

Abstract
Purpose of Review
To highlight key studies providing rationale for and utility in targeting glycolysis for the treatment of hematological malignancies.
Recent Findings
Several therapeutic strategies are capitalizing on the diagnostic utility of FDG-PET that relies on increased glycolysis and glucose utilization in tumor cells. While aerobic glycolysis was initially proposed by Warburg to be due to mitochondrial impairment, recent studies have shown a preferential switch to glycolysis in tumor cells with functional mitochondria. Increased glucose consumption can be advantageous for a tumor cell through stimulation of cellular biosynthetic, energetic, and pro-survival pathways. We now have a greater appreciation for the utilization of glucose in specific metabolic pathways that in some aspects can be complemented with other nutrients such as glutamine. Targeting glucose consumption for the treatment of hematological malignancies seems to be a promising field that will require characterization of tumor cell specific targets to inhibit glucose uptake and/or glycolysis. It is imperative to further our understanding of the tumor cell metabolome to target cellular bioenergetics in the treatment of cancer.
Summary
Targeting the glycolytic pathway for the treatment of hematological malignancies has sufficient rationale given the utility of FDG-PET in diagnostic imaging. Further research is required in developing tumor cell specific therapeutics.
Keywords: Glucose, hematologic malignancies, glycolysis
Introduction
Hematological malignancies account for approximately ten percent of diagnosed cancers (1). These cancers of lymphoid and myeloid origin, while having diverse genomic profiles, demonstrate deregulation of core molecular and biochemical pathways. In the nineties, alkylating agents, antimetabolites, anthracyclines, topoisomerase inhibitors, anti-microtubule drugs and steroids formed the spectrum of available therapeutics. The emergence of newer targeted therapeutics like imatinib, bortezomib, and rituximab coupled with advances in autologous stem cell transplantation have tremendously increased the average lifespan of patients living with these diseases. What continues to be a caveat in the treatment of all cancers is the development of resistance and the emergence of a more aggressive cancer. Therefore, the discovery of novel approaches targeting pathways that become increasingly important during disease progression is necessary.
The Warburg Effect
Nearly 80 years, ago Otto Warburg made the seminal observation that tumor cells consume surprisingly high amounts of glucose and use the less efficient glycolytic pathway (as depicted in Figure 1) to generate adenosine triphosphate (ATP) even in the presence of oxygen (2). Normal cells preferentially use the mitochondrial tricarboxylic acid (TCA) cycle for the oxidative degradation of glucose and generation of ATP, resorting to glycolysis only under conditions of oxygen deprivation such as under muscle fatigue. In the mid 1800s, Louis Pasteur studied yeast fermentation processes and demonstrated the ability of oxygen to inhibit glycolysis, facilitating mitochondrial oxidative degradation of glucose (3). While Warburg believed the preferential use of glycolysis even in the presence of oxygen by tumor cells was due to defects in the mitochondrial respiratory pathways required for oxidation of glucose, recent studies have shown that tumor cells do contain functional mitochondria (4) yet still produce excessive lactate, suggesting that the enhanced glycolytic flux may confer a growth advantage.
Figure 1. Metabolic alterations promoting tumor cell aerobic glycolysis and associated therapeutic targets.
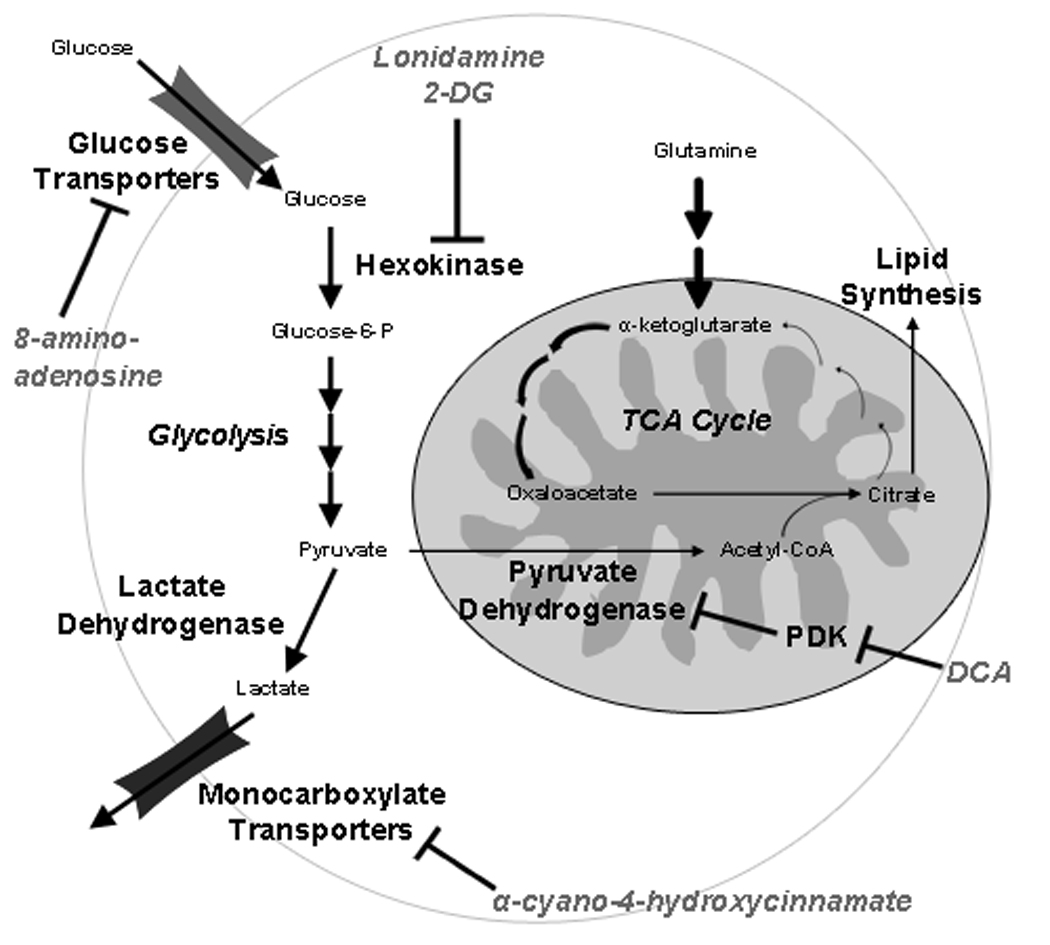
Tumor cells increase glucose consumption through enhanced expression and activity of glucose transporters and hexokinase. Hexokinase carries out the initial phosphorylation of glucose, which is required to retain glucose molecules within the cell. In normal cells, glucose is converted to pyruvate in a series of reactions in the cytosol. Pyruvate, the end product of these reactions, enters the mitochondria and is converted to acetyl-CoA, which is used in the tricarboxylic acid cycle (TCA) cycle to drive ATP synthesis via oxidative phosphorylation. In tumor cells, glucose-derived pyruvate is preferentially converted to lactate in the cytosol and subsequently extruded from the cell through monocarboxylate transporters. Pyruvate molecules which do enter the mitochondria in this context are used largely to fuel a truncated TCA cycle wherein citrate is siphoned out of the mitochondria to stimulate fatty acid biosynthesis. Glutamine can then be used to replenish subsequent metabolites in the TCA cycle through conversion to α-ketoglutarate. Specific enzyme targets for lonidamine, 2-deoxyglucose (2-DG), 8-amino-adenosine, dichloroacetate (DCA), and α-cyano-4-hydroxycinnamate are indicated. Bold arrows indicate enhanced activity in tumor cells.
In support of this notion, interference with lactate dehydrogenase activity (the enzyme responsible for conversion of lactate to pyruvate) in tumor cells forces a reversion to glucose catabolism via oxidative phosphorylation and results in reduced tumorigenicity (4). Forcing tumor cells to revert to the use of oxidative metabolism by overexpression of mitochondrial frataxin, a protein regulating mitochondrial iron transport and respiration (5), or with chemical inhibitors like dichloroacetate, also reduces tumor growth in mouse xenograft studies (6). Recently, requisite events preceding the switch from oxidative phosphorylation (OXPHOS) to aerobic glycolysis in neoplastic cells have been shown to involve expression of the embryonic form of pyruvate kinase, which appears to be required for tumor formation. This isoform is uniquely regulated by tyrosine kinase activity and is thought to divert glucose to anabolic processes, thus facilitating tumor growth (7). Several studies have demonstrated the role of mitochondrial uncoupling proteins in limiting catabolism of pyruvate in the TCA cycle and increasing fatty acid oxidation thereby promoting the Warburg effect (8, 9). Progressive oncogenic transformation by serial transduction of a set of viral oncogenes in an in vitro cell line model of tumorigenesis was also found to correlate with a progressive switch to glycolysis and increasing sensitivity to glycolytic pathway inhibitors (10). Several mechanisms have now been shown to contribute to the Warburg effect i.e. mitochondrial defects, adaptation of the cancer cells to hypoxic microenvironments, oncogenic signals and abnormal expression of metabolic enzymes (11). The intimate association between transcriptional control of glycolytic genes and the activity of classical oncogenes and tumor suppressors, including myc, p53, N and K Ras and Hif1α further underscores the prevalence of de-regulated cellular metabolism in cancer cells (12). These observations have stimulated a renewed interest in strategies that target metabolism and cellular bioenergetics unique to cancer cells.
Benefits of enhanced glycolysis
A high glycolytic rate and enhanced glucose uptake can provide several benefits to a proliferating tumor cell. Although OXPHOS generates more ATP per molecule of glucose, glycolysis can provide ATP at a higher rate provided glucose supply is unlimited (13, 14). In addition glucose can provide intermediates such as ribose sugar for nucleotide synthesis and NADPH used in the reductive biosynthesis of lipids and fats through the oxidative pentose phosphate pathway. In tumor cells much of the carbon that enters the TCA cycle is extruded as citrate resulting in a truncated TCA cycle that is used for synthesis of lipids and fatty acids (15). Glucose can contribute to maintaining mitochondrial integrity by promoting the association of hexokinase II with the mitochondria, thereby preventing the release of cytochrome C (16, 17) as well as by regulating various effectors of cell death i.e. regulation of pro-survival Mcl-1 (18), pro-apoptotic BAD (19), and pro-apoptotic Bax (20). Lactate, the by-product of glycolysis, is thought to promote tumor invasion and metastasis via degradation of extracellular matrices (21). In summary, glucose plays a critical role in sustaining tumor cell growth, thus providing a rationale for the development of therapeutic strategies to preferentially kill cancer cells by targeting glycolysis.
Hematopoietic malignancies and glucose utilization
While most of our knowledge on deregulated tumor cell metabolism comes from solid tumors, large-scale gene-expression analyses reveal the selective upregulation of genes encoding constituents of the glycolytic pathway in hematopoietic malignancies (22). Acute lymphoblastic leukemia cells demonstrate upregulation of genes facilitating glycolysis such as GLUT 1, GLUT4 and monocarboxylic acid transporter SLC16A2 (23). A study on the molecular pathogenesis of chronic myelogenous leukemia showed that transformation of normal hematopoietic cells with the BCR-ABL oncogene results in increased glucose metabolism and intracellular ROS levels, which is likely mediated through AKT/mTOR signaling (24). In certain leukemia cell lines, it has been demonstrated that co-culture with mesenchymal stem cells induces the expression of uncoupling protein 2 which further exacerbates the glycolytic phenotype of these cells (25). Probably the most crucial evidence pointing towards the preferential utilization of glycolysis and excessive glucose consumption in hematopoietic malignancies lies in the successful imaging of these tumors through 18fluoro-deoxyglucose positron emission tomography (FDG-PET) (26, 27). While proliferating primary lymphocytes also utilize the glycolytic pathway to convert 90% of glucose derived carbon to lactate (28, 29), their tumor cell counterparts consume higher amounts of glucose for the very same reasons outlined above. PET has primarily been used in the detection and diagnostic staging of Hodgkin’s disease, aggressive non-Hodgkin’s lymphomas and in multiple myeloma (26, 30)
Irrespective of whether altered metabolism and increased glucose consumption in hematopoietic malignancies are a cause or consequence of the disease, the fundamental role of glucose in maintaining energy homeostasis and apoptotic resistance provides sufficient rationale to explore inhibition of glucose utilization as a treatment strategy for these cancers.
Targeting Glycolysis in the Clinic
The wealth of compelling data pointing to glycolytic inhibition as a viable therapeutic strategy in cancer combined with the relevance of the “Warburg effect” to myriad forms of malignancy have led to the development of numerous compounds targeting this critical growth-related pathway. In addition to the ability to increase tumor cell sensitivity to a variety of traditional chemotherapeutics, interference with glucose metabolism has also recently been shown to induce a cytoprotective effect in nonmalignant tissue (31). Achieving both these benefits through the inclusion of anti-glycolytic agents in combinatorial drug regimens could result in a significant widening of the therapeutic window associated with traditional chemotherapeutics. Despite existing evidence for the glucose avidity of certain hematological malignancies, nearly all glycolysis-targeted therapies have been tested exclusively in solid tumors (32). Therefore, the following overview of the clinical development of three of the most prominent glycolytic inhibitors (as depicted in Figure 1) will focus on the successes and failures in this class of neoplasms with the understanding that expansion to the treatment of hematopoietic malignancies remains plausible, if not promising.
Lonidamine
Lonidamine, 1-(2,4-dichlorobenzyl)-1-H-indazole-3-carboxylic acid, is a glycolysis-targeting compound which has exited clinical trials and gained approval in Europe for the treatment of various solid tumors. Lonidamine acts as an inhibitor of hexokinase and exhibits selectivity for the mitochondria-bound form of the enzyme, which appears to play a prominent role in increasing glycolytic flux in both proliferating normal and neoplastic cells (33, 34). Binding of hexokinase to the outer mitochondrial membrane enhances the activity of the enzyme by modifying its interaction with a necessary substrate in two ways: first, by ensuring a constant source of ATP generated by the electron transport chain, and second, by increasing the affinity of hexokinase for the ATP molecule (35). Three clinical trials have recently come to completion utilizing lonidamine in combination with other chemotherapeutics, with the authors of these studies reporting varying degrees of success. In a phase II clinical trial evaluating the administration of both lonidamine and diazepam to patients with recurrent glioblastoma multiforme, 50% of patients exhibited disease stabilization following treatment including one case in which progression did not occur for 12 months (36). Authors of a phase II study designed to examine the efficacy of paclitaxel, cisplatin, and lonidamine co-administration to patients with advanced ovarian cancer observed an 80% overall response rate, including 40% complete responses and 40% partial responses (37). However, this trial was designed as a single-arm, uncontrolled study making the contribution of lonidamine to the overall success difficult to ascertain. A larger phase III study completed in previously untreated breast cancer patients revealed a 9% increase in overall response rate in patients receiving epirubicin plus lonidamine in comparison with epriubicin alone which bordered on reaching statistical significance (38). In comparing toxicity results between these three studies, all reported the occurrence of myalgia and the phase III study effectively linked this occurrence to the addition of lonidamine to the therapeutic regimen.
2-Deoxy-D-glucose
The most widely employed compound to investigate glycolytic inhibition in in vitro studies, 2-deoxy-D-glucose (2DG) has exhibited broad activity in tumor cell lines as a single agent and in combination with other chemotherapeutics (24, 39). This anti-metabolite with nearly complete structural identity to glucose is taken up into cells via glucose transporters and subsequently phosphorylated by hexokinase. However, as this molecule is not a recognizable substrate for the next enzyme in the glycolytic pathway, phosphoglucose isomerase, phosphorylated 2DG accumulates to high levels in the cytosol and inhibits hexokinase activity. Unlike lonidamine, this drug does not appear to display selectivity for different hexokinase isoforms and derives its selectivity (however minimal) for tumors simply through increased glucose consumption rates. According to the NIH website clinicaltrials.gov, there are currently three clinical trials utilizing this drug which are ongoing or have recently reached completion. Authors of a phase I trial conducted by Threshold Pharmaceuticals employing 2DG in combination with docetaxel in patients with advanced solid tumors observed disease stabilization in 6 of 18 evaluable patients and one partial response (40). However, grade 3 hyperglycemia limited dose escalation above 88 mg/kg in certain patients. This observation is particularly concerning given previous animal studies in which much higher dosages of 500–2000 mg/kg were required to achieve tumor growth inhibition in xenograft tumors in immuno-compromised mice (41). This compound has also been shown to stimulate Akt phosphorylation in vitro and antagonize the anti-tumor activity of a radio-immunotherapeutic in vivo, effects which further degrade the therapeutic value of 2DG (42, 43). It was recently announced that Threshold Pharmaceuticals is no longer pursuing clinical development of this compound.
Dichloroacetate
An alternative strategy to blocking flux through the glycolytic pathway by directly targeting individual constituent enzymes consists of forcing the entry of pyruvate into the mitochondria for oxidative catabolism in the TCA cycle, thus antagonizing rapid conversion to lactate in the cytosol. Pyruvate dehydrogenase is a mitochondrial enzyme which converts pyruvate to acetyl CoA. The activity of this enzyme is negatively regulated by pyruvate dehydrogenase kinase (PDK), the expression of which is cooperatively induced by HIF1α and c-myc (44). Dichloroacetate (DCA) is a small molecule inhibitor of PDK which can effectively induce a metabolic switch from aerobic glycolysis to glucose oxidation, decreasing mitochondrial hyperpolarization and rendering tumor cells more sensitive to apoptosis induction (6). This compound has previously been used in the clinic for the treatment of lactic acidosis and exhibits an appealing toxicity profile when dosed chronically, making it an ideal candidate for cancer treatment (45). Currently, a phase I trial in Canada is accepting patients with recurrent or metastatic solid tumors.
Future Directions for Anti-Glycolytic Therapies
While many compounds targeting glycolysis have generated much enthusiasm due to in vitro potency, widespread successes have not yet been realized in in vivo settings. A recurring theme in clinical trials investigating compounds within this class of drugs is the high prevalence of dose-limiting toxicities, presumably due to on-target effects in nonmalignant tissue. Therefore, it appears that a significant hurdle that must be overcome in targeting tumor cell metabolism is increasing the selectivity of these pharmaceuticals for malignant tissue versus normal. The fact that lonidamine displays an inherent selectivity for tumor (or at least actively proliferating) cells seems to at least partially account for its success in the clinical arena. Identification of tumor-specific metabolic alterations should provide a therapeutic window wide enough to substantially inhibit glucose utilization by malignant cells without impairing the metabolism of normal cells. The monocarboxylate transporters, which function to export cytoplasmic lactate, have generated interest recently as potential targets due to their relatively selective upregulation in a variety of tumors (46, 47). Additionally, our lab has shown that the purine nucleoside analogue 8-amino-adenosine (8-NH2-Ado) is capable of interfering with glucose transporter (GLUT) localization to the plasma membrane in myeloma cells, representing yet another level at which tumor selectivity may be achieved (Submitted paper: Shanmugam M, McBrayer S et al. unpublished data). With current drug development efforts aimed at improving selectivity, glycolysis inhibition as a strategy for cancer treatment may experience a broader utility in the future.
In addition to identifying tumor-specific glycolytic targets, it will also be imperative to identify new means of capitalizing on the metabolic stress induced by this therapeutic approach. A critical cellular process that is initiated to counteract metabolic stress and could provide resistance to glycolysis inhibition in vivo is autophagy. Sequestration of intracellular organelles and their subsequent breakdown through the autophagic pathway provides metabolic substrates such as amino acids which can be used as a reserve fuel source for tumor cells (48, 49). We have demonstrated in our studies that myeloma cells can resist apoptosis following 8-NH2-Ado-induced glucose deprivation through induction of autophagy (Submitted paper: Shanmugam M, McBrayer S et al. unpublished data). Consistent with this observation, co-treatment of cells with inhibitors of this pathway results in a synergistic apoptotic response. Therefore, evaluating the relationship between various glycolytic inhibitors and autophagy inhibitors could produce marked increases in therapeutic efficacy due to the potentiation of cytostatic effects and conversion to cytotoxic outcomes.
Conclusions
While considering targeting the glycolytic pathway in the treatment of cancer one must take into account compensatory contributions of other cellular metabolites. This is particularly important given the fact that specific TCA cycle intermediates generated by glucose metabolism can also be generated by glutaminolysis. Therapeutics targeting the glycolytic pathway have been shown to synergize with specific therapeutics (41). Further characterization of other classes of drugs that may synergize with the inhibition of glycolysis will aid in enhancing sensitivity to current therapeutics. Targeting the very basis for clinical imaging of cancer (i.e. glucose uptake) can provide new therapeutic options that may be less prone to the development of resistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Laurent Degos DCL, Bob Lowenberg, editors. Textbook of Malignant Hematology. 2 ed. Taylor and Francis; 2005. [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956 Feb 24;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Stryer L. Biochemistry. 3rd ed. New York: W.H. Freeman Company; 1988. [Google Scholar]
- 4.Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006 Jun;9(6):425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 5.Schulz TJ, Thierbach R, Voigt A, Drewes G, Mietzner B, Steinberg P, et al. Induction of oxidative metabolism by mitochondrial frataxin inhibits cancer growth: Otto Warburg revisited. J Biol Chem. 2006 Jan 13;281(2):977–981. doi: 10.1074/jbc.M511064200. [DOI] [PubMed] [Google Scholar]
- 6.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007 Jan;11(1):37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 7. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008 Mar 13;452(7184):230–233. doi: 10.1038/nature06734. Describes the role of the embryonic PKM2 isoform of pyruvate kinase in promoting aerobic glycolysis in tumor cells.
- 8. Pecqueur C, Bui T, Gelly C, Hauchard J, Barbot C, Bouillaud F, et al. Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 2008 Jan;22(1):9–18. doi: 10.1096/fj.07-8945com. This study demonstrates the role of uncoupling protein-2 in facilitating the switch from fatty acid oxidation to glucose metabolism in proliferating cells.
- 9. Derdak Z, Mark NM, Beldi G, Robson SC, Wands JR, Baffy G. The mitochondrial uncoupling protein-2 promotes chemoresistance in cancer cells. Cancer Res. 2008 Apr 15;68(8):2813–2819. doi: 10.1158/0008-5472.CAN-08-0053. This study investigates the molecular basis for mitochondrial uncoupling protein-2 to promote chemoresistance in cancer cells.
- 10.Ramanathan A, Wang C, Schreiber SL. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci U S A. 2005 Apr 26;102(17):5992–5997. doi: 10.1073/pnas.0502267102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006 Aug 7;25(34):4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 12.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000 Jul 21;275(29):21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 13.Guppy M, Greiner E, Brand K. The role of the Crabtree effect and an endogenous fuel in the energy metabolism of resting and proliferating thymocytes. Eur J Biochem. 1993 Feb 15;212(1):95–99. doi: 10.1111/j.1432-1033.1993.tb17637.x. [DOI] [PubMed] [Google Scholar]
- 14.Bui T, Thompson CB. Cancer's sweet tooth. Cancer Cell. 2006 Jun;9(6):419–420. doi: 10.1016/j.ccr.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 15.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007 Dec 4;104(49):19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006 Aug 7;25(34):4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001 Jun 1;15(11):1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, et al. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007 Jun;27(12):4328–4339. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003 Aug 21;424(6951):952–956. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- 20.Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003 Oct;23(20):7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004 Nov;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 22.Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004 Dec;84(6):1014–1020. doi: 10.1016/j.ygeno.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 23.Boag JM, Beesley AH, Firth MJ, Freitas JR, Ford J, Hoffmann K, et al. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia. 2006 Oct;20(10):1731–1737. doi: 10.1038/sj.leu.2404365. [DOI] [PubMed] [Google Scholar]
- 24.Kim JH, Chu SC, Gramlich JL, Pride YB, Babendreier E, Chauhan D, et al. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005 Feb 15;105(4):1717–1723. doi: 10.1182/blood-2004-03-0849. [DOI] [PubMed] [Google Scholar]
- 25. Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M. The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation. Cancer Res. 2008 Jul 1;68(13):5198–5205. doi: 10.1158/0008-5472.CAN-08-0555. Demonstrates how leukemia cells co-cultured with bone marrow stromal cells up-regulate expression of uncoupling protein 2 correlating with enhanced aerobic glycolysis.
- 26.Durie BG, Waxman AD, D'Agnolo A, Williams CM. Whole-body (18)F-FDG PET identifies high-risk myeloma. J Nucl Med. 2002 Nov;43(11):1457–1463. [PubMed] [Google Scholar]
- 27.Bredella MA, Steinbach L, Caputo G, Segall G, Hawkins R. Value of FDG PET in the assessment of patients with multiple myeloma. AJR Am J Roentgenol. 2005 Apr;184(4):1199–1204. doi: 10.2214/ajr.184.4.01841199. [DOI] [PubMed] [Google Scholar]
- 28.Hedeskov CJ. Early effects of phytohaemagglutinin on glucose metabolism of normal human lymphocytes. Biochem J. 1968 Nov;110(2):373–380. doi: 10.1042/bj1100373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roos D, Loos JA. Changes in the carbohydrate metabolism of mitogenically stimulated human peripheral lymphocytes. II. Relative importance of glycolysis and oxidative phosphorylation on phytohaemagglutinin stimulation. Exp Cell Res. 1973 Mar 15;77(1):127–135. doi: 10.1016/0014-4827(73)90561-2. [DOI] [PubMed] [Google Scholar]
- 30. Hutchings M, Specht L. PET/CT in the management of haematological malignancies. Eur J Haematol. 2008 May;80(5):369–380. doi: 10.1111/j.1600-0609.2008.01051.x. Describes the utility of PET in defining staging, prognosis and response criteria of specific hematological malignancies.
- 31. Raffaghello L, Lee C, Safdie FM, Wei M, Madia F, Bianchi G, et al. Starvation-dependent differential stress resistance protects normal but not cancer cells against high-dose chemotherapy. Proc Natl Acad Sci U S A. 2008 Jun 17;105(24):8215–8220. doi: 10.1073/pnas.0708100105. This article demonstrates that glucose deprivation protects normal glia cells but not glioma cell lines from etoposide- and hydrogen peroxide-induced cell death.
- 32. Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin Investig Drugs. 2008 Oct;17(10):1533–1545. doi: 10.1517/13543784.17.10.1533. This excellent review details the roles of key enzymes in the glycolytic pathway and the development of small molecule inhibitors of these enzymes for cancer therapy.
- 33.Nista A, De Martino C, Malorni W, Marcante ML, Silvestrini B, Floridi A. Effect of lonidamine on the aerobic glycolysis of normal and phytohemagglutinin-stimulated human peripheral blood lymphocytes. Exp Mol Pathol. 1985 Apr;42(2):194–205. doi: 10.1016/0014-4800(85)90027-9. [DOI] [PubMed] [Google Scholar]
- 34.Floridi A, Paggi MG, D'Atri S, De Martino C, Marcante ML, Silvestrini B, et al. Effect of lonidamine on the energy metabolism of Ehrlich ascites tumor cells. Cancer Res. 1981 Nov;41(11 Pt 1):4661–4666. [PubMed] [Google Scholar]
- 35.Bustamante E, Pedersen PL. Mitochondrial hexokinase of rat hepatoma cells in culture: solubilization and kinetic properties. Biochemistry. 1980 Oct 28;19(22):4972–4977. doi: 10.1021/bi00563a006. [DOI] [PubMed] [Google Scholar]
- 36.Oudard S, Carpentier A, Banu E, Fauchon F, Celerier D, Poupon MF, et al. Phase II study of lonidamine and diazepam in the treatment of recurrent glioblastoma multiforme. J Neurooncol. 2003 May;63(1):81–86. doi: 10.1023/a:1023756707900. [DOI] [PubMed] [Google Scholar]
- 37.De Lena M, Lorusso V, Latorre A, Fanizza G, Gargano G, Caporusso L, et al. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur J Cancer. 2001 Feb;37(3):364–368. doi: 10.1016/s0959-8049(00)00400-7. [DOI] [PubMed] [Google Scholar]
- 38.Berruti A, Bitossi R, Gorzegno G, Bottini A, Alquati P, De Matteis A, et al. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: final results of a phase III study with a factorial design. J Clin Oncol. 2002 Oct 15;20(20):4150–4159. doi: 10.1200/JCO.2002.08.012. [DOI] [PubMed] [Google Scholar]
- 39. Hulleman E, Kazemier KM, Holleman A, VanderWeele DJ, Rudin CM, Broekhuis MJ, et al. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood. 2009 Feb 26;113(9):2014–2021. doi: 10.1182/blood-2008-05-157842. This article demonstrates that glucocorticoid-resistant ALL cell lines exhibit higher rates of glucose consumption and that these cell lines as well as resistant primary ALL cells are sensitized to prednisolone by lonidamine or 2-DG co-treatments.
- 40.Raez LE, Langmuir VK, Papadopoulus K, Ricart A, Rocha-Lima CM, Rosenblatt J, et al. Phase I trial of glycolitic inhibition with 2-deoxyglucose and docetaxel for patients with solid tumors. AACR Meeting Abstracts. 2006 April 1;2006(1):121-d-2. 2006. [Google Scholar]
- 41.Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF, e, et al. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004 Jan 1;64(1):31–34. doi: 10.1158/0008-5472.can-03-3294. [DOI] [PubMed] [Google Scholar]
- 42.Zhong D, Liu X, Schafer-Hales K, Marcus AI, Khuri FR, Sun SY, et al. 2-Deoxyglucose induces Akt phosphorylation via a mechanism independent of LKB1/AMP-activated protein kinase signaling activation or glycolysis inhibition. Mol Cancer Ther. 2008 Apr;7(4):809–817. doi: 10.1158/1535-7163.MCT-07-0559. [DOI] [PubMed] [Google Scholar]
- 43.Dearling JL, Qureshi U, Begent RH, Pedley RB. Combining radioimmunotherapy with antihypoxia therapy 2-deoxy-D-glucose results in reduction of therapeutic efficacy. Clin Cancer Res. 2007 Mar 15;13(6):1903–1910. doi: 10.1158/1078-0432.CCR-06-2094. [DOI] [PubMed] [Google Scholar]
- 44.Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007 Nov;27(21):7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006 May;117(5):1519–1531. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- 46. Jacques Pouyssegur JC, Karine IIc, Renaud LeFloch, Daniele Roux, Christiane Brahimi-Horn, Nathalie MMazure, editors. Carbonic Anydrases and MCTs. Denver, CO. Philadelphia (PA): AACR; 2009. Apr, Tumor cell resistance to hypoxic and acidic stresses. Key roles of HIF-induced BNIPs; pp. 18–22. This study shows that simultaneous inhibition of lactate transporters MCT-1 and MCT-4 blocks tumor growth and that HIF1α-mediated upregulation of MCT-4 promotes tumor cell chemoresistance.
- 47. Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008 Dec;118(12):3930–3942. doi: 10.1172/JCI36843. The authors of this study propose a fascinating model of whole-tumor metabolism involving lactate exported by hypoxic tumor cells being utilized as a fuel source for well-oxygenated cells.
- 48.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006 Jul;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005 Jan 28;120(2):237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]