

Abstract
Mitochondria are a primary source as well a principal target of reactive oxygen species within cells. Using immunofluorescence microscopy, we have found that a number of mitochondrial matrix proteins are normally undetectable in formaldehyde fixed cells permeabilized with the cholesterol-binding detergent saponin. However, exogenous or endogenous oxidative stress applied prior to fixation altered the permeability of mitochondria, rendering these matrix proteins accessible to antibodies. Electron microscopy revealed a loss of matrix density and disorganization of inner-membrane cristae upon oxidative stress. Notably, the changes in permeability and in structure were rapidly reversed when the oxidative stress was relieved. The ability of reactive oxygen species to reversibly alter the permeability of the mitochondrial membrane provides a potential mechanism for communication within the cell such as between nucleus and mitochondria.
Keywords: Mitochondria, Oxidative stress, Permeability, Mitochondrial matrix, Immunofluorescence microscopy, Electron microscopy, Detergents
1. Introduction
Translocation of proteins from one cellular site to another is an important mechanism in cellular regulation. Various stimuli can trigger the movement of proteins through a variety of processes, including receptor-mediated endocytosis, nuclear import and export, and reversible covalent modifications such as phosphorylation and myristoylation. Proteins destined for mitochondria are usually imported cotranslationally, but posttranslational movement of proteins to mitochondria has been documented (Zha et al., 2000). Oxidative stress is capable of triggering translocation of proteins to mitochondria (Orsini et al., 2004; Li et al., 2005; Noh et al., 2009). The methionine sulfoxide reductases are antioxidant enzymes which are present in most organisms, from microbes to plants and animals (Weissbach et al., 2005). They can reduce free or protein bound methionine sulfoxide back to methionine, thus functioning both in repair of oxidatively damaged molecules and as scavengers of reactive oxygen and nitrogen species (Stadtman et al., 2002). In mammals, methionine sulfoxide reductase A (MsrA) is encoded by a single gene but is present both in the cytosol and the mitochondria (Hansel et al., 2002; Vougier et al., 2003). In the course of our studies on the subcellular localization of MsrA, we observed that when cells were treated with exogenous oxidants such as hydrogen peroxide, cytosolic MsrA appeared to physically translocate from the cytosol to mitochondria, as assayed by immunofluorescence microscopy. Subsequent analyses revealed that the apparent translocation was in fact due to the exposure of previously hidden epitopes on mitochondrial matrix-localized MsrA. This effect was not limited to MsrA, but could be demonstrated with a number of different matrix proteins. We also found that the effect was fully reversible, indicating that mitochondria have the capacity to reverse the oxidant-induced changes that altered their accessibility and morphology.
2. Materials and Methods
2.1 Cell lines and reagents
Mouse embryonic fibroblasts (MEF) from transgenic mice over-expressing MsrA were kindly provided by Hang Zhao (Laboratory of Biochemistry, National Heart, Lung, and Blood Institute). Generation of these mice will be described elsewhere. Early-to-middle passage (6-20) cells were grown in Dulbecco's modified Eagle's medium containing 4.5 g/L glucose, supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine, and penicillin and streptomycin and maintained in a humidified 5% CO2 atmosphere. Glucose oxidase (Type X-S from Aspergillus niger; # G7141), rotenone, and antimycin A were from Sigma.
2.2 Antibodies and immunofluorescence labeling
Polyclonal antiserum against MsrA was generated from rabbits immunized with recombinant mouse MsrA purified from E. coli. This recombinant MsrA lacked the N-terminal 20 amino acids that encode the mitochondrial leader peptide. A monoclonal antibody against cytochrome c was from BD Pharmingen (# 556432) and used at 1:500 dilution. Anti-Mitochondrial Heat Shock Protein 70 monoclonal antibody was from Affinity Bioreagents (now Thermo Scientific; # MA3-028) and diluted 1:250. Alexa Fluor 488- and 594-conjugated species-specific secondary antibodies were from Invitrogen and used at a 1:500 dilution.
Cells were grown on 12 mm glass coverslips until approximately 50-75% confluent and treated as indicated. They were directly fixed for 20-30 min at room temperature with 2% formaldehyde in phosphate buffered saline (PBS). Fixation with 3.7% formaldehyde in PBS or 3.7% formaldehyde in 0.1 M sodium phosphate, pH 7.2, 150 mM NaCl gave similar results. After fixation, cells were rinsed three times with 10% FBS in PBS. Some cells were treated after fixation with 0.1% Triton X-100 for 1 min, followed by three rinses in PBS prior to rinsing in FBS/PBS. The cells were then incubated with primary antibody or serum diluted in 10% FBS/PBS containing 0.2% saponin for 1 hr at room temperature. The cells were then washed three times over a 15 min period with 10% FBS/PBS and incubated with fluorochrome-conjugated secondary antibodies (diluted in 10% FBS/PBS plus 0.2% saponin) for 1 hr at room temperature. Cells were again rinsed for 10 min with 10% FBS/PBS and one final rinse with PBS before mounting on a glass slide with Fluoromount G (Southern Biotech) containing 2.5 μg/mL DAPI (4′,6-diamidino-2-phenylindole) to stain nuclei. For treatment of cells with hydrogen peroxide, either a single bolus of hydrogen peroxide (250 μM) was added to cells or alternatively, hydrogen peroxide was generated enzymatically by glucose oxidase (25-125 mU/mL), which produces hydrogen peroxide in a more chronic manner via oxidation of glucose in the cell-culture medium. Similar results were observed for both.
Images were obtained using a LSM 510 confocal microscope (Carl Zeiss, Thornwood, NY) with a 63× 1.3 numerical aperture PlanApo oil-immersion objective. Optical sections were less than 1.5 μm thick. After acquisition, images were handled using Adobe Photoshop (Adobe Systems).
2.3 Cell Fractionation and Western blotting
MEFs were swollen in ice-cold hypotonic buffer (10 mM Hepes, pH 7.5, 5 mM MgCl2, 40 mM KCl, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail) for 30 min. Cells were disrupted by passage 5 times through a 26 gauge needle. The broken cells were centrifuged at 800 × g for 8 min to separate the pellet and the supernatant. The supernatant was removed carefully without disrupting the pellet and centrifuged at 9,000 × g for 15 min. The resulting supernatant was saved as the cytosolic fraction. The high spin pellet was washed twice with PBS for the mitochondrial fraction. Both fractions were separated by SDS-PAGE (reducing), followed by heating at 95°C for 5 min. Electrophoresis was performed on 10-20% Tris-Glycine gels (Invitrogen). Proteins were transferred onto nitrocellulose membranes (Invitrogen), and incubated with primary [MsrA, mHSP70, and α-tubulin (Santa Cruz)] and secondary [Alexa Fluor 680 goat anti-rabbit IgG (Invitrogen, A21109) and IRDye800CW (Rockland)] antibodies for 1 hr at room temperature. Membranes were washed three times with 0.1% Tween 20 in 1 × PBS, then visualized by scanning with an Odyssey infrared scanner with 700 and 800 nm channels (LI-COR Biosciences, Lincoln, NE).
2.4 Transmission electron microscopy
Cells were grown on 60 mm Permanox dishes (Nunc Nalgene) until approximately 75-90% confluent, treated as indicated, then directly fixed in 2.5% glutaraldehyde, 1% paraformaldehyde in 0.12 M sodium cacodylate buffer, pH 7.4. Cells were fixed for 30 min at room temperature, and then kept at 4°C overnight in fixative. Cells were post-fixed with osmium tetroxide, stained en bloc with uranyl acetate, ethanol dehydrated, and Epon embedded. Chemicals were from Electron Microscopy Sciences. Thin sections were cut parallel to the adherent surface, stained with uranyl acetate and lead citrate, and viewed with a JEM-1200EX electron microscope (JEOL USA) equipped with an AMT XR-60 digital camera (Advanced Microscopy Techniques).
3. Results
3.1 Apparent translocation of MsrA
While investigating the distribution of MsrA in mouse embryonic fibroblasts (MEFs) derived from transgenic mice over-expressing wild type mouse MsrA, we noticed a striking difference in its localization by immunofluorescence microscopy which depended on whether saponin (0.2%) or Triton X-100 (0.1%) was used as a permeabilizing agent on formaldehyde-fixed cells. Fig. 1A shows staining using antibodies to MsrA and to the mitochondrial intermembrane space protein cytochrome c. Specific staining of cytochrome c was evident in both punctate and tubular structures that colocalized with MitoTracker dyes, confirming their identity as mitochondria. Surprisingly, in cells fixed and permeabilized with the cholesterol-binding detergent saponin, only cytosolic MsrA could be observed, even though cell fractionation clearly identified a pool of MsrA in the mitochondrial fraction by western blotting (Fig. 2). The same lack of mitochondrial staining was also observed with antibodies to matrix proteins mtHSP70 (Fig. 1B) and ornithine transcarbamylase (data not shown). Since various post-fixation detergent treatments can unmask hidden epitopes in various organelles, such as the nucleus and mitochondria (Goldenthal et al., 1985), cells were permeabilized after fixation with the non-ionic detergent Triton X-100. Under these conditions, both MsrA and mtHSP70 (and ornithine transcarbamylase) were now readily detected in mitochondria (Fig 1C). Thus, epitope inaccessibility in the presence of saponin limits detection of mitochondrial matrix proteins, including MsrA. These results also confirm the sub-mitochondrial localization of MsrA to the matrix, as previously reported (Vougier et al., 2003)
Fig. 1.

Immunofluorescence of untreated MEFs expressing mouse MsrA. Cells were fixed in formaldehyde/PBS followed by permeabilization with saponin. Staining was with antibodies to MsrA and cytochrome c (A) or MsrA and mtHSP70 (B). Note that MsrA is restricted to the cytosol in saponin permeabilized cells and mtHSP70 is not detectable. In (C) cells were post-fixed with 0.1% Triton X-100 for 1 min prior to antibody staining. Both MsrA and mtHSP70 are visible in mitochondria. Bar = 10 μm.
Fig. 2.
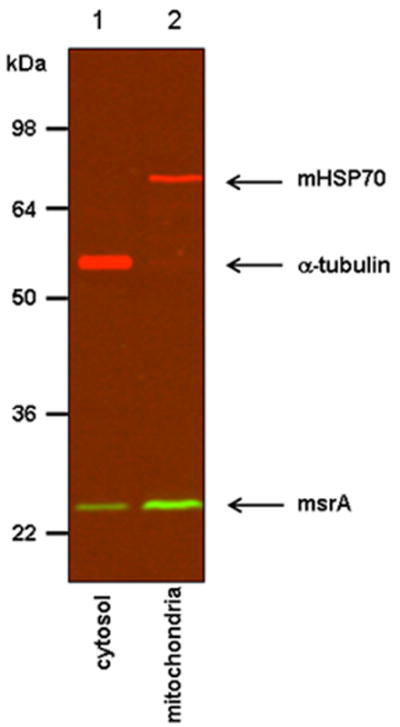
Fractionation of MEFs expressing mouse MsrA. MsrA was detected both in the cytosol (lane 1) and in the mitochondrial fraction (lane 2). The cytosolic marker (α-tubulin) and the mitochondrial marker protein (mtHSP70) are indicated and demonstrate the lack of cross-contamination between compartments
Surprisingly, when cells were incubated with exogenous oxidants such as hydrogen peroxide prior to fixation, MsrA was now detected in mitochondria with saponin permeabilization (Fig. 3A). Under these conditions, both cytosolic and mitochondrial MsrA distribution patterns were observed, consistent with its dual localization. Oxidant-induced epitope unmasking was not limited to MsrA but was also observed with a number of mitochondrial proteins normally found in the matrix, including mtHSP70 (Fig. 3B) and ornithine transcarbamylase. Thus, exposure of cells to hydrogen peroxide has the same effect on antibody access to matrix antigens as does post-fixation membrane permeabilization with Triton X-100. Similar effects were observed in transiently transfected COS and HEK293 cells, although low levels of MsrA, but not mtHSP70, could be detected in cells permeabilized with saponin (data not shown). In many cells, the levels of cytosolic MsrA appeared reduced in the presence of hydrogen peroxide whereas mitochondrial staining increased, giving the impression of translocation of MsrA from the cytosol to mitochondria. However biochemical fractionation showed no change in MsrA distribution between cytosol and mitochondrial fractions (not shown). Thus, given the above results, it is likely that in addition to unmasking of mitochondrial epitopes, extraction of a portion of cytosolic MsrA occurred upon fixation and permeabilization. This has been described for other membrane (Hannah et al., 1998) and cytosolic (Cole et al., 2008) proteins in formaldehyde-fixed cells. Thus, extraction of cytosolic MsrA may be increased in cells treated with hydrogen peroxide.
Fig. 3.

Immunofluorescence of MEFs treated with 50 mU/mL glucose oxidase for 1 h before fixing with formaldehyde/PBS followed by permeabilization with saponin. Both MsrA and mtHSP70 are detected in mitochondria and co-localize with cytochrome c. Bar = 10 μm.
3.2 Effect of endogenously generated ROS
We next determined whether oxidants generated within mitochondria had a similar effect of exposing matrix epitopes as did treatment with exogenous hydrogen peroxide. We tested the effects of rotenone and antimycin A, two electron transport inhibitors that increase the production of superoxide, primarily at electron transport complexes I and III, respectively. The superoxide is then quickly dismutated by the mitochondrial superoxide dismutase (Mn-SOD) to hydrogen peroxide (Loschen et al., 1974; Boveris and Cadenas, 1975; Gyulkhandanyan and Pennefather, 2004; Balaban et al., 2005). Exposure of MEFs to rotenone (Fig. 4A) or antimycin (Fig. 4B) for 3 hr resulted in detection of MsrA within mitochondria, duplicating the effect of exogenous hydrogen peroxide. We have not experimentally investigated the reason for the longer incubation time required with rotenone compared to exogenous hydrogen peroxide. Notably, not all of the mitochondria were similarly affected, and a number of cytochrome c positive mitochondria remained negative for MsrA. This indicates substantial heterogeneity in the response of individual mitochondria to electron transport inhibitors and/or production of ROS. It is also noteworthy that the mitochondria under these conditions were generally fragmented and rounded; this fragmentation process occurred prior to the unmasking of matrix epitopes, suggesting that alterations of the mitochondrial fission/fusion machinery are early effects of drug treatment (Sandebring et al., 2009). It is important to note that the MEFs remained viable at all time points analyzed. Hydrogen peroxide or electron chain inhibitor treatment did not result in subsequent cell death by apoptosis or necrosis when followed for 24 h, as assayed by the retention of cytochrome c within mitochondria and normal nuclei visualized by DAPI staining.
Fig. 4.

Immunofluorescence of MEFs treated with (A) 100 ?M rotenone or (B) 100 μM antimycin A for 3 h before fixation with formaldehyde/PBS followed by permeabilization with saponin. Note that not all mitochondria are labeled with antibodies to MsrA. Bar = 10 μm
3.3 The permeability changes are reversible
We next asked if the exposure of matrix epitopes under conditions of oxidative stress was reversed when the stress was removed. Cells treated with hydrogen peroxide were rinsed several times in medium without oxidant and incubated for various time periods before fixation. We found that as little as 1 h recovery rendered mitochondrial matrix proteins such as MsrA and HSP 70 generally undetectable by antibodies in cells treated with saponin after fixation (but not Triton X-100), although some residual mitochondrial staining remained in some cells (Fig. 5). Recovery was also observed in cells treated with rotenone and antimycin A, although in these cases mitochondria remained rounded upon drug washout (data not shown). This suggests that alterations in the mitochondrial fission/fusion machinery may be more sensitive to oxidative damage (Dagda et al., 2009) or do not recover as rapidly as the mitochondrial inner membrane and matrix. We have not measured mitochondrial membrane potential under these conditions to determine whether the recovery seen morphologically correlates with functional repair.
Fig. 5.

Immunofluorescence of MEFs from untreated (top panel), glucose oxidase-treated (middle panel), and cells in which glucose oxidase was washed out and were allowed to recover for 1h. Although some mitochondrial staining remains in washout cells, there is a general loss of ability to detect mitochondria-localized MsrA in post-fixed cells permeabilized with saponin, similar to untreated cells. Bar = 10 μm
3.4 Electron microscopic visualization of mitochondria
We also performed an ultrastructural examination of control and hydrogen peroxide-treated cells. Mitochondria from untreated cells contained a dense matrix and well-organized cristae, predominantly oriented transverse to the long axis of the mitochondria (Fig 6A). Mitochondria from cells treated with glucose oxidase appeared swollen with decreased electron density within the matrix. The cristae were noticeably thinner and often misoriented and/or decreased in number (Fig. 6B, which shows a range of mitochondrial phenotypes). The swollen nature of the mitochondria suggests that osmotic effects may be one cause of this morphology. In addition, intramitochondrial amorphous deposits that likely represent matrix precipitates and degenerating cristae, that is, flocculent or “woolly” densities, were common (arrows) (Costa et al., 1990; Chiba et al., 2005; Oberley et al., 2008). The outer and inner mitochondrial membranes appeared intact in hydrogen peroxide-treated cells, with no gross morphological alterations that would suggest easy access of antibodies to matrix antigens.
Fig. 6.

Representative electron micrographs from untreated (A), glucose oxidase-treated (B), and glucose-oxidase recovered (C) cells. Note the loss of matrix density as well as narrower, mis-orientated cristae that are reduced in number in glucose oxidase treated cells. Also note the general recovery of mitochondrial structure when glucose oxidase is washed out. Arrows indicate flocculent or woolly densities. In (B), note that the section is tangential to portions of the membranes of the two mitochondria at center and lower left such that the mitochondria appear to overlap with adjacent endoplasmic reticulum. Cells were treated identically as those shown in Fig. 5. Bar = 100 nm.
Recovery was also evident at the ultrastructural level, with the reappearance of an electron dense matrix and normal cristae after 1 h washout of glucose oxidase (fig., 6C). Remnants of flocculent densities could still be observed (see Fig. 6C, see arrows), although they were not as prominent, suggesting that mitochondria have the capacity to degrade misfolded or aggregated matrix proteins and oxidized lipids.
Discussion
In this study, we observed an unexpected effect of exogenous or endogenous ROS on mitochondria. Exposure to ROS caused reversible permeability changes in mitochondria, which rendered accessible normally hidden epitopes of matrix proteins. Had we relied solely on immunohistochemical localizations, we would have concluded that MsrA reversibly translocated from the cytosol to mitochondria under oxidative stress. Such translocation has been demonstrated for p66Shc, DJ-1, and sulfiredoxin (Orsini et al., 2004; Li et al., 2005; Noh et al., 2009). However, biochemical fractionation failed to confirm translocation of MsrA. This discrepancy prompted additional investigations to elucidate the basis of the discordance. While we found that it resulted from a permeabilization difference in fixed cells, the changes in mitochondrial structure and function may have relevance in aging or pathological conditions in which ROS production is increased (Costa et al., 1990; Chiba et al., 2005; Oberley et al., 2008).
The reversibility of the alterations induced by oxidative stress deserve underscoring; they are almost completely reversed within 1 h. While mitochondria remained somewhat rounded and dispersed after rotenone or antimycin A washout, the lack of antibody accessibility suggests that their inner morphology had already been repaired.
What might be the mechanistic basis for the permeabilizing effect of reactive oxygen species? After fixation, cellular membranes are commonly permeabilized for immunohistochemistry by detergents. Saponins are amphipathic glycosides that intercalate into membranes that contain cholesterol, forming 12-15 nm diameter holes that enable macromolecules, such as antibodies to penetrate. In contrast, the non-ionic detergent Triton X-100 (0.1%) removes the cell's phospholipid barriers and renders all membrane-bound compartments, including mitochondria and the nucleus permeable (Willingham, 1999).
Compared to other cellular membranes, mitochondrial membranes are cholesterol-poor, containing only 0.5-3% of that in typical membranes (van Meer G. et al., 2008), and most of the cholesterol is found in the outer mitochondrial membrane (Boyer et al., 1994; Stocco, 2002). This likely explains the failure to detect MsrA, mtHSP70, and other matrix proteins in fixed cells treated with saponin. It does not explain the ability to detect these proteins with saponin after hydrogen peroxide treatment because it is unlikely that cholesterol migrates from the outer to inner membrane under these conditions. We speculate that oxidative modification of the inner membrane of mitochondria is responsible for the increased permeability. The reduced number of cristae and their misorientation after hydrogen peroxide treatment is consistent with alteration of the inner membrane. Functional changes likely parallel these structural changes, and may alter the flux of ions, particularly potassium, across the inner mitochondrial membrane, resulting in osmotic dysregulation and matrix swelling.
The increased, reversible permeability changes of mitochondria may have biological significance. It seems reasonable to consider that molecular messages may be sent to and from the mitochondria during the period of enhanced permeability. For example, conditions that cause increased production of ROS within mitochondria may permit signaling proteins, peptides, or other molecules to move from the mitochondria to the nucleus. Exogenous oxidative stresses could similarly facilitate movement of signaling molecules from the nucleus to the mitochondria. Such communication does occur, although the molecular basis is not well-defined (Passos and Von, 2006). Chronic or excessive oxidative stress may induce an excessive or prolonged increase in mitochondrial permeability. Abnormal regulation of mitochondrial membrane permeability could thus occur during aging. These are speculations, but ones worthy of experimental investigation.
Acknowledgments
We thank Hang Zhao (National Heart, Lung, and Blood Institute) for generation of MEFs and Patricia S. Connelly (National Heart, Lung, and Blood Institute) for technical assistance with the electron microscopy. This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ. Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science. 2000;290:1761–1765. doi: 10.1126/science.290.5497.1761. [DOI] [PubMed] [Google Scholar]
- 2.Orsini F, Migliaccio E, Moroni M, Contursi C, Raker VA, Piccini D, Martin-Padura I, Pelliccia G, Trinei M, Bono M, Puri C, Tacchetti C, Ferrini M, Mannucci R, Nicoletti I, Lanfrancone L, Giorgio M, Pelicci PG. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J Biol Chem. 2004;279:25689–25695. doi: 10.1074/jbc.M401844200. [DOI] [PubMed] [Google Scholar]
- 3.Li HM, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radic Res. 2005;39:1091–1099. doi: 10.1080/10715760500260348. [DOI] [PubMed] [Google Scholar]
- 4.Noh YH, Baek JY, Jeong W, Rhee SG, Chang TS. Sulfiredoxin Translocation into Mitochondria Plays a Crucial Role in Reducing Hyperoxidized Peroxiredoxin III. J Biol Chem. 2009;284:8470–8477. doi: 10.1074/jbc.M808981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissbach H, Resnick L, Brot N. Methionine sulfoxide reductases: history and cellular role in protecting against oxidative damage. Biochim Biophys Acta. 2005;1703:203–212. doi: 10.1016/j.bbapap.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Stadtman ER, Moskovitz J, Berlett BS, Levine RL. Cyclic oxidation and reduction of protein methionine residues is an important antioxidant mechanism. Mol Cell Biochem. 2002;234-235:3–9. [PubMed] [Google Scholar]
- 7.Hansel A, Kuschel L, Hehl S, Lemke C, Agricola HJ, Hoshi T, Heinemann SH. Mitochondrial targeting of the human peptide methionine sulfoxide reductase (MSRA), an enzyme involved in the repair of oxidized proteins. FASEB J. 2002;16:911–913. doi: 10.1096/fj.01-0737fje. [DOI] [PubMed] [Google Scholar]
- 8.Vougier S, Mary J, Friguet B. Subcellular localization of methionine sulphoxide reductase A (MsrA): evidence for mitochondrial and cytosolic isoforms in rat liver cells. Biochem J. 2003;373:531–537. doi: 10.1042/BJ20030443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldenthal KL, Hedman K, Chen JW, August JT, Willingham MC. Postfixation detergent treatment for immunofluorescence suppresses localization of some integral membrane proteins. J Histochem Cytochem. 1985;33:813–820. doi: 10.1177/33.8.3894499. [DOI] [PubMed] [Google Scholar]
- 10.Hannah MJ, Weiss U, Huttner WB. Differential extraction of proteins from paraformaldehyde-fixed cells: lessons from synaptophysin and other membrane proteins. Methods. 1998;16:170–181. doi: 10.1006/meth.1998.0664. [DOI] [PubMed] [Google Scholar]
- 11.Cole NB, Dieuliis D, Leo P, Mitchell DC, Nussbaum RL. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp Cell Res. 2008;314:2076–2089. doi: 10.1016/j.yexcr.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loschen G, Azzi A, Richter C, Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974;42:68–72. doi: 10.1016/0014-5793(74)80281-4. [DOI] [PubMed] [Google Scholar]
- 13.Boveris A, Cadenas E. Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett. 1975;54:311–314. doi: 10.1016/0014-5793(75)80928-8. [DOI] [PubMed] [Google Scholar]
- 14.Gyulkhandanyan AV, Pennefather PS. Shift in the localization of sites of hydrogen peroxide production in brain mitochondria by mitochondrial stress. J Neurochem. 2004;90:405–421. doi: 10.1111/j.1471-4159.2004.02489.x. [DOI] [PubMed] [Google Scholar]
- 15.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 16.Sandebring A, Thomas KJ, Beilina A, van der BM, Cleland MM, Ahmad R, Miller DW, Zambrano I, Cowburn RF, Behbahani H, Cedazo-Minguez A, Cookson MR. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS One. 2009;4:e5701. doi: 10.1371/journal.pone.0005701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dagda RK, Cherra SJ, III, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Costa M, Shute B, Mergner WJ. Measurement of ATP synthesis and flocculent matrix densities in mitochondria as a function of ‘in vitro’ ischemia in the heart and liver of rats. Pathobiology. 1990;58:129–137. doi: 10.1159/000163574. [DOI] [PubMed] [Google Scholar]
- 19.Chiba Y, Yamashita Y, Ueno M, Fujisawa H, Hirayoshi K, Hohmura K, Tomimoto H, Akiguchi I, Satoh M, Shimada A, Hosokawa M. Cultured murine dermal fibroblast-like cells from senescence-accelerated mice as in vitro models for higher oxidative stress due to mitochondrial alterations. J Gerontol A Biol Sci Med Sci. 2005;60:1087–1098. doi: 10.1093/gerona/60.9.1087. [DOI] [PubMed] [Google Scholar]
- 20.Oberley TD, Swanlund JM, Zhang HJ, Kregel KC. Aging results in increased autophagy of mitochondria and protein nitration in rat hepatocytes following heat stress. J Histochem Cytochem. 2008;56:615–627. doi: 10.1369/jhc.2008.950873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willingham MC. Fluorescence labeling of intracellular antigens of attached or suspended tissue-culture cells. Methods Mol Biol. 1999;115:121–30. 121–130. doi: 10.1385/1-59259-213-9:121. [DOI] [PubMed] [Google Scholar]
- 22.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyer CS, Neve EP, Moore GA, Moldeus P. Effect of mitochondrial protein concentration on the efficiency of outer membrane removal by the cholesterol-selective detergent digitonin. Biochim Biophys Acta. 1994;1190:304–308. doi: 10.1016/0005-2736(94)90088-4. [DOI] [PubMed] [Google Scholar]
- 24.Stocco DM. Clinical disorders associated with abnormal cholesterol transport: mutations in the steroidogenic acute regulatory protein. Mol Cell Endocrinol. 2002;191:19–25. doi: 10.1016/s0303-7207(02)00048-5. [DOI] [PubMed] [Google Scholar]
- 25.Passos JF, Von ZT. Oxygen free radicals in cell senescence: are they signal transducers? Free Radic Res. 2006;40:1277–1283. doi: 10.1080/10715760600917151. [DOI] [PubMed] [Google Scholar]