

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit COX activity, reduce the production of retinal VEGF and neovascularization in relevant models of ocular disease. We hypothesized that COX-2 mediates VEGF production in retinal Müller cells, one of its primary sources in retinal neovascular disease. The purpose of this study was to determine the role of COX-2 and its products in VEGF expression and secretion. These studies have more clearly defined the role of COX-2 and COX-2-derived prostanoids in retinal angiogenesis.
Müller cells derived from wild-type and COX-2 null mice were exposed to hypoxia for 0–24 hours. COX-2 protein and activity were assessed by western blot analysis and GC-MS, respectively. VEGF production was assessed by ELISA. Wild-type mouse Müller cells were treated with vehicle (0.1% DMSO), 10 µM PGE2, or PGE2 + 5 µM H-89 (a PKA inhibitor), for 12 hours. VEGF production was assessed by ELISA.
Hypoxia significantly increased COX-2 protein (p ≤ 0.05) and activity (p ≤ 0.05), and VEGF production (p ≤ 0.0003). COX-2 null Müller cells produced significantly less VEGF in response to hypoxia (p ≤ 0.05). Of the prostanoids, PGE2 was significantly increased by hypoxia (p ≤ 0.02). Exogenous PGE2 significantly increased VEGF production by Müller cells (p ≤ 0.0039), and this effect was inhibited by H-89 (p ≤ 0.055).
These data demonstrate that hypoxia induces COX-2, prostanoid production, and VEGF synthesis in Müller cells, and that VEGF production is at least partially COX-2-dependent. Our study suggests that PGE2, signaling through the EP2 and/or EP4 receptor and PKA, mediates the VEGF response of Müller cells.
Keywords: angiogenesis, hypoxia, Müller cells, COX-2, prostanoids, VEGF
Introduction
Angiogenesis, the formation of new capillaries from the existing vasculature, is a process subject to exquisite regulation. Numerous pathological conditions are characterized by persistent, abnormal angiogenesis. In the eye, pathological angiogenesis, or ocular neovascularization (NV), is the leading cause of blindness in developed countries (Rahmani et al., 1999; Lee et al., 1998; Steinkuller et al., 1999). Prevalent diseases in which ocular NV is a central feature include retinopathy of prematurity (ROP), proliferative diabetic retinopathy (PDR), and age-related macular degeneration (AMD or ARMD). To effectively prevent and treat ocular NV, a more thorough understanding of the cellular and molecular mechanisms involved in the angiogenic process is necessary.
Neovascularization within the eye is often the result of ischemia, which induces tissue hypoxia (D’Amore, 1994; Casey and Li, 1997). In response to retinal hypoxia, various pro-angiogenic growth factors are produced, each mediating a number of angiogenic cell behaviors. Of the growth factors involved in retinal angiogenesis, vascular endothelial cell growth factor (VEGF) is thought to be a principal mediator (Aiello, 1997). In response to retinal hypoxia, several cell types exhibit increased VEGF production (Aiello et al., 1995; Pierce et al., 1995; Robbins et al., 1997). This has been demonstrated most consistently and dramatically in Müller cells, the predominant glial cells within the retina (Pierce et al., 1995; Robbins et al., 1997; Robbins et al., 1998). Notably, Müller cell-specific deletion of VEGF-A significantly inhibits NV in mice exposed to oxygen-induced retinopathy (Bai et al., 2009).
The cyclooxygenase (COX) enzymes are responsible for catalyzing the production of biologically active prostanoids (prostaglandins and thromboxanes) from phospholipid-derived arachidonic acid. Once the COX enzymes catalyze their production, prostanoids bind to their cognate G-protein-coupled receptors (GPCRs) on the surface of target cells. The receptors determine the extent and biological activity of the prostanoids. PGF2, PGI2, and TXA2 signal through the FP, IP, and TP receptors, respectively. PGD2 signals through the DP and CRTH2 receptors. PGE2 signals through the EP1, EP2, EP3, and EP4 receptors. Signaling through DP, IP, EP2, and EP4 stimulates the activation of adenylyl cyclase (AC), resulting in increased cyclic AMP (cAMP) production and PKA activation. Signaling through EP3 and CRTH2 results in reduced cAMP production. Signaling through FP, TP, and EP1 results in calcium mobilization (Hata and Breyer, 2004). The precise tissue-specific and cell-specific signaling pathways and the biological roles mediated by each of the prostanoid receptors have yet to be determined.
The cancer literature has demonstrated a role for COX-2 and its prostanoid metabolites in the angiogenesis that occurs during tumor growth (Kawamori et al., 1998; Williams et al., 1999; Form and Auerbach, 1983; Ziche et al., 1982). Specifically, prostanoids can induce the expression of pro-angiogenic factors such as VEGF and bFGF in a wide variety of cell types (Cheng et al., 1998; Pai et al., 2001), often in a PKA-dependent manner (Cheng et al., 1998; Höper et al., 1997; Amano et al., 2002).
COX-2 has been localized to various ocular tissues, and its expression has been found, or can be induced, in the following structures: cornea, iris, ciliary body, various cell types within the neurosensory retina, and the retinal pigment epithelium (RPE) (Yamada et al., 1999; Damm et al., 2001; Maihofner et al., 2001; Ju and Neufeld, 2002; Ershov and Bazan, 1999). The expression of the COX-2 enzyme in these ocular tissues suggests a functional role for its prostanoid products. In fact, inhibition of COX has been effective at reducing the production of VEGF and corneal, retinal, and choroidal NV in relevant models of ocular disease (Castro et al., 2004; Wilkinson-Berka et al., 2003; Sennlaub et al., 2003; Takahashi et al., 2004; Hu et al., 2005; Ayalasomayajula et al., 2003; Ayalasomayajula et al., 2004; Amrite et al., 2006; Yanni et al., 2010). However, although various groups have demonstrated the efficacy of COX inhibition at reducing the production of VEGF and NV, little work has been done to determine which of the COX-2 derived prostanoid(s) is (are) involved in mediating VEGF production, which stimulate(s) angiogenic endothelial cell behaviors, and results in ocular angiogenesis.
In this study, we used an in vitro assay of hypoxia-induced VEGF production combined with cells isolated from COX-2 null and wild-type mice to more precisely determine the role of COX-2 and its products in stimulating mouse Müller cell VEGF production. These studies are significant because VEGF is the growth factor that stimulates the pathological angiogenesis characteristic of ROP, PDR, AMD, and a number of other blinding conditions. These studies have further defined the roles of COX-2 and COX-2-derived prostanoids in this discrete aspect of retinal angiogenesis.
Materials and Methods
Isolation and Culture of Primary Mouse Müller Cells
Primary Müller cell cultures were established from P7 wild-type C57/129 mouse pups and COX-2 null mouse pups on the same background (breeders were a generous gift from Dr. Sudhansu Dey, Cincinnati Children's Hospital Medical Center), according to well-established methods (Hicks and Curtois, 1990). Briefly, enucleated eyes were placed in soaking medium, Dulbecco’s Modified Eagle Medium (DMEM; HyClone; Logan, UT) supplemented with 1X Antibiotic/Antimycotic Solution (Sigma; St. Louis, MO), overnight. The following day, eyes were incubated in digestion buffer, which is comprised of soaking medium plus 0.1% trypsin and 70 U/ml collagenase, for 20 minutes at 37°C. Retinas were then dissected, triturated, plated, and grown in DMEM supplemented with 10% fetal bovine serum and 1X Antibiotic/Antimycotic Solution. Cultures were maintained at 37°C in a 5% CO2/95% air (20.9% oxygen) atmosphere (normoxia) in a humidified incubator (NuAire; Plymouth, MN). The identification of Müller cells was confirmed by immunocytochemical staining with a monoclonal antibody against vimentin (Affinity Bioreagents; Golden, CO), an intermediate filament protein normally expressed in Müller cells, and with a monoclonal antibody against cellular retinaldehyde binding protein (CRALBP; Affinity Bioreagents). In order to eliminate RPE contamination, the following steps were taken: the neurosensory retinas were extruded from the eyecup, separating them from underlying tissues (RPE, choroid, sclera) at the time of isolation; residual RPE contamination was visualized and the RPE was removed at this stage; the cells were subjected to a brief spin (3,000 RPM for 5 minutes) causing the RPE to overlay the pellet created by neurosensory retinal cells, and the RPE were removed prior to plating. The use of CRALBP as a Müller cell marker is well-documented in the literature (Monnin et al., 2007; Kuzmanovic et al., 2003). Passages three to six were used for experiments. Normoxic and hypoxic conditions (< 1.0% oxygen) were generated with an Isotemp 3-gas Laboratory CO2 Incubator with O2 control (Kendro Laboratory Products; Asheville, NC) and a Proox Model 110 Gas Oxygen Controller (BioSpherix; Lacona, NY). Appropriate humidity and 5% CO2 were maintained.
Western Blot Analysis
Wild-type and COX-2 null mouse Müller cells at 70% subconfluency were exposed to hypoxia for 0, 2, 6, 12, 18, or 24 hours. The lysates were matched for protein concentration, and proteins were resolved by 10% SDS-PAGE minigels (Bio-Rad; Hercules, CA) and transferred to 0.2 µ M nitrocellulose membranes (Bio-Rad). Membranes were blocked in TBS containing 0.1% Tween-20 (Sigma) and 1% BSA (Sigma) overnight at 4°C. The blots were incubated with antibodies recognizing COX-1 and COX-2 (Cayman; Ann Arbor, MI) for one hour, followed by an anti-rabbit IgG HRP antibody (Promega; Madison, WI) for 45 minutes, at room temperature. Following thorough washings, the proteins were visualized with enhanced chemiluminiscence (ECL; Amersham; Piscataway, NJ). Membranes were stripped and reprobed for β-actin (Sigma). Digitized images of Western blots were quantified using Adobe Photoshop CS2. Raw densitometric values were normalized against internal control (β-actin).
Müller Cell Prostanoid Measurement
Wild-type mouse Müller cells at 70% subconfluency were treated with 0.5% serum medium and exposed to either normoxia or hypoxia for 0, 2, 6, 12, 18, or 24 hours. Conditioned medium was collected and the lipid soluble prostanoids were reverse-phase, solid-phase extracted using Sep-Pak C18 columns (Waters; Milford, MA) and nitrogen-evaporated. O-methoxyamine derivatives were formed by treatment with 2% methoxyamine-HCl in water at room temperature for 30 minutes. Compounds were extracted with ethyl acetate and subsequently converted to pentaflurobenzyl esters. The compounds were chromatographed on TLC plates with ethyl acetate/methanol. The compounds were then converted to trimethylsilyl ether derivatives and analyzed by negative ion chemical ionization mass spectrometry coupled with a gas chromatography system (Agilent Technologies; Palo Alto, CA). The amount of each prostanoid (ng/ml) in culture medium was normalized to total protein concentration (mg/ml) of cell lysates using a BCA assay (Pierce; Rockford, IL). Thus, any variation in prostanoid production due to differences in cell densities was resolved.
Hypoxic Induction of VEGF in Müller Cells
Wild-type and COX-2 null mouse Müller cells at 70% subconfluency were exposed to hypoxia for 0, 2, 6, 12, 18, or 24 hours. Culture medium from experimental dishes was collected and assayed for VEGF protein concentration with a colorimetric sandwich ELISA kit (R&D Systems; Minneapolis, MN) according to the manufacturer’s instructions. The assay recognizes the 164 amino acid residue splice variant of mouse VEGF. Cells were washed with cold calcium- and magnesium-free PBS (Invitrogen Corporation; Carlsbad, CA) and lysed with cold lysis buffer (Promega). The amount of VEGF (pg/ml) in culture medium was normalized to total protein concentration (mg/ml) of cell lysates using a BCA assay (Pierce). Thus, any variation in VEGF production due to differences in cell densities was resolved. Values were normalized to those of wild-type cells exposed to hypoxia for 2 hours.
PGE2 and H-89 Treatment of Müller Cells
Wild-type mouse Müller cells at 70% subconfluency were serum-starved for 6 hours, and then pre-treated with 5 µM H-89 (Cayman Chemical; Ann Arbor, MI) for 1 hour. Following pre-treatment, the cells received fresh serum-free medium with H-89, and were treated with 10 µM PGE2 (Cayman Chemical) for 12 hours. Culture medium from experimental dishes was collected and assayed for VEGF protein concentration as described above. Values were normalized to those of vehicle-treated cells.
Statistical Analysis
Data were analyzed with commercial software (JMP; SAS Institute; Cary, NC). Analysis of variance (ANOVA) with Dunnet’s post-hoc analyses and t tests were used to analyze parametric data.
Results
Effect of Hypoxia on COX-2 Protein and Activity
Wild-type mouse Müller cells exposed to hypoxia showed increased levels of the COX-2 protein (Figure 1). COX-2 protein was absent in the COX-2 null cells, as expected (Figure 1). The densitometry of the COX-2 bands (normalized against the densitometry of the β-actin bands) was quantified, and demonstrated that COX-2 was significantly (p ≤ 0.005) induced beginning 2 hours after hypoxia exposure and remained elevated through 18 hours. COX-2 was maximally and significantly increased after 6 and 12 hours in hypoxia (Figure 2). COX-1 is the isoform of COX classically recognized as being constitutively active (DuBois et al., 1998). Importantly, hypoxia did not lead to increased COX-1 in wild-type cells (Figure 1). Nor did hypoxia stimulate COX-1 in COX-2 null cells, indicating that COX-1 was not compensating for the absence of the COX-2 enzyme (Figure 1). Due to the fact that COX-2 is an enzyme, an increase in the level of the protein does not necessarily indicate that activity is increased. In order to assess COX-2 activity, we assessed the concentration of PGE2 in the conditioned medium from hypoxia-treated cells. We chose to look at PGE2 as a surrogate marker of COX-2 activity, because COX-2-derived PGE2 is the prostanoid product most consistently upregulated in angiogenic tumor models (Wang and DuBois, 2004). Consistent with an increase in COX-2 protein, COX-2 activity, as demonstrated by increased PGE2 production, is likewise significantly increased (* p ≤ 0.02; † p ≤ 0.05; ‡ p ≤ 0.05) by hypoxia (Figure 3).
Figure 1.
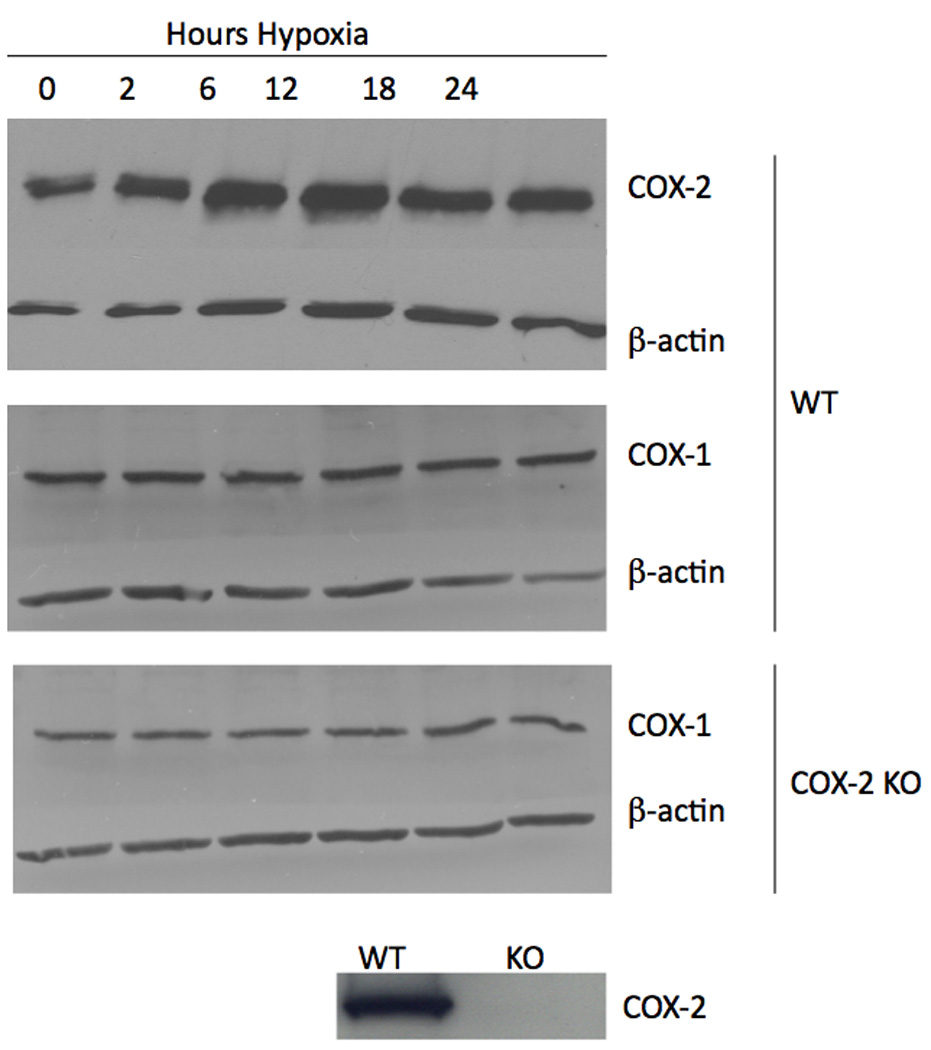
The effect of hypoxia on COX-1 and COX-2 in mouse Müller cells. Wild-type mouse Müller cells demonstrated an increase in COX-2 upon exposure to hypoxia. Neither wild-type nor COX-2 null mouse Müller cells demonstrated an increase in COX-1 upon exposure to hypoxia. Additionally, COX-1 did not appear to be compensating for genetic deletion of the COX-2 gene in null cells.
Figure 2.
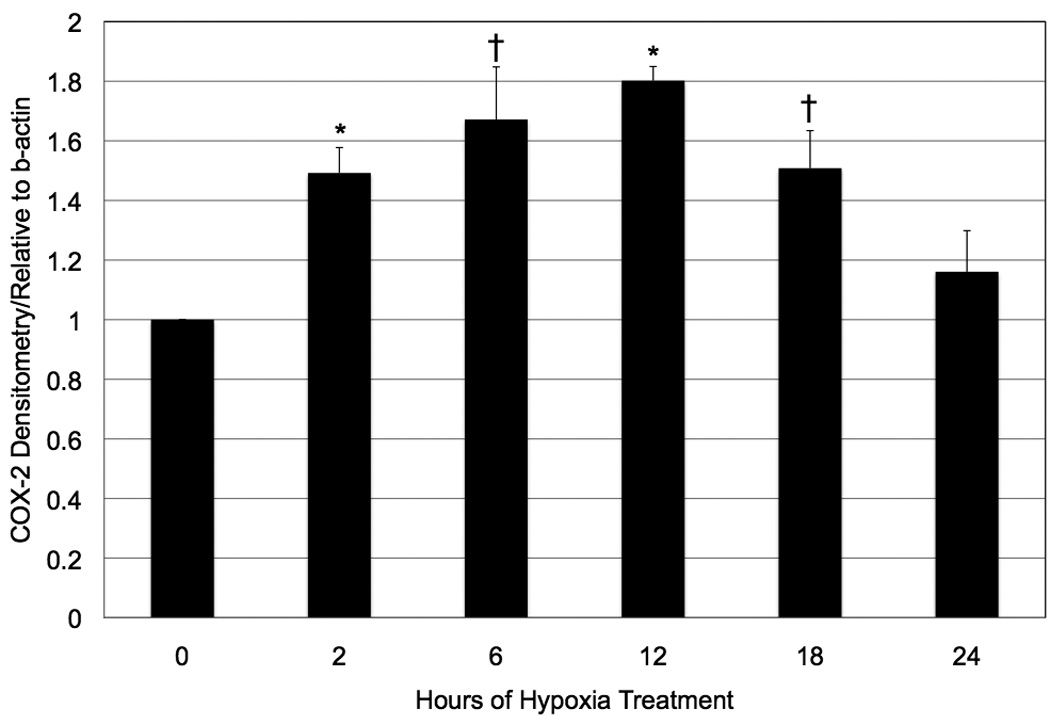
The effect of hypoxia on COX-2 in wild-type mouse Müller cells. COX-2 western blots were subjected to densitometric analysis. COX-2 (relative to beta-actin) was significantly increased after 2 hours of hypoxia, and remained elevated throughout 18 hours (* p < 0.001; † p < 0.005). Each bar represents the mean, normalized to 0 hours of treatment + SD.
Figure 3.
The effect of hypoxia on COX-2 activity in wild-type mouse Müller cells. Consistent with increased COX-2 protein, COX-2 activity was likewise significantly increased (* p ≤ 0.01; † p ≤ 0.025; ‡ p ≤ 0.05) by hypoxia, as determined by the production of COX-2-derived PGE2. Each bar represents mean ± SD.
Effect of Hypoxia on VEGF Production
We have previously demonstrated the effect of hypoxia on Müller cell production of VEGF (Werdich and Penn, 2006). We chose to look at a time course of VEGF production by hypoxic Müller cells because we hypothesized that COX-2-derived prostanoids mediate VEGF production in these cells, and we wanted to empirically determine the time course, and therefore the likelihood, of these events being mechanistically linked. VEGF levels were significantly (p ≤ 0.0003) increased over time in hypoxia, peaking at 24 hours (Figure 4). Hypoxic treatment lasting longer than 24 hours led to cell death, and hence, later time points were not examined.
Figure 4.
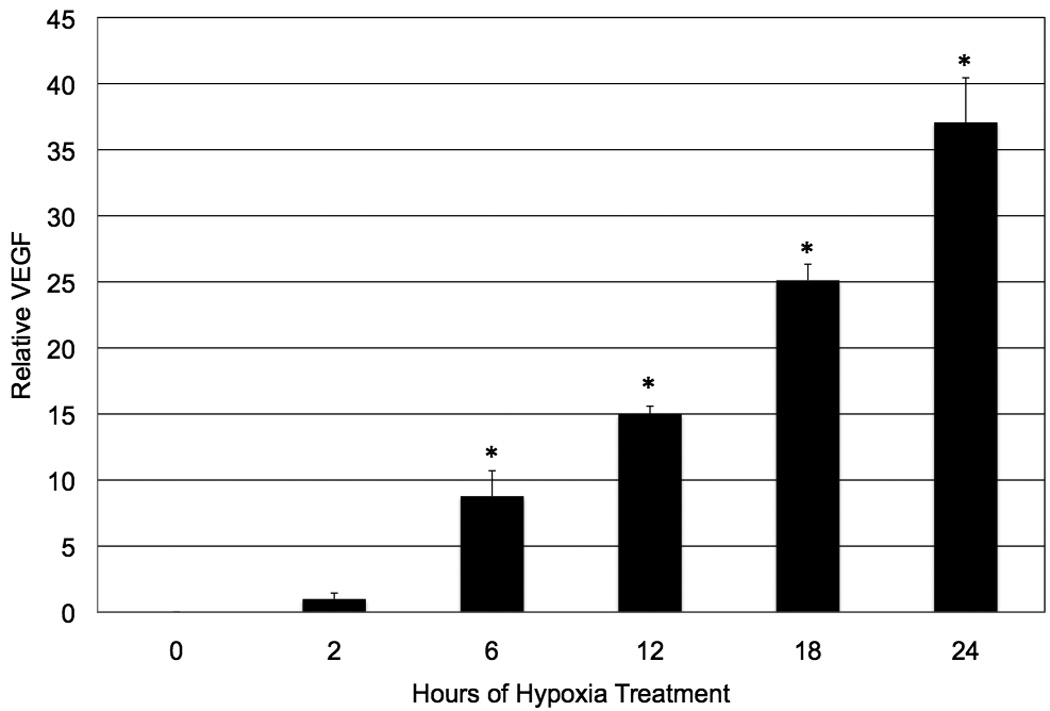
The effect of hypoxia on wild-type mouse Müller cell production of VEGF. Hypoxia significantly increased VEGF production in wild-type mouse Müller cells (* p ≤ 0.0003), with a time course that lagged behind that of hypoxia-induced COX-2 protein and activity, suggesting that some degree of VEGF production may be stimulated by COX-2-derived prostanoids. Each bar represents the mean (pg VEGF/mg protein), normalized to 2 hours of treatment ± SD.
Effect of COX-2 Deletion on VEGF Protein
In order to determine the COX-2-dependent effect on hypoxia-induced VEGF production by mouse Müller cells, we cultured wild-type and COX-2 null mouse Müller cells, exposed them to hypoxia for increasing periods of time, and assessed VEGF level. VEGF production was reduced in hypoxic COX-2 null cells, compared to wild-type cells, at every time point. This effect was statistically significant (p ≤ 0.05) after 12, 18, and 24 hours of hypoxic treatment (Figure 5). These data demonstrate that VEGF production by mouse Müller cells is at least partially dependent on COX-2 and COX-2-derived bioactive prostanoids. It should be mentioned that there is a smaller fold increase in VEGF in Figure 5 than in Figure 4. This is because Figure 4 was conducted under conditions that optimized PGE2 and VEGF production, while Figure 5 evaluated VEGF alone.
Figure 5.

The effect of COX-2 deletion on VEGF protein. VEGF production was reduced in hypoxia-treated COX-2 null cells, compared to wild-type cells, at every time point. This effect was statistically significant (* p ≤ 0.05) after 12, 18, and 24 hours of hypoxic treatment, indicating that VEGF production is partially COX-2-dependent. Each bar represents the mean (pg/ VEGF/mg protein), normalized to 2 hours of treatment in WT cells ± SD.
Effect of Hypoxia on Prostanoid Production
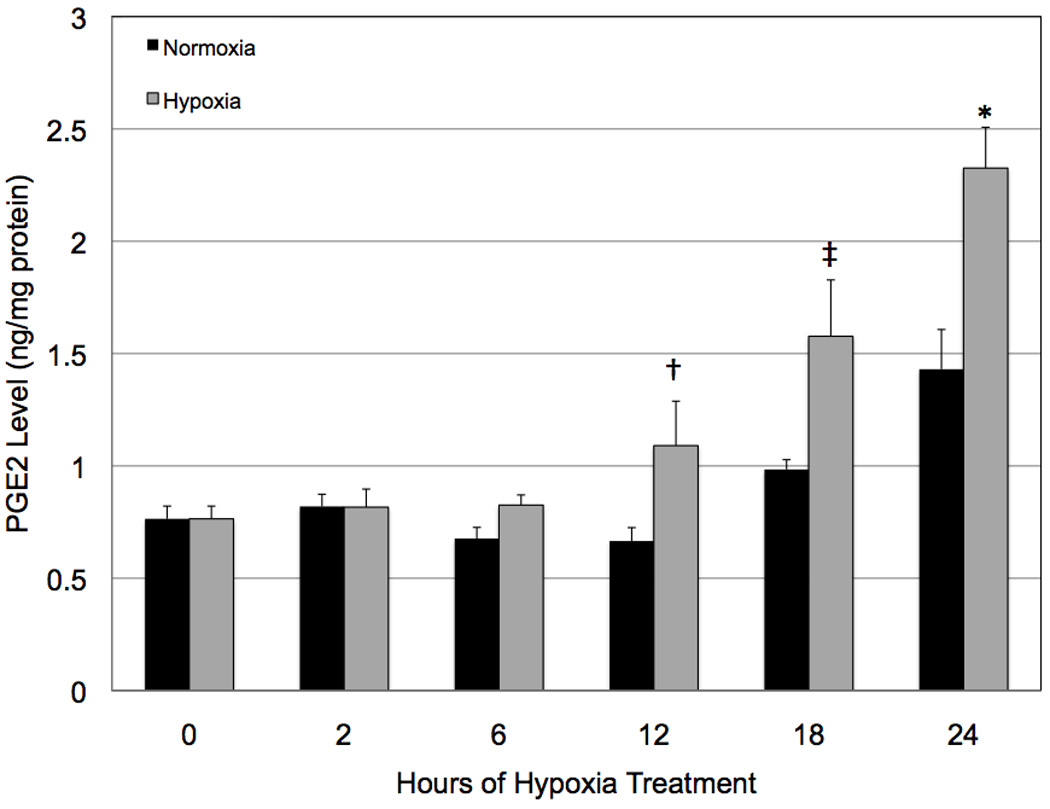
In order to more clearly define which of the prostanoids might have been influencing hypoxic VEGF production, we analyzed prostanoid production in cells that had been maintained in normoxia and hypoxia for 24 hours. We chose to examine prostanoid production at this time point because the cells remained viable and demonstrated maximal VEGF production. Hypoxic treatment significantly increased (* p < 0.02) levels of PGE2. Although levels of PGF2, PGI2, and TXA2 also were increased by hypoxia, the results were not statistically significant (Figure 6). We performed the same survey in COX-2 null Müller cells and, as expected, baseline (normoxia) prostanoid production was dramatically reduced and the cells failed to demonstrate hypoxia-reactivity (data not shown). These data demonstrate that several of the prostanoids are increased in the wild-type response to hypoxia, and suggest that PGE2, particularly, may play a significant role in VEGF production by hypoxic mouse Müller cells.
Figure 6.
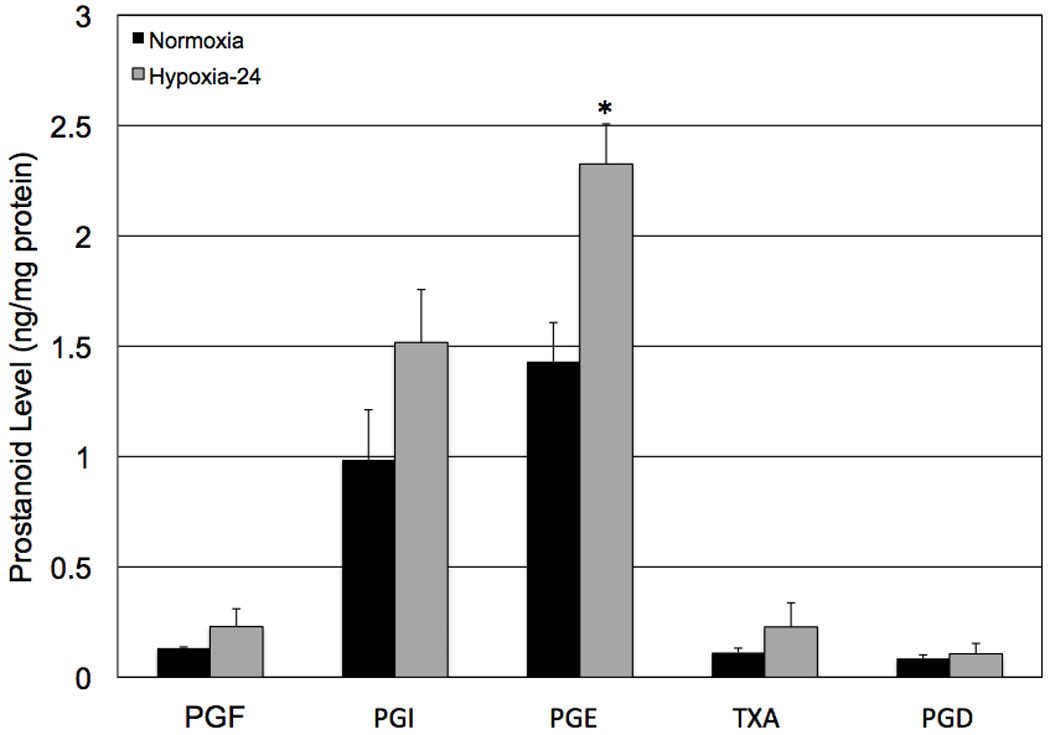
The effect of hypoxia on prostanoid production by wild-type mouse Müller cells. Twenty four hours of hypoxia led to significantly increased (* p < 0.02) levels of PGE2. Although levels of PGF2, PGI2, and TXA2 were increased by hypoxia, the results were not significant. Each bar represents mean ± SD.
Effect of PGE2 and PKA Inhibitor H-89 on Prostanoid Production
In order to more clearly define the role of COX-2-derived PGE2 on VEGF production, Müller cells were treated with a stable analog of PGE2 and their ability to produce VEGF was assessed (Figure 7). We chose to use wild-type cells because the COX-2 null cells do not produce measurable quantities of VEGF following overnight serum-starvation and subsequent treatment with a low serum concentration, as required in this assay. We chose to use a stable analog of PGE2 (10 µM dinoprostone) because our GC-MS data (Figure 6) indicated that this prostanoid was significantly increased by hypoxia. We chose to treat the cells for 12 hours because there was a 6–12 hour lag time between maximal COX-2 induction and maximal VEGF production, and we believe that these two events are mechanistically linked and that 12 hours may be needed for COX-2 and the prostanoids to affect the VEGF transcription and translation machinery. Dinoprostone significantly (* p ≤ 0.0039) increased VEGF production. The PGE2-induced increase in VEGF was completely inhibited († p ≤ 0.0055) by treatment with the PKA inhibitor H-89. These data suggest that the effect of PGE2 on VEGF induction is mediated by the EP2 and/or EP4 receptors, receptors known to signal through the PKA pathway (Regan, 2003).
Figure 7.
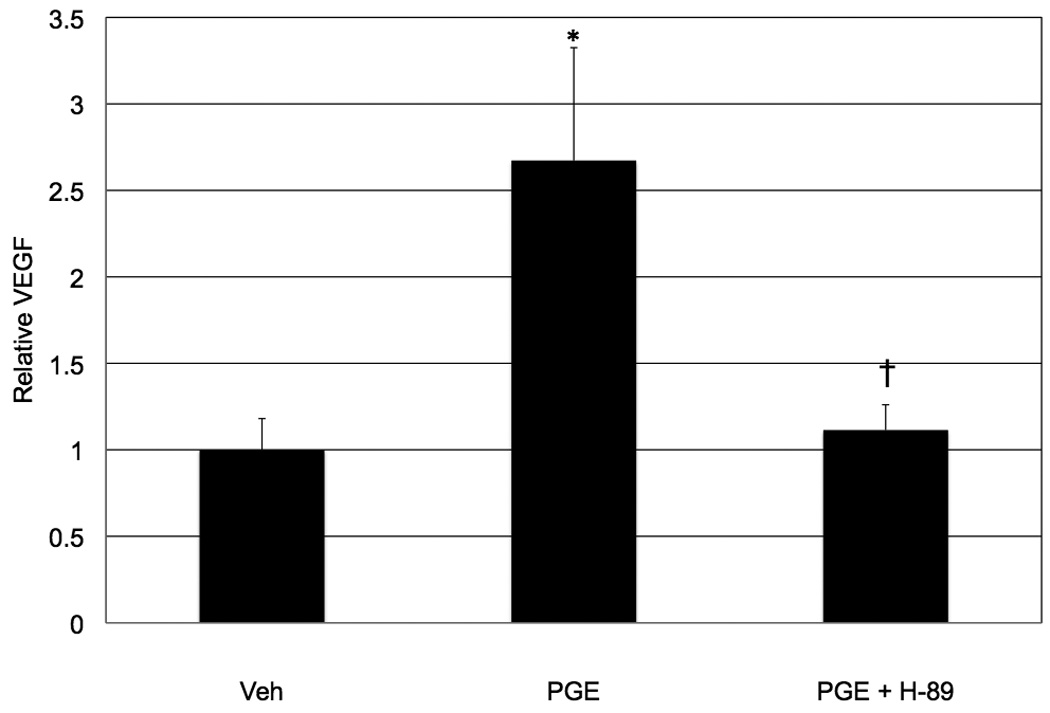
The effect of PGE2 and PKA inhibition on VEGF production by wild-type mouse Müller cells. Twelve hours of PGE2 (10 µM) treatment significantly (* p ≤ 0.0039) increased VEGF production by Müller cells. The PGE2-induced increase in VEGF was completely inhibited († p ≤ 0.0055) by treatment with 5 µM H-89, a PKA inhibitor. These data suggest that the effect of PGE2 on VEGF induction is mediated by the EP2 and/or EP4 receptors. Each bar represents the mean (pg VEGF/mg protein), normalized to vehicle-treated cells ± SD.
Discussion
Recent studies indicate that inhibiting the COX enzymes is an effective means by which to inhibit VEGF production and NV in relevant models of ocular disease (Castro et al., 2004; Wilkinson-Berka et al., 2003; Sennlaub et al., 2003; Takahashi et al., 2004; Hu et al., 2005; Ayalasomayajula et al., 2003; Ayalasomayajula et al., 2004; Amrite et al., 2006; Yanni et al., 2010). COX inhibitors have demonstrated efficacy in corneal, retinal, and choroidal models of angiogenesis. Although these studies have demonstrated the efficacy of COX inhibition at reducing the production of VEGF and NV, little work has been done to determine the mechanism by which COX-2 and its prostanoid products mediate VEGF production and the resultant ocular angiogenesis. In this study, we used an in vitro assay of hypoxia-induced VEGF production to examine the role of COX-2 and the prostanoids in mediating VEGF production by mouse retinal Müller cells, the cells that most consistently and dramatically increase production of VEGF in response to angiogenic stimulation, as a necessary first step in the process of defining a more specific therapeutic target in the treatment of retinal NV (Pierce et al., 1995; Robbins et al., 1997; Robbins et al., 1998; Bai et al., 2009).
It is well known that retinal ischemia-induced hypoxia is one driving force behind retinal NV (D’Amore, 1994; Casey and Li, 1997). It is also known that hypoxic challenge induces COX-2 in various cell types, including vascular endothelial cells, corneal epithelial cells, and various tumor cell lines (Demasi et al., 2003; Demasi et al., 2004; Bonazzi et al., 2000; Schmedtje et al., 1997; Csiki et al., 2006; Kaida et al., 2006). We looked at the COX-2 response of mouse Müller cells exposed to hypoxia for increasing periods of time. In response to hypoxia, wild-type mouse Müller cells demonstrated significantly increased levels of COX-2 and prostanoid production (Figure 1, Figure 3 and Figure 6). These data agree with the findings of others (referenced above) and were expected; COX-2 is an immediate early gene, rapidly and transiently induced by a variety of stimuli (Hla et al., 1999). Herein, we have also shown that wild-type mouse Müller cells demonstrated significantly increased levels of VEGF in response to hypoxia (Figure 4), and VEGF levels were increased with a temporal sequence that lagged behind COX-2 induction and activity. The time sequence of VEGF production by hypoxic mouse Müller cells is consistent with the hypothesis that there are COX-2-dependent aspects of VEGF production.
Previous studies have demonstrated that COX-2 is involved in mediating growth factor production in various cell types, and in response to various stimuli (Cheng et al., 1998; Pai et al., 2001). The present study has shown that COX-2 was upregulated and activated by hypoxia, and VEGF levels were increased, in an in vitro assay of hypoxia-induced VEGF production. We have also shown that, compared to wild-type cells, COX-2 null mouse Müller cells treated with hypoxia produce significantly less VEGF (Figure 5). This finding suggests that COX-2 and its prostanoid products play a role in the VEGF response of Müller cells. COX-1 does not appear to contribute to this behavior. Preliminary, non-optimized experiments have demonstrated that COX-2 null cells produce nearly undetectable levels of PGE2 in hypoxia. Thus, COX-1 is not significantly contributing to PGE2 production (data not shown). Our findings demonstrate that: 1) COX-1 protein is not increased by hypoxia (Figure 1), and 2) COX-1 activity is not increased by hypoxia (as demonstrated by the lack of PGE2 production in COX-2 null cells). Furthermore, we have previously shown that COX-1 fails to mediate hypoxia-induced VEGF production. Wild-type Müller were cells treated with SC-560, a COX-1-selective inhibitor, and placed in hypoxia (Yanni, et al., 2010). Inhibiting the COX-1 enzyme failed to inhibit VEGF production by Müller cells. These data suggest that the portion of hypoxia-induced VEGF that is COX-dependent is mediated by the COX-2 isoform and COX-2-derived prostanoids. These data agree with the findings of others; researchers studying angiogenesis related to various cancers and other neovascularizing conditions have demonstrated, using pharmacological and genetic manipulation of COX-2, that COX-2 inhibition resulted in reduced VEGF production, in vitro and in vivo (Gallo et al., 2001; Williams et al., 2000; Abdelrahim et al., 2005; Takahashi et al., 2003). Of more relevance to the eye, NSAID use has been effective at reducing the production of VEGF and NV in relevant models of ocular disease (Castro et al., 2004; Wilkinson-Berka et al., 2003; Sennlaub et al., 2003; Takahashi et al., 2004; Hu et al., 2005; Ayalasomayajula et al., 2003; Ayalasomayajula et al., 2004; Amrite et al., 2006; Yanni et al., 2010). Furthermore, several investigators have used COX-2 null mice to investigate the involvement of COX-2 in pathological ocular angiogenesis. In 2006, Cryan et al. used COX-2 null mice in a mouse model of oxygen-induced retinopathy, which models human ROP. In this model, the COX-2 null mice demonstrated a trend towards reduced neovascularization (23.5% reduction; Cryan et al., 2006). Experience tells us that this experiment used inadequate numbers of mice (n = 6), which may explain why the trend did not quite reach statistical significance. More recently, Attaran-Rezaei et al. used COX-2 null mice obtained from the Penn laboratory, and subjected them to a model of laser-induced CNV, which produces sub-retinal NV like that occurring in human AMD. In this model, choroidal neovascular lesions were significantly (p = 0.0014) smaller in the COX-2 null mice. COX-2 null mice demonstrated a 57.8% reduction in the size of CNV lesions. This information will be presented at the 2010 annual Association for Research in Vision and Ophthalmology meeting (Kasra Attaran-Rezaei, personal communication).
Our findings have significant implications for conditions characterized by retinal NV. Retinal hypoxia leads to increased production of VEGF, and VEGF is thought to be a principal mediator of the angiogenesis that occurs in retinal NV (Aiello, 1997). The Müller cells most consistently and dramatically increase production of VEGF in response to retinal hypoxia (Pierce et al., 1995; Robbins et al., 1997; Robbins et al., 1998; Bai et al., 2009). We have shown that genetic inhibition of COX-2 and the resultant reduction in prostanoid synthesis led to a significant reduction in hypoxia-induced VEGF production by Müller cells. However, COX-2 activity leads to the production of five bioactive prostanoid products. Thus, our results led us to investigate which of the prostanoids were involved in VEGF production by mouse Müller cells, in order to define a more selective chemotherapeutic target for the treatment and management of retinal NV.
COX-2 activity results in the formation of five biologically active prostanoids (PGD2, PGE2, PGF2, PGI2, TXA2). In response to hypoxia, wild-type mouse Müller cells demonstrated significantly increased levels of PGE2 (Figure 6), suggesting that this prostanoid might play a role in mediating VEGF production. In fact, treating Müller cells with a stable analog of PGE2, dinoprostone, significantly increased VEGF production (Figure 7). Of relevance to our focus, Cheng et al., using pure, primary cultures of rat Müller cells, demonstrated that PGE2 significantly induced VEGF mRNA, with maximal VEGF mRNA noted 2 hours post-prostaglandin treatment (Ziche et al., 1982). Our PGE2 data confirms what has been shown in the literature – that PGE2 stimulates VEGF production in widely diverse cell types (Hatazawa et al., 2007; Wang X et al., 2007; Bradbury et al., 2005; Abdel-Majid et al., 2004; Eibl et al., 2003; Yanni et al., 2009). PGE2 affects a wide range of physiological and pathological processes by binding to distinct cell surface G-protein-coupled receptors (GPCRs). PGE2 binds and activates one (or more) prostaglandin E (EP) receptors: EP1, EP2, EP3, and EP4 (Hata and Breyer, 2004). The receptors demonstrate distinct, as well as opposing, effects on intracellular signaling events. Of these receptors, EP2 and EP4 couple to Gs and mediate a rise in cAMP concentration and subsequent PKA activity. There have been numerous reports in the literature demonstrating a role of these two receptors, and downstream PKA activation, in mediating PGE2-induced angiogenic cell behaviors Hatazawa et al., 2007; Wang X et al., 2007; Bradbury et al., 2005; Yanni et al., 2009). It has been established that prostanoid receptors coupled to increased cAMP activate PKA, leading to phosphorylation of CREB, among other transcription factors. Phosphorylated CREB, for example, attracts CREB-binding protein (CBP), and the CREB-CBP protein complex binds to the CREB response element (CRE) on the VEGF gene, stimulating transcription. This over-simplified model may be the primary mechanism by which the EP2 and/or EP4 receptors affect VEGF production in other cell types (Muller et al., 2006). However, prostanoid receptors activate widely different intracellular signaling pathways in both tissue- and cell-specific manners. We sought to determine whether PGE2, signaling through PKA, was involved in VEGF production in our model system. Of particular interest, in our model system, hypoxia led to a 2.5-fold increase in PKA activity (data not shown). As shown in Figure 7, the PKA inhibitor virtually abolished PGE2-induced VEGF production in mouse Müller cells. These findings agree with a large body of published findings (Cheng et al., 1998; Höper et al, 1997; Amano et al., 2002; Amano et al., 2001). Our data suggest that PGE2 signals through the EP2 and/or EP4 receptor(s), activating PKA, which ultimately leads to VEGF production by Müller cells. These findings are significant because they more clearly define the involvement of specific prostanoids and prostanoid receptors in Müller cell angiogenic activity, and they point to a potential therapeutic target for the treatment of retinal conditions characterized by increased VEGF: namely, PKA.
Although we have focused our attention on PGE2 and its cognate receptors, it is possible that one or more of the other prostanoids, specifically PGI2 or PGF2, both of which were stimulated by hypoxia (Figure 6), play a role in mediating retinal NV. These prostanoids signal through the IP and FP receptors, respectively. A review of these receptors implicates both of them in angiogenic cell behaviors and tumor progression (Wang MT et al., 2007). Additionally, the IP receptor is predominantly coupled to Gs, meaning that its activation leads to increased cAMP production and PKA activation. In the future, it will be important to determine whether all prostanoids that demonstrate pro-angiogenic activity in Müller cells signal through PKA, and affect VEGF production similarly. To more clearly define the specific role(s) of the EP2 and EP4 receptors, as well as the IP and FP receptors in retinal NV, we have planned studies using pharmacological agents as well as genetically modified mice and cells derived from their retinas.
Preliminary studies conducted in our lab suggested that the effect of PGE2 on retinal angiogenesis was mediated by the EP4 receptor (Yanni et al., 2009). This hypothesis was based on the following preliminary findings: 1) Müller cells derived from EP1 and EP3 knockout mice failed to demonstrated reduced angiogenic cell behavior, as measured by hypoxia-induced VEGF production; 2) a proprietary EP2 antagonist failed to inhibit hypoxia-induced VEGF production by Müller cells; and 3) specific agonists of EP1–3 failed to elicit a VEGF response in COX-2 knockout Müller cells. To our knowledge, our study was the first to examine and demonstrate a role for the EP4 receptor in retinal angiogenesis.
We have, for the first time, used COX-2 null mouse Müller cells to show that COX-2 and at least one of its prostanoid products, PGE2, is involved in VEGF production. Our study implicates PGE2, signaling through the EP2 and/or EP4 receptor(s), in mediating the VEGF response of the cells. Figure 8 depicts our working model, illustrating how COX-2 activation leads to VEGF production in these cells. This information may provide a more selective chemotherapeutic target for the prevention and/or treatment of conditions in which retinal angiogenesis is a pathologic feature.
Figure 8.
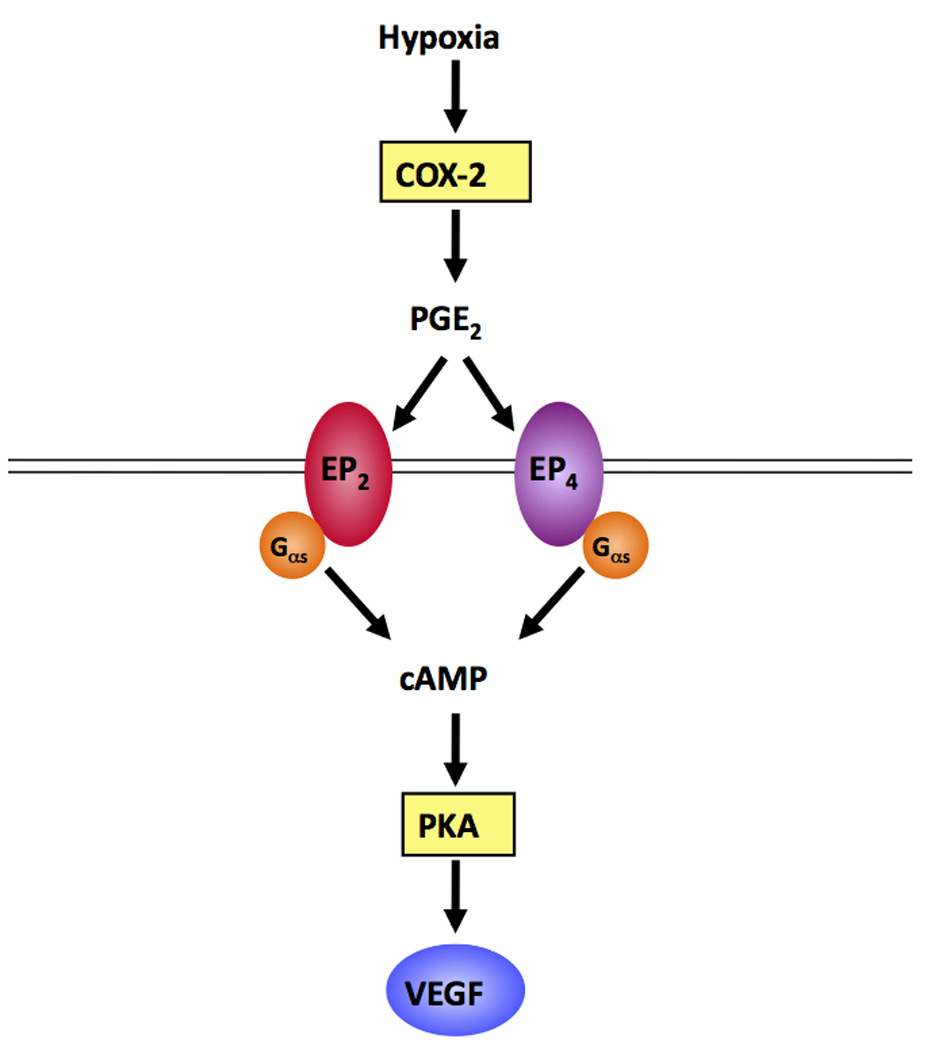
A flowchart demonstrating the way that we hypothesize COX-2-mediates hypoxia-induced VEGF production by Müller cells. In response to hypoxia, COX-2 is up-regulated, leading to increased production of pro-angiogenic PGE2. PGE2 binds to the EP2 and/or EP4 receptors, activating G proteins that couple to increased cAMP production. cAMP activates PKA, which is involved in mediating VEGF production.
Acknowledgements
The authors would like to thank Dr. Ginger Milne and Stephanie Sanchez of the Vanderbilt University Eicosanoid Core Laboratory and Dr. Rong Yang for experimental assistance.
Supported by NIH EY07533, NIH EY01826, an Unrestricted Grant from Research to Prevent Blindness, Inc., and a Research to Prevent Blindness Senior Scientific Investigator Award to JSP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Majid RM, Marshall JS. Prostaglandin E2 induces degranulation-independent production of vascular endothelial growth factor by human mast cells. J. Immunol. 2004;172:1227–1236. doi: 10.4049/jimmunol.172.2.1227. [DOI] [PubMed] [Google Scholar]
- Abdelrahim M, Safe S. Cyclooxygenase-2 inhibitors decrease vascular endothelial growth factor expression in colon cancer cells by enhanced degradation of Sp1 and Sp4 proteins. Mol. Pharmacol. 2005;68:317–329. doi: 10.1124/mol.105.011825. [DOI] [PubMed] [Google Scholar]
- Aiello LP, Northrup JM, Keyt BA, Takagi H, Iwamoto MA. Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch. Ophthalomol. 1995;113:1538–1544. doi: 10.1001/archopht.1995.01100120068012. [DOI] [PubMed] [Google Scholar]
- Aiello LP. Vascular endothelial growth factor: 20th-century mechanisms, 21st-century therapies. Invest. Ophthalmol. Vis. Sci. 1997;38:1647–1652. [PubMed] [Google Scholar]
- Amano H, Ando K, Minamida S, Hayashi I, Ogino M, Yamashina S, Yoshimura H, Majima M. Adenylate cyclase/protein kinase A signaling pathway enhances angiogenesis through induction of vascular endothelial growth factor in vivo. Jpn. J. Pharmacol. 2001;87:181–188. doi: 10.1254/jjp.87.181. [DOI] [PubMed] [Google Scholar]
- Amano H, Haysahi I, Yoshida S, Yoshimura H, Majima M. Cyclooxygenase-2 and adenylate cyclase/protein kinase A signaling pathway enhances angiogenesis through induction of vascular endothelial growth factor in rat sponge implants. Hum. Cell. 2002;15:13–24. doi: 10.1111/j.1749-0774.2002.tb00095.x. [DOI] [PubMed] [Google Scholar]
- Amrite AC, Ayalasomayajula SP, Cheruvu NPS, Kompella UB. Single periocular injection of celecoxib-PLGA microparticles inhibits diabetes-induced elevations in retinal PGE2, VEGF, and vascular leakage. Invest. Ophthalmol. Vis. Sci. 2006;47:1149–1160. doi: 10.1167/iovs.05-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayalasomayajula SP, Kompella UB. Celecoxib, a selective cyclooxygenase-2 inhibitor, inhibits retinal vascular endothelial growth factor expression and vascular leakage in a streptozotocin-induced diabetic rat model. Eur. J. Pharmacol. 2003;458:283–289. doi: 10.1016/s0014-2999(02)02793-0. [DOI] [PubMed] [Google Scholar]
- Ayalasomayajula SP, Amrite AC, Kompella UB. Inhibition of cyclooxygenase −2, but not cyclooxygenase-1, reduces prostaglandin E2 secretion from diabetic rat retinas. Eur. J. Pharmacol. 2004;498:275–278. doi: 10.1016/j.ejphar.2004.07.046. [DOI] [PubMed] [Google Scholar]
- Bai Y, Ma JX, Guo J, Wang J, Zhu M, Chen Y, Le YZ. Müller cell-derived VEGF is significant contributor to retinal neovascularization. J. Pathol. 2009;219:446–454. doi: 10.1002/path.2611. [DOI] [PubMed] [Google Scholar]
- Bonazzi A, Mastyugin V, Mieyal PA, Dunn MW. Laniado-Schwartzman M. Regulation of cyclooxygenase-2 by hypoxia and peroxisome proliferators in the corneal epithelium. J. Biol. Chem. 2000;275:2837–2844. doi: 10.1074/jbc.275.4.2837. [DOI] [PubMed] [Google Scholar]
- Bradbury D, Clarke D, Seedhouse C, Corbett L, Stocks J, Knox A. Vascular endothelial growth factor induction by prostaglandin E2 in human airway smooth muscle cells is mediated by E prostanoid EP2/EP4 receptors and SP-1 transcription factor binding sites. J. Biol Chem. 2005;280:29993–30000. doi: 10.1074/jbc.M414530200. [DOI] [PubMed] [Google Scholar]
- Casey R, Li WW. Factors controlling ocular angiogenesis. Am. J. Ophthalmol. 1997;124:521–529. doi: 10.1016/s0002-9394(14)70868-2. [DOI] [PubMed] [Google Scholar]
- Castro MR, Lutz D, Edelman JL. Effect of COX inhibitors on VEGF-induced retinal vascular leakage and experimental corneal and choroidal neovascularization. Exp. Eye Res. 2004;79:275–285. doi: 10.1016/j.exer.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Cheng T, Cao W, Wen R, Steinberg RH, LaVail MM. Prostaglandin E2 induces vascular endothelial growth factor and basic fibroblast growth factor mRNA expression in cultured rat Muller cells. Invest. Ophthalmol. Vis. Sci. 1998;39:581–591. [PubMed] [Google Scholar]
- Cryan LM, Pidgeon GP, Fitzgerald DJ, O'Brien CJ. COX-2 protects against thrombosis of the retinal vasculature in a mouse model of proliferative retinopathy. Mol. Vis. 2006;12:405–414. [PubMed] [Google Scholar]
- Csiki I, Yanagisawa K, Haruki N, Nadaf S, Morrow JD, Johnson DH, Carbone DP. Thioredoxin-1 modulates transcription of cyclooxygenase-2 via hypoxia-inducible factor-1 in non-small cell lung cancer. Cancer Res. 2006;66:143–150. doi: 10.1158/0008-5472.CAN-05-1357. [DOI] [PubMed] [Google Scholar]
- D’Amore PA. Mechanisms of retinal and choroidal angiogenesis. Invest. Ophthalmol. Vis. Sci. 1994;35:3974–3979. [PubMed] [Google Scholar]
- Damm J, Rau T, Maihofner C, Pahl A, Brune K. Constitutive expression and localization of COX-1 and COX-2 in rabbit iris and ciliary body. Exp. Eye Res. 2001;72:611–621. doi: 10.1006/exer.2001.0977. [DOI] [PubMed] [Google Scholar]
- Demasi M, Cleland LG, Cook-Johnson RJ, Caughey GE, James MJ. Effects of hypoxia on monocyte inflammatory mediator production: dissociation between changes in cyclooxygenase-2 expression and eicosanoid synthesis. J. Biol. Chem. 2003;278:38607–38616. doi: 10.1074/jbc.M305944200. [DOI] [PubMed] [Google Scholar]
- Demasi M, Cleland LG, Cook-Johnson RJ, James MJ. Effects of hypoxia on the expression and activity of cyclooxygenase 2 in fibroblast-like synoviocytes: Interactions with monocyte-derived soluble mediators. Arthritis Rheum. 2004;50:2441–2449. doi: 10.1002/art.20429. [DOI] [PubMed] [Google Scholar]
- Diaz-Flores L, Gutierrez R, Varela H. Angiogenesis: an update. Histol. Histopathol. 1994;9:807–843. [PubMed] [Google Scholar]
- DuBois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- Eibl G, Bruemmer D, Okada Y, Duffy JP, Law RE, Reber HA, Hines OJ. PGE(2) is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2003;306:887–897. doi: 10.1016/s0006-291x(03)01079-9. [DOI] [PubMed] [Google Scholar]
- Ershov AV, Bazan NG. Induction of cyclooxygenase-2 gene expression in retinal pigment epithelium cells by photoreceptor rod outer segment phagocytosis and growth factors. J. Neurosci. Res. 1999;58:254–261. [PubMed] [Google Scholar]
- Form DM, Auerbach R. PGE2 and angiogenesis. Proc. Soc. Exp. Biol. Med. 1983;172:214–218. doi: 10.3181/00379727-172-41548. [DOI] [PubMed] [Google Scholar]
- Gallo O, Franchi A, Magnelli L, Sardi I, Vannacci A, Boddi V, Chiarugi V, Masini E. Cyclooxygenase-2 pathway correlates with VEGF expression in head and neck cancer. Implications for tumor angiogenesis and metastasis. Neoplasia. 2001;3:53–61. doi: 10.1038/sj.neo.7900127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Hatazawa R, Tanigami M, Izumi N, Kamei K, Tanaka A, Takeuchi K. Prostaglandin E2 stimulates VEGF expression in primary rat gastric fibroblasts through EP4 receptors. Inflammopharmacology. 2007;15:214–217. doi: 10.1007/s10787-007-1595-z. [DOI] [PubMed] [Google Scholar]
- Hicks D, Curtois Y. The growth and behaviour of rat retinal Müller cells in vitro. 1. An improved method for isolation and culture. Exp. Eye Res. 1990;51:119–129. doi: 10.1016/0014-4835(90)90063-z. [DOI] [PubMed] [Google Scholar]
- Hla T, Bishop-Bailey D, Liu CH, Schaefers HJ, Trifan OC. Cyclooxygenase-1 and −2 isoenzymes. Int. J. Biochem. Cell Biol. 1999;31:551–557. doi: 10.1016/s1357-2725(98)00152-6. [DOI] [PubMed] [Google Scholar]
- Höper MM, Voelkel NF, Bates TO, Allard JD, Horan M, Shepherd D, Tuder RM. Prostaglandins induce vascular endothelial growth factor in a human monocytic cell line and rat lungs via cAMP. Am. J. Respir. Cell Mol. Biol. 1997;17:748–756. doi: 10.1165/ajrcmb.17.6.2888. [DOI] [PubMed] [Google Scholar]
- Hu W, Criswell MH, Ottlecz A, Cornell TL, Danis RP, Lambrou GN, Ciulla TA. Oral administration of lumiracoxib reduces choroidal neovascular membrane development in the rat laser-trauma model. Retina. 2005;25:1054–1064. doi: 10.1097/00006982-200512000-00015. [DOI] [PubMed] [Google Scholar]
- Ju W-K, Neufeld AH. Cellular localization of cyclooxygenase-1 and cyclooxygenase-2 in the normal mouse, rat, and human retina. J. Comp. Neurol. 2002;452:392–399. doi: 10.1002/cne.10400. [DOI] [PubMed] [Google Scholar]
- Kaida A, Qualtrough D, Williams AC, Paraskeva C. Direct transcriptional up-regulation of cyclooxygenase-2 by hypoxia-inducible factor (HIF)-1 promotes colorectal tumor cell survival and enhances HIF-1 transcriptional activity during hypoxia. Cancer Res. 2006;66:6683–6691. doi: 10.1158/0008-5472.CAN-06-0425. [DOI] [PubMed] [Google Scholar]
- Kawamori T, Rao CV, Seibert K, Reddy BS. Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res. 1998;58:409–412. [PubMed] [Google Scholar]
- Kuzmanovic M, Dudley VJ, Sarthy VP. GFAP promoter drives Müller cell-specific expression in transgenic mice. Invest. Ophthalmol. Vis. Sci. 2003;44:3606–3613. doi: 10.1167/iovs.02-1265. [DOI] [PubMed] [Google Scholar]
- Lee P, Wang CC, Adamis AP. Ocular neovascularization: an epidemiologic review. Surv. Ophthalmol. 1998;43:245–269. doi: 10.1016/s0039-6257(98)00035-6. [DOI] [PubMed] [Google Scholar]
- Maihofner C, Schlotzer-Schrehardt U, Guhring H, Zeilhofer HU, Naumann GO, Pahl A, Mardin C, Tamm ER, Brune K. Expression of cyclooxygenase-1 and −2 in normal and glaucomatous human eyes. Invest. Ophthalmol. Vis. Sci. 2001;42:2616–2624. [PubMed] [Google Scholar]
- Monnin J, Morand-Villeneuve N, Michel G, Hicks D, Versaux-Botteri C. Production of neurospheres from mammalian Müller cells in culture. Neurosci. Lett. 2007;421:22–26. doi: 10.1016/j.neulet.2007.04.073. [DOI] [PubMed] [Google Scholar]
- Pai R, Szabo IL, Soreghan BA, Atay S, Kawanaka H, Tarnawski AS. PGE(2) stimulates VEGF expression in endothelial cells via ERK2/JNK1 signaling pathways. Biochem. Biophys. Res. Commun. 2001;286:923–928. doi: 10.1006/bbrc.2001.5494. [DOI] [PubMed] [Google Scholar]
- Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LE. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc. Natl. Acad. Sci. USA. 1995;92:905–909. doi: 10.1073/pnas.92.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani B, Tielsch JM, Kat J, Gottsch J, Quigley H, Javitt J, Sommer A. The cause-specific prevalence of visual impairment in an urban population. The Baltimore eye survey. Ophthalmology. 1999;103:1721–1726. doi: 10.1016/s0161-6420(96)30435-1. [DOI] [PubMed] [Google Scholar]
- Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–153. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Robbins SG, Conaway JR, Ford BL, Roberto KA, Penn JS. Detection of VEGF protein in vascular and non-vascular cells of the normal and oxygen-injured retina. Growth Factors. 1997;14:229–241. doi: 10.3109/08977199709021522. [DOI] [PubMed] [Google Scholar]
- Robbins SG, Rajaratnam VS, Penn JS. Evidence for upregulation of vascular endothelial cell growth factor by hypoxia. Growth Factors. 1998;16:1–9. doi: 10.3109/08977199809017487. [DOI] [PubMed] [Google Scholar]
- Schmedtje JF, Ji YS, Liu WL, DuBois RN, Runge MS. Hypoxia induces cyclooxygenase-2 via the NF-kappa B p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 1997;272:601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- Sennlaub F, Valamanesh F, Vazquez-Tello A, El-Asrar AM, Checchin D, Brault S, Gobeil F, Beauchamp MH, Mwaikambo B, Curtois Y, Geboes K, Varma DR, Lachapelle P, Ong H, Behar-Cohen F, Chemtob S. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation. 2003;108:198–204. doi: 10.1161/01.CIR.0000080735.93327.00. [DOI] [PubMed] [Google Scholar]
- Steinkuller PG, Du L, Gilbert C, Foster A, Collins ML, Coats DK. Childhood blindness. J. AAPOS. 1999;3:26–32. doi: 10.1016/s1091-8531(99)70091-1. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Yanagi Y, Tamaki Y, Uchida S, Muranaka K. COX-2-selective inhibitor, etodolac, suppresses neovascularization in a mice model. Biochem. Biophys. Res. Commun. 2004;325:461–466. doi: 10.1016/j.bbrc.2004.10.054. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Saishin Y, Saishin Y, Mori K, Ando A, Yamamoto S, Oshima Y, Nambu H, Melia MB, Bingaman DP, Campochiaro PA. Topical nepafenac inhibits ocular neovascularization. Invest. Ophthalmol. Vis. Sci. 2003;44:409–415. doi: 10.1167/iovs.02-0346. [DOI] [PubMed] [Google Scholar]
- Wang D, DuBois RN. Cycooxygenase 2-derived prostaglandin E2 regulates the angiogenic switch. Proc. Natl. Acad. Sci. USA. 2004;101:415–416. doi: 10.1073/pnas.0307640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MT, Honn KV, Nie D. Cyclooxygenases, prostanoids, and tumor progress,ion. Cancer Metastasis Rev. 2007;26:525–534. doi: 10.1007/s10555-007-9096-5. [DOI] [PubMed] [Google Scholar]
- Wang X, Klein RD. Prostaglandin E2 induces vascular endothelial growth factor secretion in prostate cancer cells through EP2 receptor-mediated cAMP pathway. Mol. Carcinog. 2007;46:912–923. doi: 10.1002/mc.20320. [DOI] [PubMed] [Google Scholar]
- Werdich XQ, Penn JS. Specific involvement of SRC family kinase activation in the pathogenesis of retinal neovascularization. Invest. Ophthalmol. Vis. Sci. 2006;47:5047–5056. doi: 10.1167/iovs.05-1343. [DOI] [PubMed] [Google Scholar]
- Wilkinson-Berka JL, Alousis NS, Kelly DJ. Gilbert RE. COX-2 inhibition and retinal angiogenesis in a mouse model of retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 2003;44:974–979. doi: 10.1167/iovs.02-0392. [DOI] [PubMed] [Google Scholar]
- Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer and development. Oncogene. 1999;18:7908–7916. doi: 10.1038/sj.onc.1203286. [DOI] [PubMed] [Google Scholar]
- Williams CS, Tsujii M, Reese J, Dey SK, DuBois RN. Host cyclooxygenase-2 modulates carcinoma growth. J. Clin. Invest. 2000;105:1589–1594. doi: 10.1172/JCI9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Kawai M, Kawai Y, Mashima Y. The effect of selective cyclooxygenase-2 inhibitor on corneal angiogenesis in the rat. Curr. Eye Res. 1999;19:300–304. doi: 10.1076/ceyr.19.4.300.5301. [DOI] [PubMed] [Google Scholar]
- Yanni SE, Barnett JM, Clark ML, Penn JS. The role of PGE2 receptor EP4 in pathologic ocular angiogenesis. Invest. Ophthalmol. Vis. Sci. 2009;50:5479–5486. doi: 10.1167/iovs.09-3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanni SE, Clark ML, Yang R, Bingaman DP, Penn JS. The Effects of Nepafenac and Amfenac on Retinal Angiogenesis. Brain Res. Bull. 2010;81:310–319. doi: 10.1016/j.brainresbull.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziche M, Jones J, Gullino PM. Role of prostaglandin E1 and copper in angiogenesis. J. Natl. Cancer Inst. 1982;69:475–482. [PubMed] [Google Scholar]