

Abstract
Vascular barrier regulation is intimately linked to alterations in the distribution and configuration of the endothelial cell (EC) cytoskeleton in response to angiogenic and edemagenic agonists. Critical actin cytoskeletal rearrangement includes spatially-directed increases in myosin light chain (MLC) phosphorylation, catalyzed by Ca2+/calmodulin-dependent non-muscle myosin light chain kinase variants (nmMLCK1- and -2), as well as association of nmMLCK with the actin-binding protein, cortactin. As these associations have proven difficult to quantify in a spatially-specific manner, we now describe the utility of intensity correlation image analysis and the intensity correlation quotient (ICQ) to quantify colocalization in fixed and live-cell imaging assays in human pulmonary artery EC. From baseline ICQ values averaging 0.216 reflecting colocalization of cortactin-DsRed with EGFP-nmMLCK fusion proteins in resting EC, thrombin-induced EC contraction significantly reduced cortactin-DsRed-EGFP-nmMLCK colocalization (nmMLCK1: ICQ = 0.118; nmMLCK2: ICQ = 0.091) whereas the potent EC barrier-protective agonist, sphingosine 1-phosphate (S1P), significantly increased nmMLCK-cortactin colocalization within lamellipodia (nmMLCK1: ICQ = 0.275; nmMLCK2: ICQ = 0.334). Over-expression of a cortactin-DsRed mutant fusion protein lacking the SH3 domain, known to be essential for cortactin-nmMLCK association, reduced baseline and S1P-mediated live-cell colocalization with each nmMLCK variant (nmMLCK1: ICQ = 0.160; nmMLCK2: ICQ = 0.157). Similarly, expression of a truncated EGFP-nmMLCK2 mutant lacking cortactin- and actin-binding domains, markedly reduced basal localization in lamellipodia and abolished colocalization with cortactin-DsRed in lamellipodia after S1P (ICQ = −0.148). These data provide insights into the molecular basis for vascular barrier-regulatory cytoskeletal responses and support the utility of sophisticated imaging analyses and methodological assessment to quantify the critical nmMLCK and cortactin interaction during vascular barrier regulation.
Keywords: cytoskeleton, permeability, F-actin, intensity correlation image analysis, immunofluorescence, lamellipodia, sphingosine 1-phosphate
Introduction
The microvascular endothelium provides a semipermeable barrier that regulates the passage of circulating vascular nutrients and fluids and the extravascular interstitium of all organs. Perturbation in the integrity of this monolayer during tumor angiogenesis or inflammation occurs in response to edemagenic agents such as thrombin, histamine, proinflammatory cytokines and activated leukocytes (Dudek and Garcia, 2001; Garcia et al., 1986; Mehta and Malik, 2006; Petrache et al., 2001) and results in fluid leakage, organ edema and dysfunction. Restoration of endothelial cell (EC) integrity is promoted by natural angiogenic agents such as angiopoietin-1 (Thurston et al., 1999), hepatocyte growth factor (HGF) (Liu et al., 2002; Singleton et al., 2007), and sphingosine-1-phosphate (S1P), a lipid signaling molecule secreted by circulating platelets and erythrocytes (Garcia et al., 2001; McVerry and Garcia, 2005; Schaphorst et al., 2003). Both barrier-disruptive and barrier-restorative processes are now recognized as extensively dependent on dynamic rearrangements of the endothelial cytoskeleton. For example, the potent edemagenic agonist, thrombin, induces formation of thick actin stress fibers that contract EC and disrupt paracellular junctions (Dudek and Garcia, 2001; Garcia et al., 1986), whereas S1P reorganizes the cytoskeleton into dense peripheral bands that restore EC intercellular junctional integrity (Dudek et al., 2004; Garcia et al., 2001; Shikata et al., 2003).
Tensile force generation and contraction of actin filaments in non-muscle cells, such as EC, are mediated by phosphorylation of the regulatory myosin light chain (MLC) subunit on Thr18/Ser19, which catalyzes the ratcheting of actin-myosin bonds resulting in intracellular tension development. This enzymatic reaction involves the multi-dimensional non-muscle isoform of myosin light chain kinase (nmMLCK), a 1914 amino acid (210 kDa) Ca2+/calmodulin-dependent, actin-binding protein encoded by the MYLK gene, which also encodes the 1091 amino acid (108 kDa) smooth muscle isoform as well as the 19 kDa protein known as kinase-related protein (KRP) or telokin (Supplemental Figure 1A). In addition to smMLCK and KRP, we previously identified five splice variants compared to the longest variant (nmMLCK1), with nmMLCK1 and nmMLCK2 being the most abundant isoform variants in many tissues including endothelium (Supplemental Figure 1A) (Birukov et al., 2001; Lazar and Garcia, 1999).
Thrombin increases nmMLCK activity and results in profound cytoskeletal rearrangement, loss of cortical actin, and rapid and dramatic formation of transcellular stress fibers resulting in increased transendothelial permeability (Dudek and Garcia, 2001). Interestingly, ligation of barrier-enhancing receptors including S1PR1 and c-Met (Dudek et al., 2004; Garcia et al., 2001; Liu et al., 2002) as well as others (Finigan et al., 2005; Singleton et al., 2006), results in recruitment of key signaling molecules and their targets such as p60src, c-Abl, nmMLCK, and the actin- and nmMLCK-binding protein, cortactin, to lipid rafts (Zhao et al., 2009). These molecular interactions result in dynamic activation of nmMLCK and dramatic spatially-distinct localization of nmMLCK and nmMLCK binding partners, such as the actin-binding protein, cortactin, within cortical actomyosin rings, events intimately linked to enhanced paracellular junctional integrity and EC barrier enhancement (Dudek et al., 2004; Garcia et al., 2001).
Unfortunately, the inability to quantify nmMLCK association with cortactin in a spatially-specific manner has proven to be a major limitation to interrogating the molecular mechanisms underlying cytoskeleton-driven EC barrier regulation. We now report the utility of intensity correlation image analysis and the intensity correlation quotient (ICQ) (Brittain et al., 2009; Li et al., 2004; Racz, 2008) to quantify the colocalization of cortactin with nmMLCK1 and -2 isoforms in fixed and live-cell assays under conditions of EC barrier enhancement and disruption. Our quantitative results indicate that robust thrombin-induced EC contraction reduces colocalization of cortactin with nmMLCK fusion proteins whereas the potent barrier-protective agonist, S1P, increased colocalization of nmMLCK and cortactin within barrier-enhancing lamellipodia. Our imaging analyses in live-cell assays confirm our earlier biochemical studies (Dudek et al., 2002; Dudek et al., 2004) and demonstrate cortactin-nmMLCK association to require the SH3 domain of cortactin (Supplemental Figure 1B) as well as the cortactin- and actin-binding domains of nmMLCK. Together, these data provide insights into the molecular basis for vascular barrier-regulatory cytoskeletal responses and support the utility of sophisticated imaging analyses and methodological assessment to quantify the critical interaction that occurs between nmMLCK and cortactin during EC barrier regulation.
Materials and Methods
Reagents and Antibodies
Reagent chemicals, including S1P and thrombin, were obtained from Sigma (St. Louis, MO) unless otherwise specified. Laboratory grade paraformaldehyde was purchased from Fisher Scientific (Fair Lawn, NJ). Cortactin monoclonal antibody 4F11 was purchased from Upstate Biotechnology (Lake Placid, NY). The vector pAcGFP1/Actin was purchased from Clontech (Mountain View, CA) while all other fluorescent dye-labeled reagents and Prolong Gold with DAPI were obtained from Molecular Probes (Eugene, OR). Sterile Dulbecco’s phosphate buffered saline (D-PBS) was purchased from Mediatech (Herndon, VA), trypsin was purchased from Invitrogen (Grand Island, NY), and all other cell culture reagents were purchased from Lonza (Walkersville, MD). HMVEC-L transfection reagent was purchased from Amaxa Biosystems (Gaithersburg, MD).
DNA Constructs
The nmMLCK constructs used were wild-type nmMLCK1 and -2 and an N-terminal fragment of nmMLCK1 consisting of the first 496 amino acids (Supplemental Figure 1A). The open reading frames for wild-type nmMLCK1 and -2 were subcloned from the mammalian expression vectors pJM1 and pJM2 (Moitra et al., 2008; Wadgaonkar et al., 2003), respectively, as XmaI-EcoRI fragments into BspEI-EcoRI digested pEGFP-C1 vector (Clontech, Mountain View, CA). The resulting plasmids were sequence-verified on both strands and represent the CMV-promoter-driven mammalian expression vectors for EGFP-tagged nmMLCK1 and nmMLCK2. The 496-aa N-terminal fragment of nmMLCK2 was subcloned from the pGEX-6P-3 vector (Amersham Biosciences, Pittsburgh, PA) as an EcoRI blunt ended fragment (XhoI digestion and fill-in) into an EcoRI blunt ended digestion (BamHI digestion and fill-in) of pEGFP-C1. The resulting plasmid was verified by sequencing of both cDNA strands and represents the vector for the EGFP-tagged N-terminal fragment of nmMLCK2 (EGFP-nmMLCK2Nterm). Rat cortactin constructs in pDsRed-N1 were generous gifts from Drs. Mark McNiven (Mayo Clinic) and H. Clive Palfrey (University of Chicago) and represented full-length wild-type cortactin and cortactinΔSH3, consisting of amino acids 1–450 (McNiven et al., 2000) (Supplemental Figure 1B).
Cell Culture
Human pulmonary artery endothelial cells (EC) obtained from Lonza (Basel, Switzerland) were cultured in complete growth medium consisting of Endothelial Growth Medium-2 (EGM-2) with 10% fetal bovine serum and incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air as we have previously described (Dudek et al., 2004; Garcia et al., 2001). Endothelial cells were utilized at passages 5–9 and on the day prior to experimentation, cell culture medium was changed to EGM-2/2% FBS.
Transfection
The Amaxa nucleofection system was applied to EC for transient transfection of plasmid DNA (Amaxa Biosystems, Gaithersburg, MD). On the day preceding transfection, EC were seeded at ~70–80% confluence in complete medium in T-75 flasks (Corning, Lowell, MA). On the day of transfection, cells were washed twice in sterile D-PBS, harvested in 0.05% trypsin, diluted into 10 ml culture medium, and counted with a hemacytometer. As per manufacturer instructions, 500,000 cells were pelleted at 200g in 4°C for 10 min, the supernatant completely aspirated, and cells were resuspended in 100 μL of HMVEC-L kit nucleofection solution. Two μg of total plasmid DNA were added to the mixture either as 2 μg of EGFP-nmMLCK construct alone or a combination of 1 μg pEGFP-C1/nmMLCK DNA or pAcGFP1/Actin and 1 μg pDsRed-N1/cortactin DNA. The mixture was immediately transferred to a manufacturer’s cuvette, transfected with program S-5 in a Nucleofector™ I machine, followed by the addition of 500 μL of pre-warmed, equilibrated complete culture medium. The mixture was transferred to a 35 mm culture dish containing 1 ml complete medium and a sterile, gelatin-coated 25-mm glass coverslip (for eventual live-cell imaging). The cuvette was quickly washed once more with 500 μL of complete medium, which was subsequently added to the same 35 mm culture dish. Transfected cells were allowed to recover in complete medium for ~6–8 hrs, and then the culture medium was changed to EGM-2/2% FBS and incubated with transfected cells overnight followed by live-cell imaging or immunofluorescence staining approximately 24–36 hrs after nucleofection, a time when peak EGFP-nmMLCK expression has occurred.
Indirect Immunofluorescence
Colocalization of the nmMLCK isoform with endogenous cortactin was measured and indirect immunofluorescence (Burkhardt et al., 1997; Burkhardt et al., 1993) of cortactin assayed. EC transfected with one of the three EGFP-nmMLCK constructs were added to a 12-well plate containing a gelatin-coated 18 mm glass coverslip in each well and allowed to recover in complete medium for 6–8 hours. Transfected EC were stimulated at 37°C with either 1 u/ml thrombin for 10 min, 1 μM S1P for 15 or 30 min, or 1 u/ml thrombin for 10 min followed by 1 μM S1P for 30 min. Coverslips were dipped in sterile D-PBS and transferred to wells containing 1 ml of 4% paraformaldehyde/PBS, pH 7.4. Following fixation for 20 min at room temperature, cells were washed and unreacted aldehyde groups were quenched in 50 mM NH4Cl/PBS, pH 7.4 (three 5-min washes) and blocked and permeabilized in 0.25% fish skin gelatin/0.01% saponin/0.1% NaN3/PBS, pH 7.4, (blocking solution) for 30 min at RT. Cortactin monoclonal antibody 4F11 was diluted 1:100 in blocking solution and incubated with cells for 2 hrs at RT. Cells were washed thrice in blocking solution, then incubated with goat antimouse-AlexaFlour633 diluted 1:100 in blocking solution for 1 hr in the dark. Following three washes in blocking buffer, actin was stained by incubating the cells in 5 units/ml phalloidin-rhodamine/blocking solution for 30 min at RT in the dark. Finally, cells were washed x3 for 5 min each in blocking solution, then mounted in 10 μL of Prolong Gold with DAPI and cured overnight at RT in the dark. Cells were imaged on a Leica TCS SP5 AOTF laser-scanning confocal microscopy system scanning at 400 Hz with Ar 488 nm, He/Ne 561 nm, and He/Ne 633 nm lasers, a Leica DMI 6000 microscope, and an HCX PL APO CS 63X NA1.4 oil objective lens. Laser power was set to 6% illumination for the argon laser, 10% for the He/Ne 561 nm laser, and 15% for the He/Ne 633 nm laser. Emission bandwidths were set to 498–545nm for EGFP signal, 570nm–625nm for rhodamine signal, and 643–790 nm for AlexaFluor633 signal. Twelve-bit 512×512 images were acquired sequentially scan line-by-scan line and with a line average setting of 16 with Leica LAS AF software and detected with a photomultiplier tube. All post-acquisition image processing was performed with the MBF_ImageJ software bundle, version 1.39 (Tony Collins, McMaster University, http://www.macbiophotonics.ca/imagej/ and Wayne Rasband, NIH, http://rsb.info.nih.gov/ij/).
Colocalization Quantification
Colocalization was quantified by intensity correlation analysis in which colocalization is defined as the synchronous increase or decrease in fluorescence intensities, as predicted when labeled proteins are part of the same molecular complex or organelle (Bolte and Cordelieres, 2006; Khanna et al., 2006; Li et al., 2004). The overall difference of pixel intensities from the mean intensity of a defined region of interest from a single channel was assumed to equal zero, such that Σn pixels (Gi−Gav) = 0 and Σn pixels (Ri−Rav) = 0, in which Gi and Ri represent the green and red intensities of an individual pixel and Gav and Rav represent the average pixel intensity for each green and red image. The product of the two equalities should tend to equal a zero value when intensity distributions are entirely random, Σn pixels (Gi−Gav) (Ri−Rav) ≈0. When two signals vary dependently, as predicted when two proteins are part of the same molecular complex, the mean deviation product trends toward a positive value, Σn pixels(Gi−Gav)(Ri−Rav)>0. When two signals vary independently, the mean deviation products tends toward a negative value, Σn pixels (Gi−Gav) (Ri−Rav) <0. The intensity correlation quotient (ICQ) was devised as a single coefficient to account for the covariance of mean deviation products in a defined intracellular area (Bolte and Cordelieres, 2006; Khanna et al., 2006; Li et al., 2004). The ICQ is the ratio of the number of positive (Gi−Gav) (Ri−Rav) values to the total number of pixels with mean deviation products. By subtracting 0.5 from the resulting ratio, the ICQ is finally corrected to a scale of −0.5 for complete segregation, 0.0 for randomness, and +0.5 for complete colocalization. ICQ values were calculated from the intensity correlation analysis (ICA) application incorporated into the MBF_ImageJ bundle. A range of 4–20 paired images were evaluated for each experimental treatment.
For each image, background signal was subtracted by drawing a region of interest (ROI) within an area unoccupied by cells and the average fluorescence intensity within that region was subtracted from the entire image. Following background subtraction, three ICQ values were calculated for each cell. First, an ICQ between an EGFP-nmMLCK (green image) and immunofluorescent endogenous cortactin (red image) within an entire cell was calculated. A ROI was drawn around an entire cell in its paired green and red images and the ROI dimensions were saved. Both images were automatically thresheld with an isodata algorithm incorporated into ImageJ software and the ICQ calculated. Second, an ICQ was calculated for colocalization within individually selected lamellipodia in the nmMLCK image, and ROIs were drawn around thresheld lamellipodia. Lamellipodia were defined as a flat (0.1–0.2 μm in thickness), membrane–enclosed and highly dynamic leaflet of cytoplasm usually spanning 1–5 μm in length. Peripheral ruffles and leaflets were counted as lamellipodia while filopodia and isolated microspikes were excluded. These ROIs were transferred to the red image and ICQs were calculated within these lamellipodia ROIs. Third, colocalization was quantified for areas outside of lamellipodia, which we have termed “cytoplasm.” Pixels within the previously drawn lamellipodia ROIs were subtracted from paired green and red images. The saved ROI originally drawn around the entire cell was reapplied to paired green and red images lacking their lamellipodia. Within this ROI, colocalization between thresheld pixels in paired green and red images was calculated. The cellular, lamellipodia, and cytoplasm ICQ values were averaged within each compartment category, and statistical significance was determined with the non-parametric Wold-Wolfowitz runs test.
Quantification of Lamellipodia Localization
Endothelial cells transfected with EGFP-nmMLCK constructs and immunostained for endogenous cortactin were used in these calculations. The EGFP-nmMLCK images were background-subtracted and ROIs drawn around individual cells. All areas outside the cell were cleared in order to best visualize the leading edges of lamellipodia, and the fluorescence intensity within an entire cell was summed. Cell images were automatically thresheld by ImageJ with an isodata algorithm. Thresheld pixels in lamellipodia were selected by drawing regions of interest and the fluorescence intensities within the selected lamellipodia regions were summed. The percentage of EGFP-nmMLCK in lamellipodia was calculated from the ratio of the summed fluorescence intensities within selected lamellipodia to the summed fluorescence intensity of the entire cell.
Live Endothelial Cell Imaging
Prior to imaging, 25 mm gelatin-coated coverslips containing transfected EC were transferred to a recording chamber (ALA Scientific Instruments, Westbury, NY), then overlayed with 2 ml bath solution (phenol red-free EBM/2% FBS), and the recording chamber was maintained on heating stage at 37°C. All live-cell imaging was performed with a Leica TCS SP5 AOTF laser-scanning confocal system equipped with an active resonance scanner set to 8000 Hz, Ar 488-nm plus He/Ne 561-nm lasers, a Leica DMI 6000 microscope, and an HCX PL APO CS 63X NA1.4 oil objective lens. Laser power was set to 6% illumination for the argon laser and 10% for the He/Ne laser. Emission bandwidths were set to 498–555nm for EGFP signal and 570nm–730nm for DsRed signal, and twelve-bit 512×512 images were acquired every six seconds and sequentially scan line-by-scan line with Leica LAS AF software and detected with a photomultiplier tube. Movies were recorded pre-stimulation (6 min), during a 15-min thrombin (1 U/ml) application, and during 1 μM S1P stimulation (30–45 min). Data were subsequently processed with ImageJ with a Kalman stack filter to remove high gain noise and background subtracted. ROIs were drawn around lamellipodia over the course of the entire movie and ICQs were calculated at every time point for each lamellipodium with a customized ImageJ macro. Reported data represent time points of peak colocalization.
Statistical Analysis
Data are presented as group means ± SE. Statistical significance was determined with the non-parametric Wold-Wolfowitz runs test. This statistical test was selected because it allows for the evaluation of two different kinds of statistical assumptions, binomial to determine if colocalization is present or not, and Poisson to assess for the randomness of this colocalization. In all cases, we defined statistical significance as p < 0.05.
Results
Spatial Distribution of nmMLCK1 and nmMLCK2 in Human Lung Endothelium
EGFP-labeled nmMLCK constructs were ectopically expressed in EC to detect differential patterns of distribution between the two splice variants which vary by the deletion of exon 11 (69 amino acids) in nmMLCK2 (Supplemental Figure 1A). We noted largely identical distributions of nmMLCK1 and -2 in unstimulated, sub-confluent cells with both EGFP-nmMLCK1 and -2 fusion proteins prominently residing at the leading edges of lamellipodia and along fine actin fibers that transverse the cell (Figures 1A & 1B). Transfection with a unique EGFP-nmMLCK2 deletion mutant construct which lacks both the actin- and cortactin-binding domains (includes only the initial 496 aa) was utilized to explore the functional roles of these domains in determining nmMLCK cellular localization. Interestingly, this N-terminal nmMLCK2 deletion mutant associates weakly with cytoskeletal actin fibers (Figures 1C, 1F, 1I, 1L), possibly via actin-binding proteins that associate with the putative SH2- or SH3-binding domains contained within the 496 aa nmMLCK fragment (Supplemental Figure 1A). However, EGFP-nmMLCK2Nterm is nearly absent in lamellipodia (Figure 1C, 1I, 1L), suggesting that the actin-binding and/or cortactin-binding domains are required for nmMLCK localization to lamellipodia.
Figure 1. Distribution of nmMLCK variants and mutant fusion proteins in human lung endothelium.

EGFP conjugates of the nmMLCK1 and nmMLCK2 variants, as well as the 496-aa N-terminal fragment of nmMLCK2 were ectopically expressed in human lung EC and incubated with either vehicle (Panels A, B, C), thrombin for 10 min (Panels D, E, F), S1P for 30 min (Panels G, H, I), or thrombin for 10 min and followed by S1P for 30 min (Panels J, K, L). EC were then fixed, immunostained, and imaged as described in Methods section. In unstimulated cells (Panels A, B, C), EGFP-nmMLCK1 and -2 lie along thin fibers transversing the cell as well as strongly localized within lamellipodia (arrows). The N-terminal fragment of nmMLCK2 localizes to broad actin fibers but fails to localize within lamellipodia (Panel C). Thrombin challenge causes EC retraction and disappearance of lamellipodia and redistribution of both nmMLCK variants as well as nmMLCK2Nterm to thick actin stress fibers (arrows, Panels D, E, F). S1P results in translocation of some nmMLCK to spatially distinct actin cortical bands (Panels G, H, J, K) along with ruffling lamellipodia (arrows). The N-terminal fragment of nmMLCK2 is largely absent from lamellipodia (Panels I and L).
EC expressing full length EGFP-nmMLCK1 and -2 fusion proteins or the truncated EGFP-nmMLCK2Nterm protein, were next challenged with either the barrier-disrupting agent, thrombin, or the EC barrier-enhancing agonist, S1P. Both agonists serve to activate nmMLCK and to initiate dynamic cytoskeletal rearrangement of actomyosin fibers with dramatic agonist-specific alterations in the intracellular distribution of nmMLCK (Dudek and Garcia, 2001; Dudek et al., 2004). Thrombin induces cell contraction and disappearance of lamellipodia with all three nmMLCK fusion proteins associated with the newly formed thick, prominent cytoplasmic actin stress fibers (Figures 1D–1F). In contrast, S1P induced formation of dense cortical actin rings to stabilize EC (Garcia et al., 2001), and the translocation of both EGFP-nmMLCK1 and -2 fusion proteins to these spatially distinct actin cortical bands and to actively ruffling lamellipodia (Figures 1G, 1H, 1J, 1K). The EGFP-nmMLCK2Nterm fusion protein localized partially to peripheral cortical bands after S1P but failed to localize within lamellipodia (Figures 1I & 1L), suggesting that the actin-binding and/or cortactin-binding domains are again necessary for targeting to lamellipodia but not for localization to peripheral dense actin bands.
To investigate whether amino acids encoded by exon 11, deleted in nmMLCK2 (Birukov et al., 2001; Lazar and Garcia, 1999), exert an effect on nmMLCK1 localization, we quantified the relative proportion of each EGFP-nmMLCK construct found in lamellipodia, measured as the ratio of fluorescence intensity in lamellipodia against the fluorescence intensity of the entire cell. After transfection, cells were plated at subconfluence in order to view clearly definable lamellipodia suitable for quantification. However, this subconfluent plating results in EC lamellipodia which are much larger in size than that seen in confluent cells and comparable in size to that seen in S1P-stimulated cells, although prominent cortical actin ring formation remains absent. In untreated control cells, on average 9.7% of EGFP-nmMLCK1 localized to lamellipodia (Figure 2A) whereas 13.7% of EGFP-nmMLCK2 was found in the same compartment (Figure 2B). Thrombin markedly reduced the proportion of either nmMLCK isoform in the cell periphery to <2% (Figures 2A & 2B). Both EGFP-nmMLCK1- and -2 localize to lamellipodia following S1P stimulation (8.9% and 11.8%, respectively) albeit at slightly less proportions than seen in untreated subconfluent cells (Figures 2A & 2B); however, these differences did not reach statistical significance. S1P rescue of thrombin-treated EC restored EGFP-nmMLCK2 localization to lamellipodia to 11.5% but only to 5.1% for EGFP-nmMLCK1. Surprisingly, untreated subconfluent EC had a higher proportion of each nmMLCK isoform in lamellipodia than did both S1P-stimulated treatments suggesting that the formation of dense cortical bands may modestly reduce the amount of nmMLCK available for lamellipodia formation and growth. Compared to EGFP-nmMLCK1, there were statistically significant greater levels of EGFP-nmMLCK2 found in lamellipodia of untreated and S1P-rescued cells (p < 0.05), suggesting that exon 11 may encode amino acids which create protein-protein interactions for nmMLCK1 that differ from nmMLCK2 in subconfluent EC and in cells recovering from barrier disruption, possibly via tyrosine phosphorylation at Y464 and Y471 in nmMLCK1 (Birukov et al., 2001). In all cases, less than 2% of the total cellular EGFP-nmMLCK2Nterm localized to lamellipodia (Figure 2C). Clearly, the actin-binding domain and/or cortactin-binding domain are essential for significant translocation of nmMLCK to lamellipodia.
Figure 2. Relative proportion of nmMLCK isoforms in human lung endothelial lamellipodia.

Human lung EC were treated as described in Figure 1, and the percentage of total spatially–specific cellular distribution of nmMLCK isoforms determined (see Methods) after vehicle, thrombin for 10 min, S1P for 30 min, or thrombin for 10 min followed by S1P for 30 min. Panel A: The percentage of total cellular EGFP-nmMLCK1 localized to lamellipodia is indicated for each condition. * p<0.05; N = 4–18 per condition. Panel B: The percentage of total cellular EGFP-nmMLCK2 localized to lamellipodia is indicated for each condition. * p<0.05; N = 7–17 per condition. Panel C: The percentage of total cellular EGFP-nmMLCK2Nterm localized to lamellipodia is indicated for each condition. Less than 2% is localized to lamellipodia in each condition. N = 10–14 per condition.
Distribution of Cortactin in Human Lung Endothelium and Quantitative Colocalization with nmMLCK in Fixed Human Lung Endothelium
The association of the actin- and nmMLCK-binding cytoskeletal effector, cortactin, with both nmMLCK1 and -2 variants has been previously demonstrated with purified protein preparations in vitro (Dudek et al., 2002). To more fully characterize the cortactin-nmMLCK association in situ, we utilized intensity correlation analysis to quantitatively describe the colocalized distribution of nmMLCK and cortactin in human lung EC, a methodology which presumes that if two proteins are part of the same molecular complex, their fluorescence intensities should rise and fall in synchrony above the mean fluorescence intensity for the entire cell or a defined region of interest (see Methods). Three regions of interest were analyzed: the entire cell, lamellipodia, and cytoplasm with colocalization occurring when the fluorescence intensities of both green and red signals rise above their mean intensities for a defined region of interest. In a pixel with colocalized signals, both Gi−Gav > 0 and Ri−Rav > 0 and the product of these differences is positive. The degree of colocalization is quantified with the intensity correlation quotient which is the ratio of the number pixels with positive mean deviation products to all thresheld pixels within a region of interest. Peak colocalization occurs at an ICQ value of 0.5 while complete segregation of two signals occurs at ICQ = −0.5. Total random overlap of two signals occurs at ICQ = 0.
Colocalization studies of EGFP-nmMLCK1 and endogenous cortactin in lamellipodia of human lung EC demonstrated that both nmMLCK1 and cortactin distribute to lamellipodia with the fluorescence signals from both proteins overlapping as shown in yellow pixels in Figure 3Ai–iv. In these studies the lower panels, Figure 3Ai′-iv′, depict colocalization as analyzed by intensity correlation analysis with positive mean deviation products highlighted with warm colors and pixels with negative mean deviation products (and including the case when both GiGav < 0 and Ri−Rav < 0) are represented with cool colors. Endogenous cortactin and EGFP-nmMLCK1 colocalize almost exclusively in lamellipodia in unstimulated and S1P-stimulated cells (Figure 3Ai′–iv′), with average ICQ values from multiple independent experiments shown in Figure 3B. There were no statistically significant differences in colocalization in lamellipodia between control and S1P-stimulated EC for nmMLCK1. The low ICQ values for the cytoplasm indicate that nmMLCK1 and cortactin association is almost entirely random in this compartment (Figure 3B). Segregation of cortactin and nmMLCK1 occurs in the peripheral cortical cytoskeletal bands as highlighted by blue pixels in Figure 3Ai′–iv′. Thrombin-challenged EC exhibit lamellipodial retraction (Figure 3Aii & ii′) and redistribution of nmMLCK1 to thick actin stress fibers and cortactin to the cytoplasm. ICQ values (Figure 3B) mirror lamellipodia retraction by thrombin which completely ablates colocalization of EGFP-nmMLCK1 and cortactin in the cell periphery and dramatically reduces ICQ values and colocalization in the cytoplasm and cell as a whole. Thrombin induces a statistically significant reduction in peripheral as well as whole-cell colocalization of nmMLCK1 with cortactin when compared to unstimulated (lamellipodia, p <0.001) and S1P-stimulated cells (lamellipodia, p <0.001).
Figure 3. Colocalization of EGFP-nmMLCK1 and endogenous cortactin in human lung endothelial lamellipodia.

Panel A. EC overexpressing EGFP-nmMLCK1 were incubated with either vehicle (i), thrombin for 10 min (ii), S1P for 30 min (iii), or thrombin for 10 min followed by S1P for 30 min (iv) at 37°C, then fixed and immunostained for cortactin (red). Yellow indicates areas of colocalization. The lower panels, i′-iv′, depict colocalization of endogenous cortactin with nmMLCK1. Warm colors indicate pixels with positive mean deviation products, where cortactin and nmMLCK1 fluorescence intensities rose in synchrony above the cellular average intensity, thus indicating colocalization. Blue pixels represent regions with negative mean deviation products in which the cortactin and nmMLCK1 fluorescence intensities varied independently of each other. Shown are representative images selected from 4–20 images for each treatment. Panel B: The mean ICQ values for cortactin and EGFP-nmMLCK1 colocalization were calculated as described in Methods for the entire cell, the cytoplasm, and the lamellipodia under each condition [vehicle (No stim), thrombin for 10 min, S1P for 30 min, or thrombin for 10 min followed by S1P for 30 min]. Thrombin induces a statistically significant reduction in peripheral as well as whole-cell colocalization of nmMLCK1 with cortactin when compared to unstimulated (whole cell or lamellipodia, *p <0.001) and S1P-stimulated cells (whole cell or lamellipodia, *p <0.004). N = 4–20 per condition.
The colocalization of EGFP-nmMLCK2 and endogenous cortactin in lamellipodia of EC demonstrate findings very similar to that seen with nmMLCK1, with nmMLCK2 consistently found in lamellipodia in unstimulated and S1P-stimulated cells. The nmMLCK2 variant organizes into prominent stress fibers in response to thrombin (Figure 4Aii) and into dense cortical fibers in response to S1P (Figures 4Aiii & 4Aiv). As with nmMLCK1, cortactin and nmMLCK2 colocalize in lamellipodia in unstimulated (Figure 4Ai′) and S1P-treated cells (Figure 4Aiii′ & 4Aiv′) but not along nmMLCK2 stress fibers (Figure 4Aii′) and cortical dense fibers (Figure 4Aiii′–iv′) as reflected by the respective ICQ values in Figure 4B. As with nmMLCK1, the peripheral and whole-cell colocalization of EGFP-nmMLCK2 with cortactin was significantly reduced during thrombin stimulation as opposed to control or S1P treatment (Figures 4Aii–ii′ & 4B). Statistical comparisons of colocalization between the nmMLCK constructs were performed to determine whether exon 11 affected colocalization with cortactin. Interestingly, there were no statistically significant differences in lamellipodia colocalization between EGFP-nmMLCK1 and -2 among the four treatment groups, suggesting that while exon 11 may influence the amount of nmMLCK2 distributed into lamellipodia, it does not significantly influence lamellipodia colocalization between endogenous cortactin and EGFP-nmMLCK1 or -2 as measured in fixed EC.
Figure 4. Colocalization of EGFP-nmMLCK2 and endogenous cortactin in human lung endothelial lamellipodia.

Panel A. EC overexpressing EGFP-nmMLCK2 were incubated with either vehicle (i), thrombin for 10 min (ii), S1P for 30 min (iii), or thrombin for 10 min followed by S1P for 30 min (iv) at 37°C, then fixed and immunostained for cortactin (red). Yellow indicates areas of colocalization. The lower panels, i′-iv′, depict colocalization of endogenous cortactin with nmMLCK2 as described for Figure 3 above. Shown are representative images selected from 7–13 images for each treatment. Panel B: The mean ICQ values for cortactin and EGFP-nmMLCK2 colocalization were calculated as described in Methods for the entire cell, the cytoplasm, and the lamellipodia under each condition [vehicle (No stim), thrombin for 10 min, S1P for 30 min, or thrombin for 10 min followed by S1P for 30 min]. Thrombin induces a statistically significant reduction in peripheral as well as whole-cell colocalization of nmMLCK2 with cortactin when compared to unstimulated (whole cell or lamellipodia, *p <0.02) and S1P-stimulated cells (whole cell or lamellipodia, *p <0.01). N = 7–13 per condition.
As a negative control, we used a truncated nmMLCK2 construct lacking the actin- and cortactin-binding domains, EGFP-nmMLCK2Nterm, which not only distributed poorly to lamellipodia but also failed to colocalize with endogenous cortactin (Supplemental Figure 2A) with mean ICQ values consistently near zero, suggesting near random colocalization (Supplemental Figure 2B). Interestingly, EGFP-nmMLCK2Nterm still partially localized to stress fibers as well as cortical dense bands and may interact with other cytoskeletal proteins via two N-terminal putative SH3-binding sites (Supplemental Figure 1A). The truncated nmMLCK2 isoform, lacking a cortactin-binding domain but still able to localize partially to actin, serves as an effective negative control in these colocalization analyses. Of note, there were no statistically significant differences in ICQ values in thrombin-treated EC among any of the experimental conditions (nmMLCK1, nmMLCK2, or nmMLCK2Nterm), which suggests that cortactin colocalization with nmMLCK is disrupted under barrier-reducing conditions.
Colocalization Analysis of Cortactin-DsRed and Actin-GFP in Live Endothelial Cells
To characterize these protein interactions in a more dynamic fashion, next we pursued colocalization imaging studies in live cells. Such studies have the advantage of using a cell as its own control with quantitation of colocalization over time under various conditions. To our knowledge, there have been no prior published reports of intensity correlation analysis used to track dynamic colocalization of two proteins in live cells. As a positive control to characterize intensity correlation analysis in live EC, we co-expressed cortactin with its extensively characterized binding partner, actin. We selected actin as a positive control for quantification of cortactin colocalization because this interaction is well-established in the literature since the initial reports were published describing cortactin (Cosen-Binker and Kapus, 2006; Wu and Parsons, 1993). Figure 5 presents images from an extended movie (Supplemental Movie 1) depicting dynamic colocalization of cortactin-DsRed and actin-GFP in a single EC under basal, barrier disruptive (thrombin), and barrier enhancing conditions (S1P). Prior to stimulation, moderate colocalization of actin and cortactin occurred primarily in broad lamellipodia (Figure 5A), peaking at an ICQ of 0.289. After thrombin stimulation, lamellipodia retracted in association with peripheral actin-cortactin colocalization dropping more than four-fold to an ICQ of 0.070 (Figure 5B). Subsequent S1P stimulation in the same cell resulted in actin reorganization to peripheral dense bands as well as gradual lamellipodia reformation (Figures 5C–5F). Strong colocalization between actin and cortactin occurred (ICQ = 0.389) in ruffles associated with areas of lamellipodia growth as depicted in Figure 5C. Dynamic lamellipodia structures were observed for at least 35 minutes after S1P (Supplemental Movie 1), with actin-cortactin colocalization concentrated near or within these structures with ICQ values reaching peaks of 0.368, 0.369, and 0.340 (Figures 5D–5F).
Figure 5. Dynamic colocalization of actin-GFP and cortactin-DsRed with intensity correlation analysis applied to live-cell imaging.

HPAEC were cotransfected with actin-GFP and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. The panels represent images from an extended movie (see Supplemental Movie 1) depicting dynamic colocalization of cortactin-DsRed and actin-GFP in a single EC under basal conditions (A), after thrombin stimulation (B), and subsequent S1P stimulation (C–F). Yellow indicates areas of colocalization. ICQ quantitation for actin-cortactin colocalization in outlined lamellipodia is shown for each panel.
Similar colocalization was observed in two other movies (data not shown). Mean peak ICQ values during prestimulation, thrombin stimulation, and S1P stimulation were calculated from these three movies. Actin-cortactin peak ICQ was 0.311 ± 0.006 under basal conditions and dropped to 0.096 ± 0.015 after thrombin stimulation (p <0.05 vs. basal condition). When S1P subsequently was added, actin-cortactin peak colocalization increased to 0.359 ± 0.020. These studies demonstrate that colocalization between two cytoskeletal proteins can be tracked over time in living EC and that peak colocalization can be identified and quantified in highly dynamic cytoskeletal structures.
Live Endothelial Cell Imaging of EGFP-nmMLCK Variants and Cortactin-DsRed Colocalization
We applied this methodology to evaluate dynamic colocalization of nmMLCK isoforms with cortactin in live EC as well as to investigate whether the biochemically characterized interaction of nmMLCK with the cortactin SH3 domain (Dudek et al., 2002) can be interrogated in live cells. Figure 6 depicts the dynamic (live cell imaging) colocalization of EGFP-nmMLCK1 and cortactin-DsRed in lamellipodia of EC following thrombin or S1P challenge. In unstimulated cells, cortactin-DsRed localization is diffuse in the cytoplasm, and EGFP-nmMLCK1 is easily seen in stress fibers transversing the cell. Prior to stimulation (Figure 6A), cortactin-DsRed and EGFP-nmMLCK1 are moderately colocalized in lamellipodia with an ICQ = 0.284. Thrombin dramatically decreases this colocalization several fold to an average ICQ of 0.069 (Figure 6B), while subsequent S1P stimulation reverses this thrombin effect with the ICQ in lamellipodia increasing to 0.332 within 40 min (Figure 6F). Various stages of lamellipodia reformation were imaged over the course of S1P recovery and can be viewed in its entirety in Supplemental Video 2. In the earliest phase after S1P (within 5 min), fine ruffling lamellipodia formed in which EGFP-nmMLCK1 and cortactin-DsRed moderately colocalized (ICQ = 0.260) (Figure 6C, inset). The cell began to contract, and the lamellipodium retracted with a new lamellipodium later appearing 17 min after S1P stimulation (Supplemental Video 2). This lamellipodium was highly dynamic, with nmMLCK1 and cortactin moderately colocalized (Figure 6D, inset) before forming a distinct ruffle in which cortactin and nmMLCK1 eventually strongly colocalized (ICQ = 0.332) (Figure 6F, inset). Subsequent cortactin-nmMLCK1 colocalization gradually decreased over time (Figure 6G, inset).
Figure 6. Dynamic colocalization of EGFP-nmMLCK1 and cortactin-DsRed in live human lung EC.

HPAEC were cotransfected with EGFP-nmMLCK1 and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. The panels represent images from an extended movie (see Supplemental Movie 2) depicting dynamic colocalization of cortactin-DsRed and EGFP-nmMLCK1 in a single EC under basal conditions (A), after thrombin stimulation (B), and subsequent S1P stimulation (C–G). Yellow indicates areas of colocalization. ICQ quantitation for nmMLCK1-cortactin colocalization in outlined lamellipodia is shown for each panel.
Our prior studies have demonstrated that the SH3 domain of cortactin binds to two proline-rich sequences present in both nmMLCK1 and -2 (Dudek et al., 2002; Dudek et al., 2004). We next examined this interaction in live EC utilizing a cortactin-DsRed deletion mutant which lacks the SH3 domain. The cortactin-DsRed deletion mutant was co-expressed with EGFP-nmMLCK1 and colocalization of the two fusion proteins tracked via live-cell imaging (Supplemental Video 3). Lamellipodia formation and transcellular fibers were unaffected by over-expression of the cortactinΔSH3 mutant; however, compared to wild type cortactin (Figure 6A, ICQ = 0.284), cortactinΔSH3 mutant colocalization with EGFP-nmMLCK1 was reduced in lamellipodia of control and S1P-stimulated EC (Figure 7A, ICQ = 0.186 across four lamellipodia). Figure 7B demonstrates that thrombin-induced lamellipodia withdrawal and cortactinΔSH3-nmMLCK1 colocalization in the cell periphery decreased (ICQ = 0.043) to an extent comparable with wild-type cortactin and nmMLCK1. Figures 7C, 7D, and 7E illustrate the gradual redistribution of EGFP-nmMLCK1 in cortical bands and reformation of lamellipodia; however, colocalization between cortactinΔSH3-DsRed and EGFP-nmMLCK1 remained consistently lower (ICQ = 0.137–0.186; Figures 7C–7E) than that measured with wild-type cortactin and nmMLCK1 (Figures 6C–6G). These data confirm our previously published observations that the SH3 domain of cortactin is necessary for optimal interaction with nmMLCK (Dudek et al., 2002; Dudek et al., 2004).
Figure 7. Reduced colocalization of cortactinΔSH3-DsRed with EGFP-nmMLCK1 in live human lung EC.

HPAEC were cotransfected with EGFP-nmMLCK1 and cortactinΔSH3-DsRed constructs and then subjected to live cell imaging as described in Methods. The panels represent images from an extended movie (see Supplemental Movie 3) depicting dynamic colocalization of cortactinΔSH3-DsRed and EGFP-nmMLCK1 in a single EC under basal conditions (A), after thrombin stimulation (B), and subsequent S1P stimulation (C–E). Yellow indicates areas of colocalization. ICQ quantitation for nmMLCK1-cortactinΔSH3 colocalization in outlined lamellipodia is shown for each panel.
The EGFP-nmMLCK2 splice variant and cortactin-DsRed moderately colocalized within fine membrane ruffles with an ICQ =0.148 in resting EC (Figure 8A). Thrombin treatment resulted in the withdrawal of membrane ruffles and spikes of EGFP-nmMLCK2 remained, as outlined in white in Figure 8B and where cortactin-DsRed and EGFP-nmMLCK2 colocalized weakly with an ICQ = 0.094. S1P treatment led to the formation of broad ruffling lamellipodia in which cortactin-DsRed and EGFP-nmMLCK2 strongly but transiently colocalized, the full extent of which can be seen in Supplemental Video 4. Figures 8C and 8D depict ruffling lamellipodia outlined in white in which cortactin-DsRed and EGFP-nmMLCK2 colocalized strongly with peak ICQ values of 0.348 (Figure 8C) and 0.321 (Figure 8D). As with nmMLCK1, dynamic imaging studies were performed assessing EGFP-nmMLCK2 colocalization with the cortactinΔSH3-DsRed (Supplemental Video 5). S1P-induced cytoskeletal remodeling in live EC revealed reduced colocalization of cortactinΔSH3-DsRed with EGFP-nmMLCK2 (Figure 9) to an extent also comparably observed with EGFP-nmMLCK1.
Figure 8. Dynamic colocalization of EGFP-nmMLCK2 and cortactin-DsRed in lamellipodia.

HPAEC were cotransfected with EGFP-nmMLCK2 and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. The panels represent images from an extended movie (see Supplemental Movie 4) depicting dynamic colocalization of cortactin-DsRed and EGFP-nmMLCK2 in a single EC under basal conditions (A), after thrombin stimulation (B), and subsequent S1P stimulation (C–D). Yellow indicates areas of colocalization. ICQ quantitation for nmMLCK2-cortactin colocalization in outlined lamellipodia is shown for each panel.
Figure 9. Reduced colocalization of cortactinΔSH3-DsRed with EGFP-nmMLCK2 in live human lung EC.
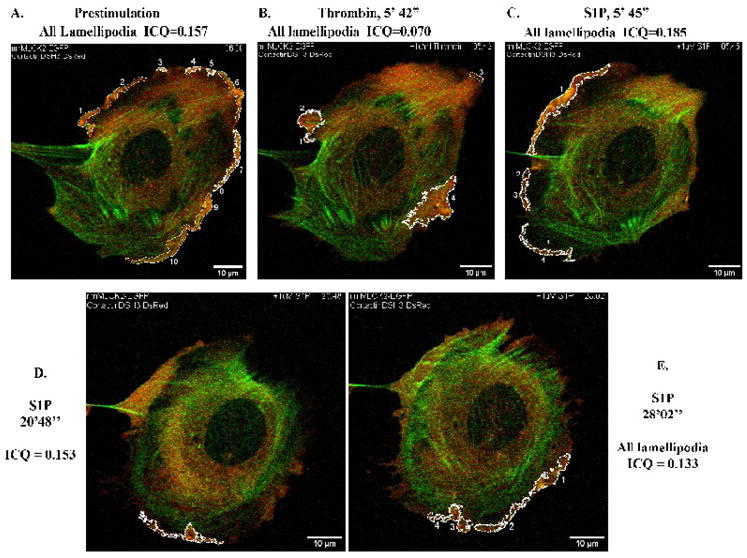
HPAEC were cotransfected with EGFP-nmMLCK2 and cortactinΔSH3-DsRed constructs and then subjected to live cell imaging as described in Methods. The panels represent images from an extended movie (see Supplemental Movie 5) depicting dynamic colocalization of cortactinΔSH3-DsRed and EGFP-nmMLCK2 in a single EC under basal conditions (A), after thrombin stimulation (B), and subsequent S1P stimulation (C–E). Yellow indicates areas of colocalization. ICQ quantitation for nmMLCK2- cortactinΔSH3 colocalization in outlined lamellipodia is shown for each panel.
Role of the nmMLCK Actin- And Cortactin-Binding Domains in Cortactin Colocalization in Lamellipodia
As noted above, the EGFP-nmMLCK2Nterm construct consists of the first 496aa of nmMLCK2 and contains several putative SH2 and SH3 domain binding sites but lacks two critical tyrosines phosphorylated by p60Src as well as actin- and cortactin-binding domains (Supplemental Figure 1). Live EC imaging (Supplemental Video 6) revealed the EGFP-nmMLCK2Nterm fusion protein to be partially localized to peripheral stress fibers under basal conditions (Figure 10A). Cortactin-DsRed localized to fine ruffles outlined in white (Figure 10A) where very little colocalization with EGFP-nmMLCK2Nterm was detected (ICQ = 0.042). Thrombin induced reorganization of EGFP-nmMLCK2Nterm into faint stress fibers while membrane ruffles retracted from the cell periphery (Figure 10B) without colocalization between cortactin and nmMLCK2Nterm (ICQ = −0.048). In the early phase of S1P-induced lamellipodia reformation, fine membrane ruffles reappeared that were similar in structure as those seen under basal conditions, with little colocalization of cortactin and nmMLCK2Nterm (Figure 10C, ICQ = 0.051). Over the course of S1P-stimulated barrier recovery (Figure 10D), ruffles gradually broadened to very dynamic lamellipodia and during this critical interval, spatial localization of cortactin and nmMLCK2Nterm began to separate as indicated by an ICQ of −0.150. These data demonstrate that deletion of the cortactin- and actin-binding regions prevents targeting of nmMLCK2Nterm to the cytoskeletal machinery producing dynamic leading edge ruffles where cortactin is concentrated. Following lamellipodia expansion (Figure 10E), colocalization became random (ICQ=0.00), suggesting that a small fraction of EGFP-nmMLCK2Nterm may have diffused into expanded lamellipodia but does not directly interact with cortactin. Interestingly, EGFP-nmMLCK2Nterm appeared to localize to sites of polymerized actin (suggested by its stress fiber-like distribution across the cells) during thrombin- and S1P-stimulated signaling, possibly via the putative SH2- and SH3-binding domains still present in this 496aa amino terminal fragment of nmMLCK. Potential binding partners to these two domains remain unidentified.
Figure 10. Deletion of the actin- and cortactin-binding domains of nmMLCK2 ablates colocalization with cortactin in S1P-stimulated live EC.

HPAEC were cotransfected with EGFP-nmMLCK2Nterm and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. The panels represent images from an extended movie (see Supplemental Movie 6) depicting dynamic colocalization of cortactin-DsRed and EGFP-nmMLCK2Nterm in a single EC under basal conditions (A), after thrombin stimulation (B), and subsequent S1P stimulation (C–E). Yellow indicates areas of colocalization. ICQ quantitation for nmMLCK2Nterm-cortactin colocalization in outlined lamellipodia is shown for each panel.
Discussion
The vascular endothelium is both a cellular target and key participant in the profound physiologic derangement which accompanies inflammatory lung injury, with vascular hyperpermeability and subsequent barrier restoration serving as key expressions of this involvement. Bioactive and biophysical stimuli alter lung EC barrier function via strikingly diverse mechanisms including multiple signaling pathways that alter the unique microenvironment of the membrane-cytoskeleton interface. Barrier-promoting pathways induce EC cytoskeletal rearrangement which result in enhanced junctional linkages between EC as well as increased linkage of the cytoskeleton with the underlying extracellular matrix (Dudek and Garcia, 2001). These events provide the conceptual underpinning for molecular targeting by permeability-reducing therapeutic strategies. Furthermore, barrier regulation is highly dependent on the exact spatial location of actin polymerization and rearrangement occurring as either barrier-disrupting cytosolic stress fibers or as a barrier-enhancing thickened cortical actin ring (Dudek and Garcia, 2001; Garcia, 2009). When actin polymerization produces transcytoplasmic stress fibers (Figure 1D–1F), the cortical actin ring disassembles and contractile tension is produced, a function of the abundant tension-generating molecular machinery (actin and myosin comprise ~16% of total EC protein). Activation of the Ca2+-calmodulin-dependent non-muscle MLCK isoform, nmMLCK, a key regulator of the EC cytoskeleton initially cloned by our laboratory (Garcia et al., 1997; Lazar and Garcia, 1999), is essential for the generation of EC centripetal tension via increases in MLC phosphorylation and leads to paracellular gap formation and increased paracellular permeability. Increased nmMLCK activity exerts a major gatekeeper function during acute lung inflammation such as following neutrophil diapedesis (Garcia et al., 1998), and mice with targeted deletion of the nmMLCK isoform or mice pretreated with small molecule nmMLCK inhibitors are protected from inflammatory lung injury (Wainwright et al., 2003). In contrast, mice over-expressing the nmMLCK isoform in vascular endothelium exhibit markedly augmented lung injury in a sexually dimorphic and age-dependent manner (Moitra et al., 2008).
Despite the essential involvement of complex cytoskeletal protein interaction in vascular barrier regulation, these interactions have proven difficult to quantify in a spatially-specific manner, which is a major limitation to interrogating the molecular mechanisms driving cytoskeleton-mediated EC barrier regulation. Recently, the technique of intensity correlation analysis has been adapted into ImageJ colocalization quantification and increasingly employed to visualize protein–protein interactions (Bolte and Cordelieres, 2006; Brittain et al., 2009; Cardone et al., 2007; Racz, 2008). In the current study, we now describe the utility of intensity correlation image analysis and the intensity correlation quotient (ICQ) to quantify the translocation of nmMLCK variants (nmMLCK1 and -2) to barrier–promoting lamellipodia and to assess the colocalized association of nmMLCK variants with the actin-binding protein, cortactin, in both fixed and live-cell imaging assays under conditions of EC barrier enhancement and disruption. Our quantitative results indicate that robust thrombin-induced EC contraction reduces colocalization of cortactin with nmMLCK fusion proteins whereas the potent barrier-protective agonist, S1P, increased colocalization of nmMLCK and cortactin within lamellipodia. Although our prior work demonstrated increased colocalization of cortactin and nmMLCK in EC after S1P relative to unstimulated controls (Dudek et al., 2004), ICQ analysis in the current study did not demonstrate a significant increase between these two conditions. We speculate that the subconfluent plating used in this current study activates EC to migrate and form monolayers in the vehicle controls in such a way as to mimic the effects of S1P, whereas the prior study was performed in confluent monolayers of presumably quiescent cells. Our current data suggest that adding S1P to already activated cells may not further increase nmMLCK-cortactin interaction within lamellipodia. Of note, the EGFP-nmMLCK2Nterm fusion protein which lacks the actin-binding and/or cortactin-binding domains is nearly absent in lamellipodia (Figure 1C, 1I, 1L), suggesting that these domains are required for nmMLCK localization to lamellipodia.
Because information is limited regarding the tissue distribution and physiological roles of the nmMLCK splice variants, we utilized intensity correlation image analysis to assess potential differences in nmMLCK1 and nmMLCK2 (the primary splice variants in EC) translocation properties in stimulated EC. Importantly, both nmMLCK1 and -2 contain several proline-rich domains which may link these variants to SH3-containing cytoskeletal signaling proteins (Birukov et al., 2001). Kinetically, nmMLCK1 or 2 do not significantly differ in Vmax or K0.5CaM, although only nmMLCK1 is capable of undergoing p60Src–catalyzed phosphorylation on Tyr464 and Tyr471, a post translational modification that significantly increases Vmax (3 fold increase) (Birukov et al., 2001). Interestingly, both tyrosine residues are encoded within the exon 11 that is spliced out in nmMLCK2 (Birukov et al., 2001). In general, we observed only subtle differences between nmMLCK1 and nmMLCK2 in terms of their subcellular distributions and colocalization with cortactin under the conditions studied. There was statistically significant greater EGFP-nmMLCK2 levels present in lamellipodia than EGFP-nmMLCK1 of untreated and S1P-rescued cells (Figure 2), suggesting that amino acids encoded within exon 11 produce protein-protein interactions for nmMLCK1 that differ from those for nmMLCK2, possibly mediated by phosphorylation on Y464 and Y471 in nmMLCK1 (Birukov et al., 2001). It is possible that tyrosine phosphorylation may factor in differentially targeting nmMLCK variants to spatially defined sites where other signaling and cytoskeletal components are assembled, particularly in lamellipodia.
Our imaging analyses in live-cell assays confirmed earlier biochemical studies (Dudek et al., 2002; Dudek et al., 2004) which demonstrated that nmMLCK association with its key cytoskeletal binding partner and essential barrier regulatory effector, cortactin, involves the SH3 domain of cortactin as well as the cortactin- and actin-binding domains of nmMLCK. Cortactin, an 80/85 kDa actin-binding and SH3-containing protein (Dudek et al., 2002), is a substrate of p60Src and is recruited to lamellipodia and membrane ruffles in response to S1P and other barrier promoting agonists (Dudek et al., 2004; Jacobson et al., 2006; Liu et al., 2002; Vouret-Craviari, 2002). The structural domains of cortactin are designed for integrating signal transduction complexes at sites of dynamic actin rearrangement (Supplemental Figure 1B) and stimulating the Arp2/3 complex (Dudek et al., 2002; Uruno, 2001; Zhan et al., 1993). The C-terminal SH3 domain (aa 496-546) binds to proline-rich sequences in numerous partners that include WASP, dynamin, ZO-1 (Cosen-Binker and Kapus, 2006), and nmMLCK (aa972-979 and aa1019-1025), which are in close proximity to the actin-binding domain of nmMLCK (Dudek et al., 2002). Although the precise significance of this spatially defined interaction remains unknown, the cortactin SH3 domain interaction with nmMLCK1 is required for peripheral MLC phosphorylation induced by S1P (Dudek et al., 2004). In an in vitro actin polymerization assay, cortactin inhibited nmMLCK1 binding to F-actin and in turn, nmMLCK1 down-regulates the enhancing effect of cortactin on Arp2/3-dependent actin polymerization (Dudek et al., 2002). These findings suggest that spatially-defined cortactin association with nmMLCK may regulate the capacity for cortactin to crosslink and stabilize the cortical actin meshwork against actin depolymerization. These data strongly indicate that cortactin-nmMLCK interaction is a critical regulator of cytoskeletal rearrangement necessary for S1P-mediated endothelial cell barrier enhancement. Since p60Src-mediated phosphorylation of either nmMLCK1 or cortactin increases their association, it is likely that nmMLCK2, which lacks the two tyrosine residues normally phosphorylated by p60Src, may interact with cortactin with dynamics distinct from nmMLCK1. Indeed, nmMLCK1 and -2 bind cortactin with differing affinities in vitro (Dudek et al., 2004), with an apparent KD of 1 μM for nmMLCK1 and an apparent KD of 0.25 μM for nmMLCK2. Interestingly, polymorphisms in the human cortactin and MLCK genes may combine to increase susceptibility to inflammatory lung disorders (Christie et al., 2008; Ma et al., 2008).
It is important to note some limitations of our current study. The intensity correlation analyses used here do not measure direct physical interactions of proteins per se, but rather, they quantitate the amount of overlap from fluorescent signals given off by the proteins of interest. It is conceivable that two proteins can be close enough to each other to have some overlap of these signals without having direct interaction. The advantage with the intensity correlation analysis used in this study is that it defines pixels as colocalized when both the green and red mean deviation products rise above their mean intensities for the defined region of interest. Other methods, such as the Manders and Pearson’s correlation coefficients, do not take covarying signals into account (Bolte and Cordelieres, 2006; Khanna et al., 2006; Li et al., 2004). The method used in our study also excludes pixels that have both green and red negative mean deviations, so potential false positives are eliminated. Another potential limitation of our study is that the colocalization analyses utilized overexpressed proteins, and these elevated expression levels may introduce some artifact. Finally, the potential influence of the endogenous nmMLCK and cortactin within these EC are not accounted for in our study.
In summary, our studies offer a quantitative methodology which accurately captures thrombin-induced EC contractile events that results in quantifiable reduction in colocalization of cortactin-DsRed with GFP-nmMLCK fusion proteins. In contrast, the potent barrier-protective agonist, S1P, increased translocation of nmMLCK and cortactin to lamellipodia and increased colocalization of these two essential cytoskeletal effectors, confirming our earlier observations that nmMLCK and cortactin rapidly colocalize at the EC periphery during S1P-induced barrier enhancement (Dudek et al., 2004) while adding useful novel information concerning the magnitude of these interactions. Live-cell assays verified that cortactin-nmMLCK association involves the SH3 domain of cortactin as well as the cortactin- and actin-binding domains within the nmMCLK molecule. Together, these data provide insights into the molecular basis for vascular barrier-regulatory cytoskeletal responses and support the utility of sophisticated imaging analyses and methodological assessment to quantify the critical interaction that occurs between nmMLCK and cortactin during S1P-induced EC barrier enhancement.
Supplementary Material
HPAEC were cotransfected with actin-GFP and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. Under basal conditions, lamellipodia are broad and actin-GFP and cortactin-DsRed colocalize across most of each lamellipodium. Thrombin immediately induces retraction of lamellipodia and no significant overlap between actin and cortactin is observed. S1P rapidly induces formation of large ruffles directed toward lamellipodia and dynamic ruffle formation persists for the entire recording. “Plumes” of actin-cortactin colocalization are also seen within the cell boundaries and these plumes may represent ruffles near the cell surface. Over the course of S1P stimulation, actin and cortactin colocalize very strongly in these ruffles.
HPAEC were cotransfected with EGFP-nmMLCK1 and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. As in Supplemental Video 1, lamellipodia are quite broad in subconfluent unstimulated EC, and nmMLCK1 and cortactin colocalize along the length of each lamellipodium. Thrombin immediately causes lamellipodia retraction and stress fiber formation and no significant colocalization between nmMLCK1 and cortactin is apparent. Small but transient lamellipodia, where nmMLCK1 and cortactin modestly colocalize, begin to form almost immediately after application of S1P. These brief lamellipodia retract for ~5 min, then reform again by ~15 min into S1P application. Cortactin and nmMLCK1 transiently colocalize in ruffles and ridges of rippling lamellipodia and strongly so in a prominent ruffle that forms at ~33′30″ and flows toward a leading edge where the ruffle expands and dissolves by ~39 min. The cell progressively contracts and appears to form a triangular cortical ring in which nmMLCK1 localizes brightly.
HPAEC were cotransfected with EGFP-nmMLCK1 and cortactinΔSH3-DsRed constructs and then subjected to live cell imaging as described in Methods. Broad lamellipodia circle an unstimulated EC in which EGFP-nmMLCK1 and cortactinΔSH3-DsRed appear to colocalize to an extent less than that seen in Supplemental Video 2. Colocalization all but disappears during thrombin stimulation, although small lamellipodia begin to re-emerge by ten minutes. Robust lamellipodia form during the first 12 min of S1P application but colocalization between nmMLCK1 and cortactinΔSH3 appears weak. The cell gradually contracts throughout the rest of the recording and nmMLCK1 reorganizes into triangular, dense cortical bands. Lamellipodia remain highly dynamic but cortactinΔSH3 and nmMLCK1 colocalize very weakly in the periphery.
HPAEC were cotransfected with EGFP-nmMLCK2 and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. EGFP-nmMLCK2 and cortactin weakly colocalize in very fine lamellipodia flutter on the cell periphery. Stress fibers appear to thicken during thrombin stimulation, and microspikes and filopodia are left on the periphery after lamellipodia withdrawal. S1P induces gradual growth of lamellipodia, and nmMLCK2 and cortactin colocalize strongly on ridges of emerging lamellipodia. A fine meshwork of cortactin and nmMLCK2 appears in the lamella near the end of the recording.
HPAEC were cotransfected with EGFP-nmMLCK2 and cortactinΔSH3-DsRed constructs and then subjected to live cell imaging as described in Methods. EGFP-nmMLCK2 modestly colocalizes with cortactinΔSH3-DsRed in an unstimulated EC. Although thrombin stimulation was unfortunately weak in this cell, the lamellipodia are briefly retracted by 5′42″, and nmMLCK2-cortactinΔSH3 colocalization at this point decreases to an ICQ of 0.070. Over the course of S1P treatment, the cell contracts and nmMLCK2 aligns along progressively thickening cortical dense bands while the cell forms very robust lamellipodia where only weak nmMLCK2-cortactinΔSH3 colocalization is observed.
HPAEC were cotransfected with EGFP-nmMLCK2Nterm and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. The 496-aa amino terminal fragment of nmMLCK2 still localizes to cytoskeletal fibers, possibly by two putative SH3 domain-binding regions in association with other actin-binding signaling proteins. Cortactin-nmMLCK2 colocalization occurs only at near-random levels.
Non-muscle MLCK and cortactin domain structures with sites of p60Src-mediated phosphorylation. Panel A depicts the multiple domains present within the 210 kDa nmMLCK1 isoform (top bar) including a 922-amino acid amino terminal domain containing six IgCam domains (gray dotted lines) and the two alternatively spliced exons, exon 3 (aa56-124) and exon 11 (aa437-505). Amino acids encoded within exon 11 include Y464 and Y471 which are two p60Src phosphorylation sites (black arrows) and are spliced out of human nmMLCK2 (middle bar). Y464 also serves as an SH2 binding site and thus potentially interacts with signaling and adaptor proteins. These sites impart potential differential signaling regulation of the nmMLCK1 variant compared to nmMLCK2. Both nmMLCK1 and -2 contain two SH3 binding sites (aa314-318, aa373-379) (thick black lines) between the second and third IgCams. The nmMLCK is identical to smooth muscle isoform smMLCK in residues 923-1914 with an actin-binding region (aa923-1031), two proline-rich binding sites for the cortactin SH3 domain (Dudek et al., 2002), an MLC binding domain (aa1280-1469), an ATP/catalytic domain (aa1471-1705), a regulatory domain containing an autoinhibitory sequence (aa1721-1741) plus a CaM-binding site (aa1740-1763), and a kinase-related protein (KRP)/myosin-binding domain (aa1761-1914) (Birukov et al., 2001). The bottom bar depicts a truncated mutant of nmMLCK2 containing the first 496 amino acids of nmMLCK2 which lacks both cortactin- and actin-binding domains, but retains the two putative SH3-binding sites (thick black lines). All nmMLCK constructs were conjugated to the carboxy terminus of EGFP. Adapted from (Cosen-Binker and Kapus, 2006), Panel B depicts the structure of cortactin with the amino terminal half of cortactin beginning with the N-terminal acidic region (NTA), which contains the DDW motif necessary for binding to the Arp3 component of the Arp2/3 complex, and followed by 6.5 tandem repeats of 37 residues responsible for F-actin binding. The carboxy terminal half of cortactin is composed of an α-helical domain of undefined function and followed by a proline-serine-threonine enriched domain containing several critical tyrosine residues phosphorylated by a number of kinases including p60Src (Y421, Y466, Y486). A C-terminal SH3 domain binds to nmMLCK as well as numerous other signal proteins.
Deletion of the actin- and cortactin-binding domains of nmMLCK2 dramatically reduces colocalization with endogenous cortactin in human lung endothelial lamellipodia. Panel A: EC overexpressing EGFP-nmMLCK2Nterm were incubated with either vehicle (i), thrombin for 10 min (ii), S1P for 30 min (iii), or thrombin for 10 min followed by S1P for 30 min (iv) at 37°C, then fixed and immunostained for cortactin (red). Yellow indicates areas of colocalization. The lower panels, i′-iv′, depict colocalization of endogenous cortactin with nmMLCK2Nterm as described for Figure 3. Shown are representative images selected from 4–10 images for each treatment. Panel B: The mean ICQ values for cortactin and EGFP-nmMLCK2Nterm colocalization were calculated as described in Methods for the entire cell, the cytoplasm, and the lamellipodia under each condition [vehicle (No stim), thrombin for 10 min, S1P for 30 min, or thrombin for 10 min followed by S1P for 30 min]. The low ICQ values describe quantitatively the nearly random colocalization between EGFP-nmMLCK2Nterm and endogenous cortactin. Lamellipodia colocalization of cortactin with nmMLCK2Nterm was significantly lower than that seen with nmMLCK1 or nmMCLK2 in unstimulated (p<0.0001) and S1P-treated (p<0.0001) EC. N = 4–10 per condition.
Acknowledgments
The authors gratefully acknowledge the expert technical assistance and contributions of Eddie Chiang and Nicholas Shank and thank Lakshmi Natarajan for continued maintenance and supply of high quality endothelial cell culture stocks. This work was supported by grants from the National Heart, Lung, and Blood Institute (Garcia, HL058064; Dudek, HL088144).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Birukov KG, et al. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60(Src) J Biol Chem. 2001;276:8567–73. doi: 10.1074/jbc.M005270200. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. Journal of Microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Brittain JM, et al. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. 2009;284:31375–90. doi: 10.1074/jbc.M109.009951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JK, et al. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. Journal of Cell Biology. 1997;139:469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JK, et al. The giant organelles in Beige and Chediak-Higashi Fibroblasts are derived from late endosomes and mature lysosomes. Journal of Experimental Medicine. 1993;178:1845–1856. doi: 10.1084/jem.178.6.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone RA, et al. The NHERF1 PDZ2 domain regulates PKA-RhoA-p38-mediated NHE1 activation and invasion in breast tumor cells. Molecular Biology of the Cell. 2007;18:1768–1780. doi: 10.1091/mbc.E06-07-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JD, et al. Variation in the myosin light chain kinase gene is associated with development of acute lung injury after major trauma. Crit Care Med. 2008;36:2794–800. doi: 10.1097/ccm.0b013e318186b843. [DOI] [PubMed] [Google Scholar]
- Cosen-Binker LI, Kapus A. Cortactin: the gray eminence of the cytoskeleton. Physiology (Bethesda) 2006;21:352–61. doi: 10.1152/physiol.00012.2006. [DOI] [PubMed] [Google Scholar]
- Dudek SM, et al. Novel interaction of cortactin with endothelial cell myosin light chain kinase. Biochem Biophys Res Commun. 2002;298:511–9. doi: 10.1016/s0006-291x(02)02492-0. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- Dudek SM, et al. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692–700. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- Finigan JH, et al. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280:17286–93. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- Garcia JG. Concepts in microvascular endothelial barrier regulation in health and disease. Microvasc Res. 2009;77:1–3. doi: 10.1016/j.mvr.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Garcia JG, et al. Myosin light chain kinase in endothelium: molecular cloning and regulation. Am J Respir Cell Mol Biol. 1997;16:489–94. doi: 10.1165/ajrcmb.16.5.9160829. [DOI] [PubMed] [Google Scholar]
- Garcia JG, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JG, et al. Thrombin-induced increase in albumin permeability across the endothelium. J Cell Physiol. 1986;128:96–104. doi: 10.1002/jcp.1041280115. [DOI] [PubMed] [Google Scholar]
- Garcia JG, et al. Adherent neutrophils activate endothelial myosin light chain kinase: role in transendothelial migration. J Appl Physiol. 1998;84:1817–21. doi: 10.1152/jappl.1998.84.5.1817. [DOI] [PubMed] [Google Scholar]
- Jacobson JR, et al. Endothelial cell barrier enhancement by ATP is mediated by the small GTPase Rac and cortactin. Am J Physiol Lung Cell Mol Physiol. 2006;291:L289–95. doi: 10.1152/ajplung.00343.2005. [DOI] [PubMed] [Google Scholar]
- Khanna R, et al. N type Ca2+ channels and RIM scaffold protein covary at the presynaptic transmitter release face but are components of independent protein complexes. Neuroscience. 2006;140:1201–8. doi: 10.1016/j.neuroscience.2006.04.053. [DOI] [PubMed] [Google Scholar]
- Lazar V, Garcia JG. A single human myosin light chain kinase gene (MLCK; MYLK) Genomics. 1999;57:256–67. doi: 10.1006/geno.1999.5774. [DOI] [PubMed] [Google Scholar]
- Li Q, et al. A Syntaxin1, Gao, and N-Type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. Journal of Neuroscience. 2004;24:4070–4081. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, et al. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. Faseb J. 2002;16:950–62. doi: 10.1096/fj.01-0870com. [DOI] [PubMed] [Google Scholar]
- Ma SF, et al. A common cortactin gene variation confers differential susceptibility to severe asthma. Genet Epidemiol. 2008;32:757–66. doi: 10.1002/gepi.20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNiven MA, et al. Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J Cell Biol. 2000;151:187–98. doi: 10.1083/jcb.151.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVerry BJ, Garcia JG. In vitro and in vivo modulation of vascular barrier integrity by sphingosine 1-phosphate: mechanistic insights. Cell Signal. 2005;17:131–9. doi: 10.1016/j.cellsig.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Moitra J, et al. A transgenic mouse with vascular endothelial over-expression of the non-muscle myosin light chain kinase-2 isoform is susceptible to inflammatory lung injury: role of sexual dimorphism and age. Transl Res. 2008;151:141–53. doi: 10.1016/j.trsl.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache I, et al. Differential effect of MLC kinase in TNF-alpha-induced endothelial cell apoptosis and barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1168–78. doi: 10.1152/ajplung.2001.280.6.L1168. [DOI] [PubMed] [Google Scholar]
- Racz I, et al. Interferon-Gamma Is a Critical Modulator of CB2 Cannabinoid Receptor Signaling during Neuropathic Pain. J Neurosci. 2008;46:12136–12145. doi: 10.1523/JNEUROSCI.3402-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaphorst KL, et al. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol. 2003;285:L258–67. doi: 10.1152/ajplung.00311.2002. [DOI] [PubMed] [Google Scholar]
- Shikata Y, et al. Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. Faseb J. 2003;17:2240–9. doi: 10.1096/fj.03-0198com. [DOI] [PubMed] [Google Scholar]
- Singleton PA, et al. Transactivation of sphingosine 1-phosphate receptors is essential for vascular barrier regulation. Novel role for hyaluronan and CD44 receptor family. J Biol Chem. 2006;281:34381–93. doi: 10.1074/jbc.M603680200. [DOI] [PubMed] [Google Scholar]
- Singleton PA, et al. CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J Biol Chem. 2007;282:30643–57. doi: 10.1074/jbc.M702573200. [DOI] [PubMed] [Google Scholar]
- Thurston G, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–4. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- Uruno T, et al. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nature Cell Biology. 2001;3:259–266. doi: 10.1038/35060051. [DOI] [PubMed] [Google Scholar]
- Vouret-Craviari V, et al. Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine-1-phosphate in endothelial cells. Journal of Cell Science. 2002;115:2475–2484. doi: 10.1242/jcs.115.12.2475. [DOI] [PubMed] [Google Scholar]
- Wadgaonkar R, et al. Mutation analysis of the non-muscle myosin light chain kinase (MLCK) deletion constructs on CV1 fibroblast contractile activity and proliferation. J Cell Biochem. 2003;88:623–34. doi: 10.1002/jcb.10370. [DOI] [PubMed] [Google Scholar]
- Wainwright MS, et al. Protein kinase involved in lung injury susceptibility: Evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc Natl Acad Sci U S A. 2003;100:6233–8. doi: 10.1073/pnas.1031595100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Parsons JT. Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol. 1993;120:1417–26. doi: 10.1083/jcb.120.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, et al. Murine cortactin is phosphorylated in response to fibroblast growth factor-1 on tyrosine residues late in the G1 phase of the BALB/c 3T3 cell cycle. J Biol Chem. 1993;268:24427–31. [PubMed] [Google Scholar]
- Zhao J, et al. Phosphotyrosine protein dynamics in cell membrane rafts of sphingosine-1-phosphate-stimulated human endothelium: role in barrier enhancement. Cell Signal. 2009;21:1945–60. doi: 10.1016/j.cellsig.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HPAEC were cotransfected with actin-GFP and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. Under basal conditions, lamellipodia are broad and actin-GFP and cortactin-DsRed colocalize across most of each lamellipodium. Thrombin immediately induces retraction of lamellipodia and no significant overlap between actin and cortactin is observed. S1P rapidly induces formation of large ruffles directed toward lamellipodia and dynamic ruffle formation persists for the entire recording. “Plumes” of actin-cortactin colocalization are also seen within the cell boundaries and these plumes may represent ruffles near the cell surface. Over the course of S1P stimulation, actin and cortactin colocalize very strongly in these ruffles.
HPAEC were cotransfected with EGFP-nmMLCK1 and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. As in Supplemental Video 1, lamellipodia are quite broad in subconfluent unstimulated EC, and nmMLCK1 and cortactin colocalize along the length of each lamellipodium. Thrombin immediately causes lamellipodia retraction and stress fiber formation and no significant colocalization between nmMLCK1 and cortactin is apparent. Small but transient lamellipodia, where nmMLCK1 and cortactin modestly colocalize, begin to form almost immediately after application of S1P. These brief lamellipodia retract for ~5 min, then reform again by ~15 min into S1P application. Cortactin and nmMLCK1 transiently colocalize in ruffles and ridges of rippling lamellipodia and strongly so in a prominent ruffle that forms at ~33′30″ and flows toward a leading edge where the ruffle expands and dissolves by ~39 min. The cell progressively contracts and appears to form a triangular cortical ring in which nmMLCK1 localizes brightly.
HPAEC were cotransfected with EGFP-nmMLCK1 and cortactinΔSH3-DsRed constructs and then subjected to live cell imaging as described in Methods. Broad lamellipodia circle an unstimulated EC in which EGFP-nmMLCK1 and cortactinΔSH3-DsRed appear to colocalize to an extent less than that seen in Supplemental Video 2. Colocalization all but disappears during thrombin stimulation, although small lamellipodia begin to re-emerge by ten minutes. Robust lamellipodia form during the first 12 min of S1P application but colocalization between nmMLCK1 and cortactinΔSH3 appears weak. The cell gradually contracts throughout the rest of the recording and nmMLCK1 reorganizes into triangular, dense cortical bands. Lamellipodia remain highly dynamic but cortactinΔSH3 and nmMLCK1 colocalize very weakly in the periphery.
HPAEC were cotransfected with EGFP-nmMLCK2 and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. EGFP-nmMLCK2 and cortactin weakly colocalize in very fine lamellipodia flutter on the cell periphery. Stress fibers appear to thicken during thrombin stimulation, and microspikes and filopodia are left on the periphery after lamellipodia withdrawal. S1P induces gradual growth of lamellipodia, and nmMLCK2 and cortactin colocalize strongly on ridges of emerging lamellipodia. A fine meshwork of cortactin and nmMLCK2 appears in the lamella near the end of the recording.
HPAEC were cotransfected with EGFP-nmMLCK2 and cortactinΔSH3-DsRed constructs and then subjected to live cell imaging as described in Methods. EGFP-nmMLCK2 modestly colocalizes with cortactinΔSH3-DsRed in an unstimulated EC. Although thrombin stimulation was unfortunately weak in this cell, the lamellipodia are briefly retracted by 5′42″, and nmMLCK2-cortactinΔSH3 colocalization at this point decreases to an ICQ of 0.070. Over the course of S1P treatment, the cell contracts and nmMLCK2 aligns along progressively thickening cortical dense bands while the cell forms very robust lamellipodia where only weak nmMLCK2-cortactinΔSH3 colocalization is observed.
HPAEC were cotransfected with EGFP-nmMLCK2Nterm and cortactin-DsRed constructs and then subjected to live cell imaging as described in Methods. The 496-aa amino terminal fragment of nmMLCK2 still localizes to cytoskeletal fibers, possibly by two putative SH3 domain-binding regions in association with other actin-binding signaling proteins. Cortactin-nmMLCK2 colocalization occurs only at near-random levels.
Non-muscle MLCK and cortactin domain structures with sites of p60Src-mediated phosphorylation. Panel A depicts the multiple domains present within the 210 kDa nmMLCK1 isoform (top bar) including a 922-amino acid amino terminal domain containing six IgCam domains (gray dotted lines) and the two alternatively spliced exons, exon 3 (aa56-124) and exon 11 (aa437-505). Amino acids encoded within exon 11 include Y464 and Y471 which are two p60Src phosphorylation sites (black arrows) and are spliced out of human nmMLCK2 (middle bar). Y464 also serves as an SH2 binding site and thus potentially interacts with signaling and adaptor proteins. These sites impart potential differential signaling regulation of the nmMLCK1 variant compared to nmMLCK2. Both nmMLCK1 and -2 contain two SH3 binding sites (aa314-318, aa373-379) (thick black lines) between the second and third IgCams. The nmMLCK is identical to smooth muscle isoform smMLCK in residues 923-1914 with an actin-binding region (aa923-1031), two proline-rich binding sites for the cortactin SH3 domain (Dudek et al., 2002), an MLC binding domain (aa1280-1469), an ATP/catalytic domain (aa1471-1705), a regulatory domain containing an autoinhibitory sequence (aa1721-1741) plus a CaM-binding site (aa1740-1763), and a kinase-related protein (KRP)/myosin-binding domain (aa1761-1914) (Birukov et al., 2001). The bottom bar depicts a truncated mutant of nmMLCK2 containing the first 496 amino acids of nmMLCK2 which lacks both cortactin- and actin-binding domains, but retains the two putative SH3-binding sites (thick black lines). All nmMLCK constructs were conjugated to the carboxy terminus of EGFP. Adapted from (Cosen-Binker and Kapus, 2006), Panel B depicts the structure of cortactin with the amino terminal half of cortactin beginning with the N-terminal acidic region (NTA), which contains the DDW motif necessary for binding to the Arp3 component of the Arp2/3 complex, and followed by 6.5 tandem repeats of 37 residues responsible for F-actin binding. The carboxy terminal half of cortactin is composed of an α-helical domain of undefined function and followed by a proline-serine-threonine enriched domain containing several critical tyrosine residues phosphorylated by a number of kinases including p60Src (Y421, Y466, Y486). A C-terminal SH3 domain binds to nmMLCK as well as numerous other signal proteins.
Deletion of the actin- and cortactin-binding domains of nmMLCK2 dramatically reduces colocalization with endogenous cortactin in human lung endothelial lamellipodia. Panel A: EC overexpressing EGFP-nmMLCK2Nterm were incubated with either vehicle (i), thrombin for 10 min (ii), S1P for 30 min (iii), or thrombin for 10 min followed by S1P for 30 min (iv) at 37°C, then fixed and immunostained for cortactin (red). Yellow indicates areas of colocalization. The lower panels, i′-iv′, depict colocalization of endogenous cortactin with nmMLCK2Nterm as described for Figure 3. Shown are representative images selected from 4–10 images for each treatment. Panel B: The mean ICQ values for cortactin and EGFP-nmMLCK2Nterm colocalization were calculated as described in Methods for the entire cell, the cytoplasm, and the lamellipodia under each condition [vehicle (No stim), thrombin for 10 min, S1P for 30 min, or thrombin for 10 min followed by S1P for 30 min]. The low ICQ values describe quantitatively the nearly random colocalization between EGFP-nmMLCK2Nterm and endogenous cortactin. Lamellipodia colocalization of cortactin with nmMLCK2Nterm was significantly lower than that seen with nmMLCK1 or nmMCLK2 in unstimulated (p<0.0001) and S1P-treated (p<0.0001) EC. N = 4–10 per condition.