

Abstract
Somatic cells in tissue culture package several copies of mitochondrial DNA (mtDNA) in aggregates known as nucleoids that appear to be remarkably stable. The clustering of multiple mtDNA genomes in a single nucleoid complex may promote the progressive age-related accumulation of deletion and point mutations in mtDNA in many somatic tissues, particularly in post-mitotic cells. In contrast, oocytes appear to have the ability to select against deleterious mutations in mtDNA, at least in mice. This fundamental difference suggests that oocytes may be better able to detect and remove defective mtDNA genomes than somatic cells, possibly due in part to the simpler organization of the mtDNA in smaller nucleoids. These observations suggest the hypothesis that a complex nucleoid structure containing several mtDNA molecules may impair the ability of the cell to select against deleterious mtDNA mutations, thereby contributing to age-related mitochondrial dysfunction.
Keywords: mitochondrial DNA, disposable soma theory, oxidative stress, oocyte mitochondria, mtDNA bottleneck
Introduction
The oxidative theory of aging has enjoyed a phoenix-like resilience in the years since its initial formulation by Harmon in 1956 (Harman, 1956) and its restatement implicating mitochondria as the mainspring of a biological clock (Harman, 1972). Successive waves of criticism have remodeled the landscape without eroding the underlying concept that progressive mitochondrial dysfunction is a hallmark of the aging process. Still, a comprehensive understanding of exactly how mitochondrial function degrades with age has been elusive. For many years, the most popular notion has maintained that aging results from progressive oxidative damage emanating from the mitochondrial electron transport chain. One variant of the mitochondrial oxidative stress model suggests that oxidative damage to mtDNA may lead to the formation of aberrant respiratory complexes that are increasingly likely to generate even more reactive oxygen and nitrogen species (RONS) leading, through a vicious cycle, to progressively greater mtDNA damage (Bandy and Davison, 1990). An alternative model maintains that mtDNA damage leads to decreased oxygen consumption and a corresponding reduction in further oxidative damage to mitochondrial lipids, resulting in slower turnover of mitochondria bearing mutant or damaged mtDNA genomes and, ultimately, to a progressive accumulation of defective mtDNA genomes (De Grey, 1997). This model has been referred to as the “survival of the slowest” (SOS) model. These theories have stimulated additional research, which, in many cases, has failed to support the a priori models.
The mitochondrial oxidative stress theory now faces a stringent challenge posed by several major experimental findings, as follows:
First, two laboratories have independently generated genetically engineered mouse strains that express error-prone versions of mitochondrial DNA polymerase γ (Kujoth et al., 2005; Trifunovic et al., 2004). These mice accumulate a high burden of mtDNA mutations and show several phenotypes consistent with premature aging, but do not exhibit increased oxidative stress. These models permit the conclusion that mtDNA mutations may decrease longevity independent of any effects on oxidative stress. The spectrum of mutations observed in mtDNA does not show a signature consistent with oxidative stress as a major factor in mutagenesis. Unrepaired 8-oxo-deoxyguanosine would be expected to frequently base pair with deoxyadenosine during replication, leading to G to T transversion mutations (Pinz et al., 1995). The relative lack of such mutations suggests that other mechanisms, such as the inherent error frequency of pol γ are more important sources of mtDNA mutations than oxidative stress (Zheng et al., 2006).
Second, a series of transgenic and knockout mice have been generated in an effort to reduce the mitochondrial production of RONS or to over-express enzymes expected to alleviate oxidative stress. With one exception, the mouse expressing mitochondrially-targeted catalase (Schriner et al., 2005), these mice have failed to show any significant increase in longevity (reviewed in (Jang and Remmen, 2009; Lapointe and Hekimi, ; Pérez et al., 2009; Van Remmen and Jones, 2009)).
Third, a comparison of two rodent species that differ considerably in their maximum life expectancy has found that the long-lived naked mole rat generates as much or more RONS as the shorter-lived Mus musculus (Perez et al., 2009).
Finally, an extensive series of studies in many laboratories (reviewed in (Chan, 2006)) has revealed that mitochondria in most cell types engage in rapid cycles of fission and fusion that would tend to homogenize mitochondrial lipid contents so that oxidized lipids in the mitochondrial membranes would not be expected to remain stably associated with damaged mtDNA molecules. This argues against at least one version of the SOS model.
Taken as a whole, these observations have led to considerable confusion in the field. It appears that mitochondrial oxidative stress is at most only one of several variables that contribute to aging and that mtDNA mutations may be a more important factor in age-related mitochondrial dysfunction (Wallace, 2005). We know that mtDNA mutations are commonly found in the cytochrome oxidase-deficient post-mitotic cells that accumulate with age. This phenomenon is well-established for neurons in the substantia nigra (Bender et al., 2006; Kraytsberg et al., 2006) and in skeletal muscle (Herbst et al., 2007; McKiernan et al., 2009), but the extent to which such deletions contribute to normal human aging is still disputed. The mechanism underlying the accumulation of these mtDNA mutations with age has not been established firmly.
In this article, I will present the argument that the packaging of multiple mtDNA molecules in large aggregates known as nucleoids may promote the age-related accumulation of mtDNA mutations.
The fundamental organization of mtDNA nucleoids
The discovery of mtDNA in the 1960’s strongly supported the hypothesis that mitochondria were derived from an endosymbiotic bacterium. As a bacterial chromosome is typically confined to a distinct nucleoid domain associated with the inner membrane, Nass applied the term “nucleoids” to the mtDNA nucleoprotein complexes (Nass, 1969). These mtDNA-protein structures can be visualized using PicoGreen and other DNA stains (Bereiter-Hahn and Vöth, 1996) or by immunofluorescence with anti-DNA antibodies (Iborra et al., 2004). Visualization of nucleoids is facilitated by their typical content of 5-7 mtDNA molecules packed in structures with a diameter estimated at 70 nm (Iborra et al., 2004; Legros et al., 2004). Since a single mtDNA molecule has a contour length of 5 μm, this packaging requires a considerable compaction of the DNA, which is probably largely accomplished by the DNA wrapping ability of the HMG-box protein TFAM (Antoshechkin and Bogenhagen, 1995; Kaufman et al., 2007). The protein composition of nucleoids has been studied for complexes affinity-purified using antibodies directed against major mtDNA binding proteins, mtSSB and TFAM, and for complexes cross-linked using formaldehyde (Bogenhagen et al., 2008; Wang and Bogenhagen, 2006). These studies detected most of the proteins known to be involved in mtDNA maintenance, such as DNA pol γ, the T7-like helicase, mitochondrial RNA polymerase, TFBM1, TFBM2, Terf1, and mitochondrial topoisomerase I. In addition several less well-studied proteins with helicase motifs were identified in nucleoids, such as the suv3-like helicase, DEAD box protein 28 and a novel helicase known as DHX30 which was first identified as a mitochondrial protein based on its detection in nucleoids (Wang and Bogenhagen, 2006).
While nucleoids are clearly foci for mtDNA replication and transcription, they may play an even broader role in mitochondrial biogenesis. Proteomic analysis of nucleoids also revealed a number of factors involved in protein folding and quality control including Hsp60, Hsp70, Hsp40, prohibitins 1and 2, and the complex IV assembly factor LRPPRC ((Mootha et al., 2003) also known as LRP130) along with the ClpX and Lon proteases. In independent studies, Lon has been reported to have DNA binding activity (Liu et al., 2004). Experiments using an immunoaffinity method to search for proteins interacting with a mitochondrial translation factor found nucleoid proteins co-purifying with mitochondrial ribosomes (Rorbach et al., 2008). These results suggested that nucleoids might have a layered structure with an inner core enriched in proteins involved in nucleic acid synthesis and processing and an outer zone involved in the assembly of nascent polypeptides into respiratory complexes (Bogenhagen et al., 2008). This layered structure may help explain why somatic cells tend to assemble multiple mtDNA genomes in single clusters in the first place. Large nucleoids may permit the products of several mtDNA genomes to be pooled to support assembly of respiratory complexes, thus serving as a component of a putative “unit mitochondrion” as envisioned by Capaldi and coworkers (Capaldi et al., 2002).
A nucleoid containing several mtDNA genomes and such a wide array of proteins would have a molecular mass of over 100 megadaltons, making it the largest structure in a mitochondrion, dwarfing even the massive respiratory supercomplexes (Acín-Pérez et al., 2008; Schafer et al., 2007) and pyruvate dehydrogenase (Zhou et al., 2001). Nucleoids move freely within cells, but only as passengers within the dynamically motile organelles that they sustain. They appear to be very stable and largely immobile within mitochondria, and do not readily fuse or exchange mtDNA genomes (Gilkerson et al., 2008). Other aspects of the dynamic behavior of nucleoids are not well understood. As cells grow and divide, there must be some mechanism for the fragmentation and distribution of nucleoids. This has been studied in more detail in lower eukaryotes (Kuroiwa et al., 1994), but even in these instances, the molecular mechanisms involved are poorly understood.
What is the impact of nucleoid organization on mtDNA quality control and aging?
It has long been recognized that the asexual nature of mtDNA inheritance may promote the accumulation of mtDNA mutations according to Muller’s ratchet (Muller, 1932). The organization of mtDNA in nucleoids may greatly influence the phenotypic expression of mtDNA defects. Typically, a single newly-acquired mutation in one mtDNA copy within a nucleoid is not detrimental since it can be complemented by the remaining wild-type genomes. Such complementation may even permit a mitochondrion to function relatively well when a nucleoid retains a single wild-type mtDNA molecule alongside several mutant copies. This correlates well with the general observation that mtDNA mutation rates must reach or exceed about 80% before a heteroplasmic cell shows signs of mitochondrial dysfunction (DiMauro and Schon, 2003; Taylor and Turnbull, 2005). Of course, genetic complementation between mtDNA molecules is not strictly limited to molecules co-localized in a single nucleoid, but it is reasonable to expect that complementation is less effective if mtDNA gene products must diffuse over long distances. Indeed, in many post-mitotic cells containing mitochondria in confined locations, such as neurons and cardiomyocytes, genetic complementation between nucleoids separated by large distances may be relatively ineffective.
Intra-nucleoid mtDNA genetic complementation may be an example of “antagonistic pleiotropy” as expressed by Williams (Williams, 1957), in other words, a two-edged sword. As noted above, it helps protect against the deleterious effects of relatively rare mutations. However, this same local complementation makes it difficult to selectively cull defective mtDNA genomes in somatic cells, resulting in more rapid accumulation of mutations. By analogy, we can consider that mitochondrial biogenesis is a group project in which the contributions of a few functional genomes can mask the poor performance of their ineffective collaborators. Although the gradual acquisition of mtDNA mutations with age in somatic tissues appears to be an inescapable consequence of Muller’s ratchet, new results indicate that nature has evolved a mechanism to select against deleterious mutations in the germline. How is this apparent rejuvenation reconciled with nucleoid structure?
The critical importance of the variability in germline transmission of mutant mtDNAs in human diseases has been evident since the early days of mitochondrial medicine when it was documented that the siblings and mother of a proband with a mitochondrial disorder could all exhibit different degrees of heteroplasmy (Shoffner et al., 1990). Remarkably, individual oocytes from a mother heteroplasmic for a mtDNA disease mutation can contain essentially homoplasmic mutant or wild-type mtDNA (Blok et al., 1997). Earlier work attributed such rapid generational shifts in the level of mtDNA heteroplasmy in cattle to a stage in early oocyte development characterized by a very low mtDNA copy number, which Hauswirth and Laipis termed the mtDNA bottleneck (Hauswirth and Laipis, 1985; Laipis et al., 1988). This bottleneck in mtDNA population size is estimated at 200 genomes in primordial germ cells (PGCs) in mice (Cree et al., 2008; Jenuth et al., 1996). Another recent publication reported a somewhat larger number of mtDNA molecules in PGCs and suggested that these might be organized into approximately 200 segregating units, presumably nucleoids (Cao et al., 2007).
PGCs, which are first detectable in mice as clusters of as few as eight cells during gastrulation (Ginsburg et al., 1990; McLaren, 2003), may be derived from a small segment of ooplasm, raising the possibility that there may be a highly non-random selection of oocyte mtDNAs to populate the next generation of germ cells. Non-random selection of mtDNA molecules for replication may be another significant source of mtDNA genetic variability (Wai et al., 2008). During embryonic development in the human female, the sparse population of PGCs expands to roughly 7 million primary oocytes each containing about 8000 mtDNA molecules in small, spherical mitochondria (Jansen and deBoer, 1998; Jansen and Burton, 2004). The final stages of oocyte maturation involve a further expansion of mtDNA copy numbers to about 200,000 per egg. Thus, as shown in Figure 1, embryogenesis entails a massive PCR-like amplification of the small pool of mtDNAs in the initial population of PGCs, providing an opportunity for considerable variation in the degree of heteroplasmy.
Fig. 1. The life cycle of mtDNA inheritance in the female germline.
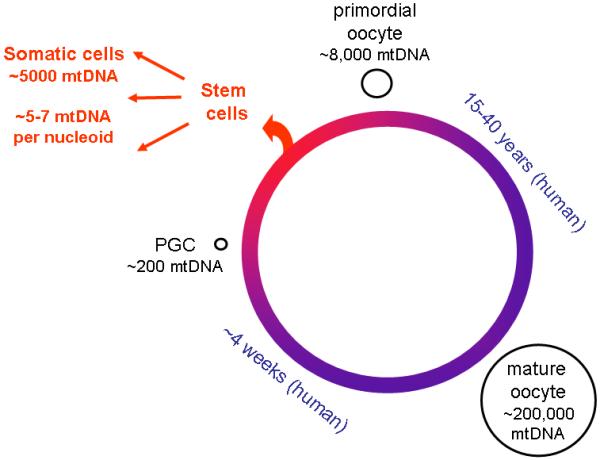
MtDNA in somatic cells is not inherited and may be organized differently than in oocytes.
While there is broad agreement that oogenesis is accompanied by rapid shifts in heteroplasmy, until recently this variance was thought to be random, incapable of filtering or selectively removing deleterious mutations (Chinnery et al., 2000; Shoubridge, 2000; Van Blerkom, 2004). This view must be modified in light of two new genetic studies documenting purifying selection against deleterious mutations (Fan et al., 2008) or non-synonymous codon changes (Stewart et al., 2008b) in the germline. These papers imply that mtDNA is subject to some sort of quality control during oogenesis, as discussed in a recent review (Stewart et al., 2008a). While the precise mechanism for this purifying selection remains uncertain, it must be remembered that it is not highly effective since mutations, even substantial deletions in an engineered mouse strain (Inoue et al., 2000), can be transmitted through the germline.
Purifying selection against mtDNA mutations could function at two levels, as discussed by Stewart et al. (Stewart et al., 2008a). First, we know that the vast majority of oocytes are lost through atresia, raising the possibility that there may be selective destruction of oocytes containing an excessive proportion of defective mitochondria (Krakauer and Mira, 1999; Tilly, 2001). In addition to selection at the cellular level, there may be selection against individual mitochondria harboring defective mtDNAs, possibly based on the inability of these mitochondria to develop a sufficient membrane potential (Δψ). Under some conditions, mitochondria with a relatively low membrane potential have been shown to be more likely to be removed by autophagy (Twig et al., 2008). Additional research will be necessary to define the mechanism of purifying selection against mutant mtDNA genomes and to determine the extent to which mtDNA quality control may contribute to oocyte atresia.
An effective mtDNA quality control process must be able to link defective genomes with stable phenotypes reflecting mitochondrial dysfunction. This is difficult to accomplish if mutant mtDNAs are genetically complemented by wild-type genomes within nucleoids or even if high rates of mitochondrial fusion and fission permit functional complementation in heteroplasmic cells. Thus, if purifying selection functions in the female germline but not in most somatic cells, this must reflect some distinctive features of mitochondrial biology in oocytes. There are at least two unusual aspects of mtDNA genome organization in oocytes that may enhance quality control. First, measurements of the total number of mitochondria and of mtDNA in oocytes indicate that, on average, each mitochondrion may harbor only a single mtDNA genome (Piko and Matsumoto, 1976). This is a statistic that has been quoted frequently in reviews (Cummins, 2002; Jansen and Burton, 2004; Van Blerkom, 2004), but it is important to remember that this has not been demonstrated formally, it is only an average value. The possibility that some organelles have several mtDNA genomes while others have none has not been eliminated. Nevertheless, it is intriguing to consider that oocytes may indeed have accomplished a “mitochondrial meiosis” (Hauswirth and Laipis, 1985) that permits quality control at the single-genome level. Second, oocyte mitochondria are generally characterized as small, rather spherical organelles (Jansen and deBoer, 1998; Piko and Matsumoto, 1976), suggesting that there is not a large interconnected mitochondrial reticulum to permit facile exchange of mitochondrial gene products in these cells. We suggest that the balance between fusion and fission may be tipped in favor of fission in oocytes. Fission and fusion cycles are necessary during later embryonic development (Chen et al., 2003), but may be rather quiescent in oocytes.
The concept that purifying selection against deleterious mtDNA mutations may operate more efficiently in oocytes containing smaller nucleoids is in accord with the “disposable soma” theory of aging (Kirkwood and Holliday, 1979). In this view, organization of mtDNA in larger nucleoids is favored in somatic cells that need to build large energy farms of respiratory complexes, but this organization makes it more difficult to eliminate mtDNA mutations, since somatic mutations are not inherited. Additional studies are required to further define these mechanisms and to explore nucleoid structure and the status of mitochondrial fission and fusion accomplished in stem cells, as suggested recently (Rajasimha et al., 2008). Such investigations may ultimately suggest a means to intervene to improve selection of healthy mtDNA genomes in oocytes as well as in somatic cells.
Acknowledgments
This work was supported by Grants NIEHS R01-ES012039 and a Senior Scholar Award from the Ellison Medical Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acín-Pérez R, Fernández-Silva P, Peleato ML, Pérez-Martos A, Enriquez JA. Respiratory Active Mitochondrial Supercomplexes. Mol Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Antoshechkin I, Bogenhagen DF. Distinct roles for two purified factors in transcription of Xenopus mitochondrial DNA. Mol Cell Biol. 1995;15:7032–7042. doi: 10.1128/mcb.15.12.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: Implications for carcinogenesis and aging? Free Radical Biol Med. 1990;8:523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Vöth M. Distribution and dynamics of mitochondrial nucleoids in animal cells in culture. Exp Biol Online. 1996;1:4. [Google Scholar]
- Blok R, Gook D, Thorbum D, Dahl H. Skewed Segregation of the mtDNA nt8993 (T-G) Mutation in Human Oocytes. Am J Human Genet. 1997;60:1495–1501. doi: 10.1086/515453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenhagen DF, Rousseau D, Burke S. The Layered Structure of Human mtDNA Nucleoids. J Biol Chem. 2008;283:3665–3675. doi: 10.1074/jbc.M708444200. [DOI] [PubMed] [Google Scholar]
- Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J-I, Yonekawa H. The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet. 2007;39:386–390. doi: 10.1038/ng1970. [DOI] [PubMed] [Google Scholar]
- Capaldi RA, Aggeler R, Gilkerson R, Hanson G, Knowles M, Marcus A, Margineantu D, Marusich M, Murray J, Oglesbee D, et al. A replicating module as the unit of mitochondrial structure and functioning. Bioc Biop Acta (BBA) - Bioenergetics. 2002;1555:192–195. doi: 10.1016/s0005-2728(02)00277-3. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial Fusion and Fission in Mammals. Ann Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery P, Johnson M, Wardell T, Singh-Kler R, Hayes C, Brown D, Taylor R, Bindoff L, Turnbull D. The Epidemiology of Pathogenic Mitochondrial DNA Mutations. Ann Neurol. 2000;48:188–193. [PubMed] [Google Scholar]
- Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, Dahl H-HM, Chinnery PF. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat Genet. 2008;40:249–254. doi: 10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- Cummins J. The Role of Maternal Mitochondria during Oogenesis, Fertilization and Embryogenesis. Reprod BioMed Online. 2002;4:176–182. doi: 10.1016/s1472-6483(10)61937-2. [DOI] [PubMed] [Google Scholar]
- De Grey ADNJ. A proposed refinement of the mitochondrial free radical theory of aging. BioEssays. 1997;19:161–166. doi: 10.1002/bies.950190211. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon E. Mitochondrial Respiratory-Chain Diseases. New England J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, MacGregor GR, Wallace DC. A Mouse Model of Mitochondrial Disease Reveals Germline Selection Against Severe mtDNA Mutations. Science. 2008;319:958–962. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson R, Schon E, Hernandez E, Davidson M. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J Cell Biol. 2008;181:1117–1128. doi: 10.1083/jcb.200712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsburg M, Snow MH, McLaren A. Primordial germ cells in the mouse embryo during gastrulation. Development. 1990;110:521–528. doi: 10.1242/dev.110.2.521. [DOI] [PubMed] [Google Scholar]
- Harman D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Hauswirth W, Laipis P. Transmission Genetics of Mammalian Mitochondria: A Molecular Model and Experimental Evidence. In: Quagliarello E, et al., editors. Achievements and Perspectives of Mitochondrial Research. Elsevier; Amsterdam: 1985. [Google Scholar]
- Herbst A, Pak JW, McKenzie D, Bua E, Bassiouni M, Aiken JM. Accumulation of Mitochondrial DNA Deletion Mutations in Aged Muscle Fibers: Evidence for a Causal Role in Muscle Fiber Loss. J Gerontol A Biol Sci Med Sci. 2007;62:235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra F, Kimura H, Cook P. The functional organization of mitochondrial genomes in human cells. BMC Biology. 2004;2:9. doi: 10.1186/1741-7007-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Nakada K, Ogura A, Isobe K, Goto Y, Nonaka I, Hayashi J-I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nature Genetics. 2000;26:176–181. doi: 10.1038/82826. [DOI] [PubMed] [Google Scholar]
- Jang YC, Remmen HV. The mitochondrial theory of aging: Insight from transgenic and knockout mouse models. Experimental Gerontology. 2009;44:256–260. doi: 10.1016/j.exger.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Jansen R, deBoer K. The bottleneck: mitochondrial imperatives in oogenesis and ovarian follicular fate. Mol Cell Endocrin. 1998;145:81–88. doi: 10.1016/s0303-7207(98)00173-7. [DOI] [PubMed] [Google Scholar]
- Jansen RPS, Burton GJ. Mitochondrial dysfunction in reproduction. Mitochondrion. 2004;4:577–600. doi: 10.1016/j.mito.2004.07.038. [DOI] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nature Genet. 1996;14:146–151. doi: 10.1038/ng1096-146. [DOI] [PubMed] [Google Scholar]
- Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA. The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TBL, Holliday R. The evolution of ageing and longevity. Proc R Soc Lond B. 1979;205:531–546. doi: 10.1098/rspb.1979.0083. [DOI] [PubMed] [Google Scholar]
- Krakauer DC, Mira A. Mitochondria and germ-cell death. Nature. 1999;400:125–126. doi: 10.1038/22026. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nature Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kuroiwa T, Ohta T, Kuroiwa H, Shigeyuki K. Molecular and cellular mechanisms of mitochondrial nuclear division and mitochondriokinesis. Microsc Res Tech. 1994;27:220–232. doi: 10.1002/jemt.1070270304. [DOI] [PubMed] [Google Scholar]
- Laipis P, Walle MVD, Hauswirth W. Unequal partitioning of bovine mitochondrial genotypes among siblings. Proc Natl Acad Sci USA. 1988;85:8107–8110. doi: 10.1073/pnas.85.21.8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J, Hekimi S. When a theory of aging ages badly. Cell Mol Life Sci. 2009 doi: 10.1007/s00018-009-0138-8. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros F, Malka F, Frachon P, Lombes A, Rojo M. Organization and dynamics of human mitochondrial DNA. J Cell Sci. 2004;117:2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- Liu T, Lu B, Lee I, Ondrovicova G, Kutejova E, Suzuki CK. DNA and RNA Binding by the Mitochondrial Lon Protease Is Regulated by Nucleotide and Protein Substrate. J Biol Chem. 2004;279:13902–13910. doi: 10.1074/jbc.M309642200. [DOI] [PubMed] [Google Scholar]
- McKiernan SH, Colman R, Lopez M, Beasley TM, Weindruch R, Aiken JM. Longitudinal analysis of early stage sarcopenia in aging rhesus monkeys. Experimental Gerontology. 2009;44:170–176. doi: 10.1016/j.exger.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren A. Primordial germ cells in the mouse. Dev Biol. 2003;262:1–15. doi: 10.1016/s0012-1606(03)00214-8. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. Some Genetic Aspects of Sex. American Naturalist. 1932;66:118–138. [Google Scholar]
- Nass M. Mitochondrial DNA I. Intramitochondrial Distribution and Structural Relations of Single- and Double-length Circular DNA. J Mol Biol. 1969;42:521–528. doi: 10.1016/0022-2836(69)90240-x. [DOI] [PubMed] [Google Scholar]
- Pérez VI, Bokov A, Remmen HV, Mele J, Ran Q, Ikeno Y, Richardson A. Is the oxidative stress theory of aging dead? Biochimica et Biophysica Acta (BBA) - General Subjects. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, Andziak B, Yang T, Edrey Y, Friguet B, et al. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc Natl Acad Sci USA. 2009;106:3059–3064. doi: 10.1073/pnas.0809620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piko L, Matsumoto L. Number of mitochondria and some properties of mitochondrial DNA in the mouse egg. Dev Biol. 1976;49:1–10. doi: 10.1016/0012-1606(76)90253-0. [DOI] [PubMed] [Google Scholar]
- Pinz KG, Shibutani S, Bogenhagen DF. Action of Mitochondrial DNA Polymerase Gamma at Sites of Base Loss or Oxidative Damage. J Biol Chem. 1995;270:9202–9206. doi: 10.1074/jbc.270.16.9202. [DOI] [PubMed] [Google Scholar]
- Rajasimha HK, Chinnery PF, Samuels DC. Selection against Pathogenic mtDNA Mutations in a Stem Cell Population Leads to the Loss of the 3243A’G Mutation in Blood. Am J Human Genet. 2008;82:333–343. doi: 10.1016/j.ajhg.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, Kuhl I, Gaisne M, Bonnefoy N, Smeitink JA, et al. The human mitochondrial ribosome recycling factor is essential for cell viability. Nucl Acids Res. 2008;36:5787–5799. doi: 10.1093/nar/gkn576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer E, Dencher NA, Vonck J, Parcej DN. Three-Dimensional Structure of the Respiratory Chain Supercomplex I1III2IV1 from Bovine Heart Mitochondria. Biochemistry. 2007;46:12579–12585. doi: 10.1021/bi700983h. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, et al. Extension of Murine Life Span by Overexpression of Catalase Targeted to Mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Shoffner JM, Lott MT, Lezza AMS, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- Shoubridge E. Mitochondrial DNA segregation in the developing embryo. Hum Reprod. 2000;15:229–234. doi: 10.1093/humrep/15.suppl_2.229. [DOI] [PubMed] [Google Scholar]
- Stewart JB, Freyer C, Elson JL, Larsson N-G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat Rev Genet. 2008a doi: 10.1038/nrg2396. advanced online publication. [DOI] [PubMed] [Google Scholar]
- Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson N-G. Strong Purifying Selection in Transmission of Mammalian Mitochondrial DNA. PLoS Biology. 2008b;6:e10. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly JL. Commuting the Death Sentence: How Oocytes Strive To Survive. Nat Rev Mol Cell Biol. 2001;2:838–848. doi: 10.1038/35099086. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink J, Rovio A, Bruder C, Bohlooly-Y M, Gidlof S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJA, Mohamed H, Wikstro JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Blerkom J. Mitochondria in human oogenesis and preimplantation embryogenesis: engines of metabolism, ionic regulation and developmental competence. Reproduction. 2004;128:269–280. doi: 10.1530/rep.1.00240. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Jones DP. Current Thoughts on the Role of Mitochondria and Free Radicals in the Biology of Aging. J Gerontol A Biol Sci Med Sci. 2009;64:171–174. doi: 10.1093/gerona/gln058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, Teoli D, Shoubridge EA. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat Genet. 2008;40:1484–1488. doi: 10.1038/ng.258. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annual Review of Genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Bogenhagen DF. Human Mitochondrial DNA Nucleoids Are Linked to Protein Folding Machinery and Metabolic Enzymes at the Mitochondrial Inner Membrane. J Biol Chem. 2006;281:25791–25802. doi: 10.1074/jbc.M604501200. [DOI] [PubMed] [Google Scholar]
- Williams GC. Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution. 1957;11:398–411. [Google Scholar]
- Zheng W, Khrapko K, Coller HA, Thilly WG, Copeland WC. Origins of human mitochondrial point mutations as DNA polymerase gamma-mediated errors. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2006;599:11–20. doi: 10.1016/j.mrfmmm.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Zhou ZH, McCarthy DB, O’Connor CM, Reed LJ, Stoops JK. The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc Natl Acad Sci USA. 2001;98:14802–14807. doi: 10.1073/pnas.011597698. [DOI] [PMC free article] [PubMed] [Google Scholar]