NFAT translocation. When parallel control samples were stained for EGR-1, a rapid synthesis and nuclear accumulation of EGR-1 after 45 min was revealed in VEGF-as well as in EGF-treated samples (Fig. 3B). These data support that the specific calcineurin-dependent activation and nuclear translocation of NFAT by VEGF might be a relevant factor for the selective activation of certain genes by VEGF, which can not be induced by some other growth factors such as EGF in endothelial cells.
Figure 3. VEGF and A23187 ionophore induce cytoplasmic-nuclear translocation of NFAT, whereas EGF fails to activate NFAT.
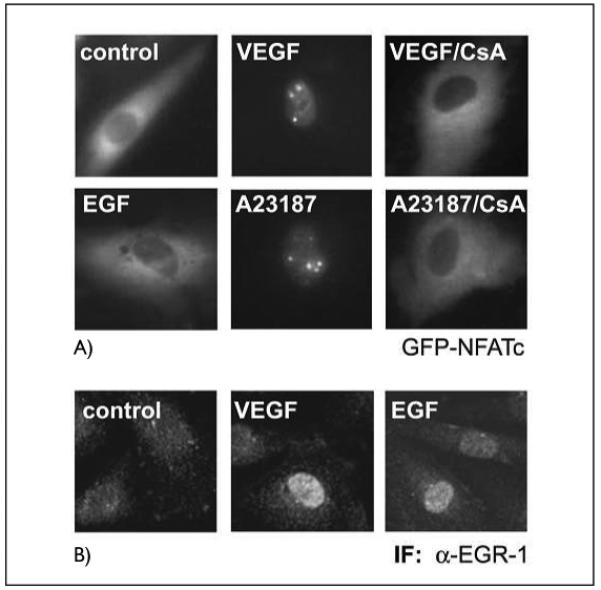
A) Cytoplasmic-nuclear transfer of a GFP-NFATc fusion protein. HUVE cells were transfected with an expression construct for a GFP-NFATc fusion protein. Twenty-four h post transfection the cells were induced with VEGF (100 ng/ml), EGF (10 ng/ml) or A23187 (2.5 μM) for 45 min. Where indicated, cells were preincubated with CsA (1 μg/ml) for 30 min prior to stimulation. Cytoplasmic-nuclear transfer was monitored in a Nikon fluorescence microscope equipped with a BM510 (B-2A) filter. B) Immunofluorescence of nuclear accumulation of EGR-1. HUVEC were treated with VEGF or EGF for 45 min, then the cells were fixed, permeabilized and stained using an anti-EGR-l antibody and a FITC-conjugated secondary antibody as described in Materials and methods.
NFAT cooperates with EGR-1 in TF gene induction
To show that the inhibition observed at the mRNA and protein level is due to reduced TF gene transcription, we further examined the effects of CsA on a TF reporter gene. HUVEC were transfected with the reporter gene containing the −330/+118 bp TF promoter element fused to a luciferase reporter construct (18). In this assay, TF promoter transactivation was upregulated five-fold when cells were treated with VEGF. Comparable to the reduction in TF mRNA and protein levels, the induction of the reporter gene by VEGF was inhibited by preincubation with CsA by about 50–60%. PMA/A23187-induced transcription was similarly affected suggesting that indeed a significant portion of the VEGF stimulation of the TF promoter can be mediated through the Ca++/calcineurin/NFAT pathway (Fig. 4A).
Figure 4. Functional and physical interaction of NFATc and EGR-1.
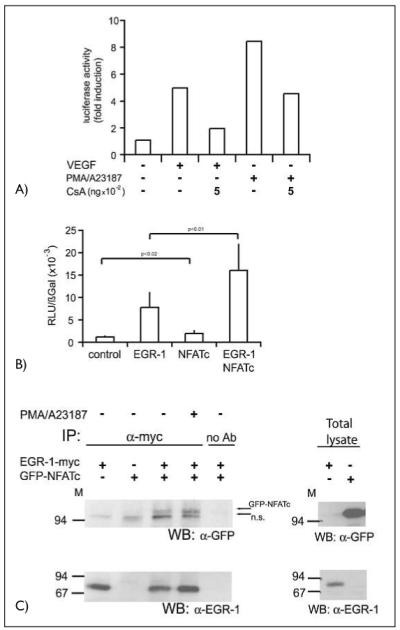
A) A reporter gene construct containing a 330-bpTF promoter element was transfected into HUVEC. Sixteen h post transfection the cells were starved in M199 medium with 2% serum for at least 8 h. The cells, in part pretreated with CsA as indicated, were then stimulated with VEGF (100 ng/ml) or a combination of PMA (10 nM) and A23187 (2.5 μM) for further 6 h. Then the cells were lysed and luciferase activity was measured using a dual-luciferase reporter assay system. A representative experiment of three performed in duplicates is shown. B) Endothelial cells were transfected with a tissue factor reporter gene construct containing the wild-type –330 bp promoter sequence. Cotransfection was performed with either an EGR-1– or an NFATc overexpression construct or a combination of both. Twenty-four h post transfection the cells were harvested, lysed and luciferase activity determined. Luciferase values were normalized to β-Gal expression from a cotransfected CMV-βGal construct. Mean values ± SD obtained from three independent experiments performed in duplicates are displayed. C) Two hundred ninety-three HEK cells were cotransfected with overexpression plasmids for myc-tagged EGR-1 and GFP-NFATc fusion protein. Cells were harvested without or after treatment with a combination of PMA (10 nM) and A23187 (2.5 μM) for 60 min. Cells and immunoprecipitations were performed with rabbit polyclonal anti-myc antibodies and protein A sepharose beads overnight. Equal aliquots of the immunoprecipitates or aliquots of the total cellular lysates were then loaded on 7.5% SDS-PAGE and probed with α-GFP or α-EGR-l antibodies. The positions of the GFP-NFATc and of a non-specific band (n.s.) are indicated by arrows.
We further analyzed the individual capacities of EGR-1 and NFAT to transactivate the TF promoter. For this purpose expression plasmids for EGR-1 or NFAT and combinations of both were transfected into HUVEC. These experiments showed that overexpression of EGR-1 alone was sufficient to transactivate the –330 bp reporter gene up to six-fold. In contrast to EGR-1, NFAT overexpression only slightly, although significantly (p < 0.02 vs. control), induced the TF promoter reporter gene. However, co-expression of both transcription factors resulted in synergistic transactivation of the promoter. In several independent reporter gene experiments on average an 11-fold upregulation of the luciferase gene driven by the tissue factor promoter was revealed (Fig. 4B). These data provide evidence for a cooperative transactivation of the TF promoter by both factors, although it appears that EGR-1 plays a predominant role in the in-vitro reporter assay.
Since the reporter gene assays revealed a functional interaction of EGR and NFAT in endothelial cells it seemed possible, that these two proteins might also interact physically with each other. To test this possibility, myc-tagged EGR-1 and the GFP-NFATc fusion protein were overexpressed in human epithelial kidney 293 cells (293 HEK). Assuming that PKC activation and intracellular Ca++ signals might lead to modifications of EGR-1 and NFAT supporting interactions, samples of the cells were also treated with PMA and calcium ionophore A23187. Co-immunoprecipitations were then performed with extracts from the un-induced and PMA/ionophore-induced 293 HEK cells. The precipitated proteins were analyzed on immunoblots with GFP- and EGR-1-specific antibodies. The obtained data show the presence of GFP-NFATc in immunoprecipitates of EGR-1-myc from uninduced and induced lysates (Fig. 4C); however, no significant increase of GFP-NFATc appeared to be present in immunoprecipitates from PMA/A23187-induced lysates. The sizes and levels of expression of EGR-1-myc and GFP-NFATc in total cell lysates are shown for control purposes in the right panel of Figure 4C. These data suggest that in addition to the binding to their respective specific binding elements in the tissue factor promoter a physical interaction between NFAT and EGR-1 provides a basis for their functional cooperation in endothelial cells. A similar interaction has been proposed to occur during the regulation of the IL-2 and TNF-α genes in T lymphocytes (34, 35).
The calcineurin inhibitor DSCR-1 is selectively upregulated by VEGF
Recent microarray data obtained by us and other laboratories have identified DSCR1, a gene encoding a protein with calcineurin inhibitory function, among the most highly VEGF-upregulated genes in endothelial cells (21, 22; B. Schweighofer and E. Hofer, unpublished data). DSCR1 spans nearly 45 kb of genomic DNA and comprises seven exons, four of which (exons 1–4) are alternative first exons (36). Since our data from Affymetrix microarray analysis had further indicated that DSCR-1 splice variant 4 (Calcipressin 1 isoform c) is, similar to TF, selectively upregulated by VEGF but not by EGF, we included DSCR1 in this study. The data obtained with real-time RT-PCR showed that VEGF strongly induces the DSCR1 exon 4 splice form (DSCR1_E4) mRNA with kinetics similar to TF up to 80-fold, whereas mRNA levels after EGF treatment are nearly unchanged (Fig. 5A). In contrast to TF induction, this upregulation was almost completely inhibited by CsA supporting that the exon 4 splice variant of the DSCR-1 gene is upregulated by VEGF predominantly via the calcineurin/NFAT pathway (Fig. 5B). A corresponding promoter element containing several NFAT binding sites has previously been identified upstream of exon 4 of the DSCR-1 gene (26). These data further support that NFAT activation is important for the upregulation of a group of genes by VEGF, which are not influenced by EGF.
Figure 5. The calcineurin inhibitor DSCRI is selectively regulated by VEGF and inhibits NFAT-dependent gene expression.

A) DSCRI_E4 mRNA levels in HUVEC in response to VEGF and EGF. HUVEC were induced with VEGF (100 ng/ml) or EGF (10 ng/ml), and total DSCRI mRNA levels were determined by real time RT-PCR. B) Inhibition of VEGF-mediated DSCR1_E4 mRNA induction by CsA. HUVEC were preincubated with CsA (I μg/ml) for 30 min and then induced with VEGF (100 ng/ml) for the times indicated. DSCR1_E4 mRNA levels were determined by real time RT-PCR. C) Inhibition of VEGF-me-diated DSCRI_E4 and TF mRNA induction by DSCRI_EI and NAB2 adenoviruses. HUVEC were infected with DSCR1_EI and NAB2 adenoviruses and further grown for 24 h. Infected cells were induced with VEGF (100 ng/ml) for 2 h. DSCR1_E4 and TF mRNA levels were determined by real-time RT-PCR. Data are displayed as % of the VEGF-induced mRNA levels. D) Inhibition of ionophore-mediated DSCRI_E4 and TF mRNA induction by DSCRI_EI and NAB2 adenoviruses. HUVEC were infected with DSCR1_EI and NAB2 adenoviruses and further grown for 24 h. Infected cells were induced with A23187 for 2 h. DSCR1_E4 and TF mRNA levels were determined by real-time RT-PCR. Data are displayed as % of the A23187-induced mRNA levels. Values were obtained from three independent experiments performed throughout A-D.
DSCR-1 in combination with the EGR-specific repressor NAB2 completely inhibits VEGF-induced TF expression
Since DSCR-1, also known as calcipressinl or MCIP1 (modulatory calcineurin interacting protein 1), has been shown to bind to and inhibit the enzymatic activity of calcineurin (23, 25), we have further constructed recombinant adenoviruses expressing DSCR1. To be able to monitor the endogenous DSCR1_E4 splice form, which is regulated by VEGF, adenoviruses for expression of DSCR1 exon 1 splice form (DSCR1_E1) were prepared. Splice form 1 is not regulated by VEGF and is only expressed at very low levels or absent from endothelial cells ([21]; B. Schweighofer and E. Hofer, unpublished data). Overexpression of DSCR1_E1 was then tested for its effects on VEGF-mediated upregulation of TF and endogenous DSCR1_E4 (Fig. 5C, D). The obtained data show that DSCR1_E1 overexpression prevented induction of DSCR1_E4 mRNA by VEGF as well as A23187 almost completely. This demonstrates that the calcineurin/NFAT pathway is predominantly important for DSCR1_E4 induction. In the case of TF mRNA the A23187-mediated upregulation was reduced by 90%, the inhibition of the VEGF regulation resulted on average in reductions of about 50% similar to the CsA inhibition. However, when adenoviruses expressing the corepressor NAB2 where used in addition to DSCR1_E1 viruses, TF mRNA induction was nearly completely inhibited. This supports the importance of both, the NFAT and EGR-1 pathways, for TF induction.
Discussion
The individual members of the endothelial-specific growth factor receptor families including the VEGF, Tie and ephrin receptors as well as some other receptors present on many different cell types, such as the EGF receptor, are receptor tyrosine kinases and possess multiple tyrosine residues in their cytoplasmic domain. These are in part phosphorylated upon activation by their specific ligands and then function as specific docking sites for molecules initiating cytoplasmic signaling cascades (2). Since the different receptor tyrosine kinases elicit to some degree overlapping, but also distinct responses in endothelial cells, part of the signals generated from the different receptors presumably differ from each other. In the case of the VEGFR-2 the cytoplasmic domain contains 19 tyrosines (1, 37, 38) and their differential degree of phosphorylation and functioning as docking sites for different signaling molecules leading to the activation of distinct signaling pathways is currently intensively investigated.
Since we have identified the TF gene as the example of a gene strongly upregulated by VEGF (6, 7), but to a lesser degree or not at all by some other tyrosine kinase receptors such as the EGF receptor (see Fig. 1), we have investigated the intracellular signals leading to this selective induction. The VEGF-A-mediated upregulation, as previously shown, is mainly mediated via VEGFR-2, since isoforms of VEGF selectively binding to VEGFR-2 can upregulate TF comparable to VEGF-A (7, 39) and P1GF, which specifically binds to VEGFR-1, only marginally affects TF ([40]; G. Schabbauer and E. Hofer, unpublished data).
TF is an important component of the repair mechanisms of vascular cells and as primary initiator of the blood coagulation cascade ensures the structural integrity of damaged vessels after inflammatory and/or mechanical injuries (8, 9). However, it seems to be a molecule of dual functions, to be also important for later stages of tissue repair such as wound healing (9, 41) and to have a regulatory role in the formation of new vessels (12, 15). TF has been directly implicated in vasculogenesis (14), as well as in tumor angiogenesis (13, 16, 41). The available evidence suggests that TF contributes to these processes by induction of intracellular signaling directly through its cytoplasmic domain and indirectly via factor Vila-dependent PAR-2 activation (9, 15, 42). In endothelial cells the duality of its function seems to be reflected by its upregulation by inflammatory mediators and by VEGF-A.
We have previously identified the transcription factor EGR-1 as a relevant factor involved in the induction of the TF gene by VEGF-A (7). Induction of TF and EGR-1 by VEGF was further dependent on the MAP kinases MEK/ERK and on PKC, since TF induction could be largely inhibited by the respective inhibitors of MEK and PKC, PD098059 and bis-indolylmaleimide (6). Overexpression of NAB2, a specific corepressor of EGR-1, strongly inhibited VEGF-A-mediated TF upregulation supporting the important role of EGR-1 (5). However, since EGR-1 is induced to a similar extent by EGF and VEGF, it seems that although EGR-1 is obviously essential for a significant part of the VEGF induction, in the form induced by EGF it is not sufficient to mediate upregulation of the endogenous TF gene. This differs from transcription studies using reporter gene assays, since in these assays cotransfections with EGR-1 expression plasmids alone can efficiently upregulate the TF promoter (5, 7).
Several lines of evidence suggested that VEGFR-2 is very effective in recruiting and activating PLC-γ and the downstream Ca++ and PKC signaling pathways (37, 43, 44) including our previous results that EGR-1 induction by VEGF is largely PKC-dependent and could be inhibited by PKC inhibitors (6). It seemed therefore plausible that the induction of the Ca++ signaling pathway leading to calcineurin activation, NFAT dephosphorylation and cytoplasmic-nuclear translocation, might be required for efficient TF upregulation. On the other hand, we speculated that EGF would insufficiently trigger the activation of this pathway. We show here that this indeed is the case. VEGF triggered the rapid cytoplasmic-nuclear shuttling of an NFAT-GFP fusion protein, whereas EGF was completely inefficient. The specific calcineurin inhibitor CsA as well as the Ca++ chelator BAPTA-AM significantly inhibited TF upregulation by VEGF to about 50%. Furthermore, the Ca++ ionophore A23187 strongly upregulated TF, demonstrating that the Ca++ signals alone can trigger TF upregulation.
Since these data supported that EGR-1 and NFAT are both contributing to full TF induction by VEGF, we investigated a potential direct cooperation between the two factors in reporter gene assays and by coimmunoprecipitation. In the reporter gene assay EGR-1 alone caused a significant upregulation and the combination of EGR-1 with NEAT displayed a synergistic effect (Fig. 4B). By itself NEAT only slightly increased promoter activity suggesting that the factor alone is not effective to cause strong TF upregulation but requires the presence of EGR-1. It is important to note that the NFAT consensus sequence within the TF promoter significantly overlaps with the NF-κB binding site (33). In case of stimulation of the cells with suboptimal doses of VEGF and TNF-α, NFAT induced by VEGF and NF-κB induced by TNF-α might additively contribute to TF expression, whereas in excess of both factors they are likely to compete for binding to the composite NF-κB/NFAT DNA binding site. Coimmunoprecipitations performed with overexpressed NFAT and EGR-1 support the idea that the functional cooperation between the factors is based on a physical interaction. It is possible that EGR-1 and NFAT by interacting with each other mutually increase their binding to the respective promoter elements or synergistically increase their potential to promote transcription initiation, as it is supported by the reporter gene studies. This hypothesis is in accordance with previously published reports, describing direct cooperative interactions and stable physical protein complexes between EGR-1 and NFAT important in the activation of the IL-2 and TNF-α promoters in T cells (34, 35, 45).
In regard of the role of the Ca++ signals for VEGF-mediated gene regulation it is of importance that recent data from differential screens and microarrays have identified the calcineurin inhibitor DSCR-1 as one of the most highly VEGF upregulated genes (21, 22). Our own Affymetrix data confirmed by RT-PCR have defined DSCR-1 and TF in atop group of the 20 most highly VEGF-upregulated genes in relative terms. Both display an over 30-fold upregulation of mRNA when compared to uninduced cells. In contrast to TF, DSCR-1 belongs also in the absolute amount of mRNA produced to the top group (Schweighofer et al., manuscript in preparation). Several splice forms of the DSCR-1 protein have been shown to bind to calcineurin and to inhibit its phosphatase activity preventing the activation of NFAT (46). The regulatory importance of DSCR-1 for Ca++ signaling is shown by the multiple disease manifestations primarily in trisomy 21 (24) connected to inappropriate expression of the protein. In addition, chronic overexpression of DSCR-1 was on the one hand linked to Alzheimer disease (47), but on the other hand protected against cardiac hypertrophy in animal models (48) supporting the importance of a disturbed NEAT/DSCR-1 balance in these diseases. The strong induction of DSCR-1 by VEGF seems to result in a negative feedback loop to limit the Ca++/calcineurin component of VEGF signaling and therefore potential harmful effects of excess and prolonged NFAT activation.
We have therefore used the DSCR-1 gene as a second example to test for the importance of the calcineurin/NFAT pathway for VEGF-mediated gene regulation. The obtained data on the one hand clearly show that NFAT is the predominant factor in VEGF-mediated DSCR1_E4 promoter regulation. DSCR1_E4 mRNA is upregulated to a similar extent by VEGF and A23187 and both inductions could be strongly inhibited by CsA or DSCR-1 overexpression. The inability of EGF to trigger DSCR1_E4 induction further suggests that the activation of the calcineurin/NFAT pathway by VEGF could be the primary basis for the induction of a wider spectrum of genes by this factor. Our data indicate that NFAT induction could be important for a significant portion of the VEGF-mediated gene regulation and provide the factor with unique properties when compared to other growth factors not triggering this pathway to the same extent. It is intriguing, that a single tyrosine residue in VEGFR-2, Y1175, was previously found to be essential for PLC-γ activation (43, 49) and downstream calcineurin/NFAT activation. Whereas certain genes, such as DSCR-1, might be predominantly regulated by NFAT, others, such as TF, may require the combination of EGR-1 with NFAT and/or possibly other factors to be determined. Based on the obtained data it might be possible that combined inhibition of both, the calcineurin/NFAT and the EGR-1 pathways might be worthwhile to be explored for more effective inhibition of VEGF-mediated gene expression and angiogenesis than it was shown already for the inhibition of the individual pathways (5, 21).
Acknowledgments
We are grateful to L. Gerace for a gift of pRSGFP-Cl-NFATc. Further we thank our colleagues from the Department of Vascular Biology and Thrombosis Research for help and discussions during the course of these studies. This work was supported by grants to E.H. of the Austrian Science Fund (NFN94–3) and the European Commission (LSH-CT-2005–518178).
Abbreviations
- BAPTA-AM
bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester
- CsA
cyclosporin A
- DSCR-1
Down syndrome critical region gene-1
- ECGS
endothelial cell growth supplement
- EGF
epidermal growth factor
- EGR-1
early growth response
- FCS
fetal calf serum
- Flk-1
fetal liver kinase-1
- GFP
green fluorescent protein
- HEK 293
human embryo kidney 293
- HUVEC
human umbilical vein endothelial cells
- IL-2
interleukin-2
- MCIP1
modulatory calcineurin-in-teracting protein 1
- NFAT
nuclear factor of activated T cells
- NF-κB
nuclear factor kappa B
- PAR-2
protease activated receptor 2
- PBS
phosphate buffered saline
- PKC
protein kinase C
- PLC-γ
phospholipase C gamma
- PMA
phorbol 12-myristate 13-acetate
- SCS
supplemented calf serum
- TGF
transforming growth factor
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
- VEGFR-2
VEGF receptor-2
References
- 1.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 2.Yancopoulos GD, Davis S, Gale NW, et al. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 4.Amin DN, Hida K, Bielenberg DR, et al. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006;66:2173–2180. doi: 10.1158/0008-5472.CAN-05-3387. [DOI] [PubMed] [Google Scholar]
- 5.Lucerna M, Mechtcheriakova D, Kadl A, et al. NAB2, a corepressor of EGR-1, inhibits vascular endothelial growth factor-mediated gene induction and angiogenic responses of endothelial cells. J Biol Chem. 2003;278:11433–11440. doi: 10.1074/jbc.M204937200. [DOI] [PubMed] [Google Scholar]
- 6.Mechtcheriakova D, Schabbauer G, Lucerna M, et al. Specificity, diversity, and convergence in VEGF and TNF-alpha signaling events leading to tissue factor upregulation via EGR-1 in endothelial cells. Faseb J. 2001;15:230–242. doi: 10.1096/fj.00-0247com. [DOI] [PubMed] [Google Scholar]
- 7.Mechtcheriakova D, Wlachos A, Holzmuller H, et al. Vascular endothelial cell growth factor-induced tissue factor expression in endothelial cells is mediated by EGR-1. Blood. 1999;93:3811–3823. [PubMed] [Google Scholar]
- 8.Martin DM, Boys CW, Ruf W. Tissue factor: molecular recognition and cofactor function. Faseb J. 1995;9:852–859. doi: 10.1096/fasebj.9.10.7615155. [DOI] [PubMed] [Google Scholar]
- 9.Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24:1015–1022. doi: 10.1161/01.ATV.0000130465.23430.74. [DOI] [PubMed] [Google Scholar]
- 10.Mackman N. Regulation of the tissue factor gene. Faseb J. 1995;9:883–889. doi: 10.1096/fasebj.9.10.7615158. [DOI] [PubMed] [Google Scholar]
- 11.Pawlinski R, Mackman N. Tissue factor, coagulation proteases, and protease-activated receptors in endotoxemia and sepsis. Crit Care Med. 2004;32:S293–297. doi: 10.1097/01.ccm.0000128445.95144.b8. [DOI] [PubMed] [Google Scholar]
- 12.Pawlinski R, Pedersen B, Erlich J, et al. Role of tissue factor in haemostasis, thrombosis, angiogenesis and inflammation: lessons from low tissue factor mice. Thromb Haemost. 2004;92:444–150. doi: 10.1160/TH04-05-0309. [DOI] [PubMed] [Google Scholar]
- 13.Contrino J, Hair G, Kreutzer DL, et al. In situ detection of tissue factor in vascular endothelial cells: correlation with the malignant phenotype of human breast disease. Nat Med. 1996;2:209–215. doi: 10.1038/nm0296-209. [DOI] [PubMed] [Google Scholar]
- 14.Carmeliet P, Mackman N, Moons L, et al. Role of tissue factor in embryonic blood vessel development. Nature. 1996;383:73–75. doi: 10.1038/383073a0. [DOI] [PubMed] [Google Scholar]
- 15.Belting M, Dorrell MI, Sandgren S, et al. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med. 2004;10:502–509. doi: 10.1038/nm1037. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Bierhaus A, Schiekofer S, et al. Tissue factor--a receptor involved in the control of cellular properties, including angiogenesis. Thromb Haemost. 2001;86:334–345. [PubMed] [Google Scholar]
- 17.Ruf W, Mueller BM. Tissue factor in cancer angiogenesis and metastasis. Curr Opin Hematol. 1996;3:379–384. doi: 10.1097/00062752-199603050-00008. [DOI] [PubMed] [Google Scholar]
- 18.Moll T, Czyz M, Holzmuller H, et al. Regulation of the tissue factor promoter in endothelial cells. Binding of NF kappa B-, AP-1-, and Spl-like transcription factors. J Biol Chem. 1995;270:3849–3857. doi: 10.1074/jbc.270.8.3849. [DOI] [PubMed] [Google Scholar]
- 19.Winsauer G, de Martin R. Resolution of inflammation: Intracellular feedback loops in the endothelium. Thromb Haemost. 2007;97:364–369. [PubMed] [Google Scholar]
- 20.Wrighton CJ, Hofer-Warbinek R, Moll T, et al. Inhibition of endothelial cell activation by adenovirus-mediated expression of I kappa B alpha, an inhibitor of the transcription factor NF-kappa B. J Exp Med. 1996;183:1013–1022. doi: 10.1084/jem.183.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minami T, Horiuchi K, Miura M, et al. Vascular endothelial growth factor- and thrombin-induced termination factor, Down syndrome critical region-1, attenuates endothelial cell proliferation and angiogenesis. J Biol Chem. 2004;279:50537–50554. doi: 10.1074/jbc.M406454200. [DOI] [PubMed] [Google Scholar]
- 22.Hesser BA, Liang XH, Camenisch G, et al. Down syndrome critical region protein 1 (DSCR1), a novel VEGF target gene that regulates expression of inflammatory markers on activated endothelial cells. Blood. 2004;104:149–158. doi: 10.1182/blood-2004-01-0273. [DOI] [PubMed] [Google Scholar]
- 23.Rothermel B, Vega RB, Yang J, et al. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem. 2000;275:8719–8725. doi: 10.1074/jbc.275.12.8719. [DOI] [PubMed] [Google Scholar]
- 24.Fuentes JJ, Genesca L, Kingsbury TJ, et al. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum Mol Genet. 2000;9:1681–1690. doi: 10.1093/hmg/9.11.1681. [DOI] [PubMed] [Google Scholar]
- 25.Vega RB, Yang J, Rothermel BA, et al. Multiple domains of MCIP1 contribute to inhibition of calcineurin activity. J Biol Chem. 2002;277:30401–30407. doi: 10.1074/jbc.M200123200. [DOI] [PubMed] [Google Scholar]
- 26.Lange AW, Molkentin JD, Yutzey KE. DSCR1 gene expression is dependent on NFATcl during cardiac valve formation and colocalizes with anomalous organ development in trisomy 16 mice. Dev Biol. 2004;266:346–360. doi: 10.1016/j.ydbio.2003.10.036. [DOI] [PubMed] [Google Scholar]
- 27.Knappik A, Pluckthun A. An improved affinity tag based on the FLAG peptide for the detection and purification of recombinant antibody fragments. Biotechniques. 1994;17:754–761. [PubMed] [Google Scholar]
- 28.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gruber F, Hufnagl P, Hofer-Warbinek R, et al. Direct binding of Nur77/NAK-1 to the plasminogen activator inhibitor 1 (PAI-1) promoter regulates TNF alpha-induced PAI-1 expression. Blood. 2003;101:3042–3048. doi: 10.1182/blood-2002-07-2331. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 31.Lucerna M, Pomyje J, Mechtcheriakova D, et al. Sustained expression of early growth response protein-1 blocks angiogenesis and tumor growth. Cancer Res. 2006;66:6708–6713. doi: 10.1158/0008-5472.CAN-05-2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kehlenbach RH, Dickmanns A, Gerace L. Nucleocytoplasmic shuttling factors including Ran and CRM1 mediate nuclear export of NEAT In vitro. J Cell Biol. 1998;141:863–874. doi: 10.1083/jcb.141.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Armesilla AL, Lorenzo E, Gomez del Arco P, et al. Vascular endothelial growth factor activates nuclear factor of activated T cells in human endothelial cells: a role for tissue factor gene expression. Mol Cell Biol. 1999;19:2032–2043. doi: 10.1128/mcb.19.3.2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Decker EL, Nehmann N, Kampen E, et al. Early growth response proteins (EGR) and nuclear factors of activated T cells (NEAT) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic Acids Res. 2003;31:911–921. doi: 10.1093/nar/gkg186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Decker EL, Skerka C, Zipfel PE. The early growth response protein (EGR-1) regulates interleukin-2 transcription by synergistic interaction with the nuclear factor of activated T cells. J Biol Chem. 1998;273:26923–26930. doi: 10.1074/jbc.273.41.26923. [DOI] [PubMed] [Google Scholar]
- 36.Fuentes JJ, Pritchard MA, Estivill X. Genomic organization, alternative splicing, and expression patterns of the DSCR1 (Down syndrome candidate region 1) gene. Genomics. 1997;44:358–361. doi: 10.1006/geno.1997.4866. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto T, Bohman S, Dixelius J, et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. Embo J. 2005;24:2342–2353. doi: 10.1038/sj.emboj.7600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmqvist K, Cross MJ, Rolny C, et al. The adaptor protein shb binds to tyrosine 1175 in vascular endothelial growth factor (VEGF) receptor-2 and regulates VEGF-dependent cellular migration. J Biol Chem. 2004;279:22267–22275. doi: 10.1074/jbc.M312729200. [DOI] [PubMed] [Google Scholar]
- 39.Meyer M, Clauss M, Lepple-Wienhues A, et al. A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. Embo J. 1999;18:363–374. doi: 10.1093/emboj/18.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clauss M, Pipp F, Issbrucker K, et al. Dissection of monocyte and endothelial activities by using VEGF-receptor specific ligands. Adv Exp Med Biol. 2003;522:75–82. doi: 10.1007/978-1-4615-0169-5_8. [DOI] [PubMed] [Google Scholar]
- 41.Nakagawa K, Zhang Y, Tsuji H, et al. The angiogenic effect of tissue factor on tumors and wounds. SeminThromb Hemost. 1998;24:207–210. doi: 10.1055/s-2007-995843. [DOI] [PubMed] [Google Scholar]
- 42.Siegbahn A, Johnell M, Sorensen BB, et al. Regulation of chemotaxis by the cytoplasmic domain of tissue factor. Thromb Haemost. 2005;93:27–34. doi: 10.1160/TH04-07-0405. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi T, Yamaguchi S, Chida K, et al. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. Embo J. 2001;20:2768–2778. doi: 10.1093/emboj/20.11.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi T, Ueno H, Shibuya M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999;18:2221–2230. doi: 10.1038/sj.onc.1202527. [DOI] [PubMed] [Google Scholar]
- 45.Alfonso-Jaume MA, Mahimkar R, Lovett DH. Cooperative interactions between NEAT (nuclear factor of activated T cells) cl and the zinc finger transcription factors Sp1/Sp3 and Egr-1 regulate MT1-MMP (membrane type 1 matrix metalloproteinase) transcription by glomerular mesangial cells. Biochem J. 2004;380:735–747. doi: 10.1042/BJ20031281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rothermel BA, Vega RB, Williams RS. The role of modulatory calcineurin-interacting proteins in calcineurin signaling. Trends Cardiovasc Med. 2003;13:15–21. doi: 10.1016/s1050-1738(02)00188-3. [DOI] [PubMed] [Google Scholar]
- 47.Ermak G, Harris CD, Davies KJ. The DSCR1 (Adapt78) isoform 1 protein calcipressin 1 inhibits calcineurin and protects against acute calcium-mediated stress damage, including transient oxidative stress. Faseb J. 2002;16:814–824. doi: 10.1096/fj.01-0846com. [DOI] [PubMed] [Google Scholar]
- 48.Vega RB, Rothermel BA, Weinheimer CJ, et al. Dual roles of modulatory calcineurin-interacting protein 1 in cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:669–674. doi: 10.1073/pnas.0237225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sakurai Y, Ohgimoto K, Kataoka Y, et al. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci USA. 2005;102:1076–1081. doi: 10.1073/pnas.0404984102. [DOI] [PMC free article] [PubMed] [Google Scholar]