

Abstract
Mitochondrial DNA is constantly exposed to oxidative injury. Due to its location close to the main site of reactive oxygen species, the inner mitochondrial membrane, mtDNA is more susceptible than nuclear DNA to oxidative damage. The accumulation of DNA damage is thought to play a critical role in the aging process and to be particularly deleterious in post-mitotic cells. Thus, DNA repair is an important mechanism for maintenance of genomic integrity. Despite the importance of mitochondria in the aging process, it was thought for many years that mitochondria lacked an enzymatic DNA repair system comparable to that in the nuclear compartment. However, it is now well established that DNA repair actively takes place in mitochondria. Oxidative DNA damage processing, base excision repair mechanisms were the first to be described in these organelles, and consequently the best understood. However, new proteins and novel DNA repair pathways, thought to be exclusively present in the nucleus, have recently been described also to be present in mitochondria. Here we review the main mitochondrial DNA repair pathways and their association with the aging process.
1. Introduction
DNA is constantly exposed to endogenous and exogenous agents that generate DNA lesions and induce DNA instability. Reactive oxygen species (ROS) are particularly important genotoxic agents, which are endogenously generated in mitochondria. They generate a large number of DNA lesions, including oxidized DNA bases, abasic sites, and single- and double- strand breaks. Many of these ROS-induced DNA lesions show mutagenic or cytotoxic effects due to mispair of bases, which may give rise to mutations upon DNA replication (Grollman and Moriya, 1993; Kavli et al., 2007). DNA damage accumulation can generate blockage of DNA replication and transcription and also chromosomal rearrangements, leading to genomic instability. In order to maintain genomic integrity, different DNA repair pathways have evolved and the particular pathway employed depends, in part, upon the type of DNA damage that is being repaired. These pathways have been extensively investigated in the nucleus. For example, bulky lesions induced by UV as well as by carcinogenic compounds are removed by the nucleotide excision repair (NER) pathway, whereas simpler lesions such as alkylation or oxidation products caused by ROS are repaired by the base excision repair (BER) pathway (Seeberg et al., 1995). Another pathway, mismatch repair (MMR), removes mismatches in the DNA, while complex lesions such as interstrand cross-links in DNA are processed by recombinational DNA repair (Bohr 2002a). Interestingly, some of these DNA repair mechanisms are highly conserved among species (Bohr, 2008; Krokan et al., 1997; Meyer et al., 2007; Soerensen et al., 2009), stressing their importance in maintenance of genome integrity.
DNA damage is believed to play an important role in the aging process. The increased susceptibility to cellular loss and functional decline observed during aging has been associated with DNA integrity (Beckman and Ames, 1999; Sastre et al., 2000; Trifunovic et al., 2004), particularly of mitochondrial DNA (mtDNA). The stability of mtDNA is constantly challenged by the endogenous production of mitochondrial ROS (mtROS), which are generated during normal electron flux through the mitochondrial electron transport. Unlike nuclear DNA, mtDNA is located in the proximity of mtROS generation sites which are located within complex I and III of the electron transport chain (Barja, 1999; Muller et al., 2004). In fact, the steady-state levels of oxidatively induced lesions observed in mtDNA can be several-fold higher than those in nDNA (Barja and Herrero, 2000; Hamilton et al., 2001; Ricther et al., 1988). Due to the mutagenic nature of many of the ROS-induced lesions, mitochondrial free radicals are thought to be an important source of mtDNA mutations and DNA instability. Studies performed over 30 years ago on mitochondrial capacity to repair UV-induced damage, via nucleotide excision repair (NER), provided the basis for the notion at that time that mitochondria lacked an enzymatic DNA repair system (Clayton et al., 1974). The higher level of oxidative lesions in mtDNA than in nuclear DNA was considered to be a consequence, in part, of the absence of DNA repair in mitochondria. However, nowadays it is well established that some DNA repair pathways actively take place in mitochondria. Furthermore, over the years characterization of known mitochondrial DNA repair pathways have improved and new DNA repair enzymes have been detected in mitochondria (Liu et al., 2008; Zheng et al., 2008; Souza-Pinto et al., 2009; Hu et al., 2005). Newly obtained knowledge on mtDNA repair pathways include the observation that repair mechanisms thought to be present only in the nucleus also take place in mitochondria.
That is the case for MMR (Mason et al., 2003; Souza-Pinto et al., 2009), and the long-patch BER (Akbari et al., 2008; Liu et al., 2008; Szcaesny et al., 2008). On the other hand, an important question still remains unsolved, whether mitochondria posses any capabilities to repair DNA lesions via the NER pathway, despite removal of UV-induced pyrimidine lesions does not take place in mitochondria.
Because BER is the main DNA repair pathway coping with oxidative lesions, most of the investigations on mitochondrial DNA repair and aging or age-related diseases focus on mtBER. Although not the only one, BER is the best-characterized DNA repair pathway in mitochondria (Bohr et al., 2007; Robertson et al., 2009). This review explores the different DNA repair pathways that have been described in mitochondria together with the experimental evidences supporting the importance of these mechanisms in the aging process as well as in the aetiology of age-related diseases such as neurological disorders.
2. Direct repair of mitochondrial DNA damage
The most direct form of DNA repair is the chemical reversal of the aberrant DNA adducts. By the use of light, photolyase can monomerize UV-light induced cyclobutane pyrimidine dimers. Photoreactivation may take place in mitochondria of lower eukaryotes, but photolyase does not seem to exist in cells of higher eukaryotes. The major direct repair (DR) protein in the mammalian nucleus is O6-methyl-guanine DNA methyl transferase (MGMT). This ‘suicide’ enzyme transfers the methyl group directly to a cysteine residue within the enzyme and thereby inactivates itself. It has been demonstrated that mitochondrial extracts from rat liver cells effectively removes O6-methyl-guanosine from mtDNA (Myers et al., 1988), and Satoh et al. (1988) have reported the removal of O6-ethyl-2’-deoxyguanosine from mtDNA of rat liver tissue in vivo. Also, mitochondrial damage induced by methylating agents is repaired in vivo in Chinese hamster ovary cells while complex alkylation damage is not (LeDoux et al., 1992). Together, these results suggest that a direct repair mechanism, corresponding to MGMT-directed repair in the nucleus, is present in mammalian mitochondria. The role of DR in aging is still to be elucidated.
3. Mitochondrial recombinational repair
Double strand breaks (DSBs) are thought to contribute to the different types of naturally occurring mtDNA rearrangements observed during aging and age-associated disease. Repair of DSBs in mtDNA may be conducted by recombination or nonhomologous end-joining (NHEJ) mechanisms, as it occurs in the nucleus. Recombination of mtDNA has been readily observed in yeast. Two proteins involved in recombination of nuclear DNA, the dsDNA binding protein Rad50 and the nuclease Mre11, have been reported to co-localize in yeast mitochondria (Sickmann et al., 2003), and several other proteins involved in recombination have also been found in yeast mitochondria. Therefore, recombination may be a crucial mechanism of maintenance of mtDNA in yeast (Graziewicz et al., 2005). Drosophila melanogaster seems to have an effective system to repair DSB induced by bleomycin (Morel et al., 2008), but the mechanism and mitochondrial proteins involved remain to be identified.
Recombination of mitochondrial DNA in mammalians is much less understood. Some types of lesions believed to be repaired by recombinational repair, such as cisplatin interstrand crosslinks, are repaired in mammalian mitochondria (LeDoux et al., 1992), whereas others, such as psoralen interstrand crosslinks, are not (Cullinane and Bohr, 1998). Recombination activity has been reported in mammalian cell cultures both in vitro and in vivo and mammalian mitochondria can rejoin blunt-ended and cohesive linearized plasmid DNA at a low level (Lakshmipathy and Campbell, 1999a). Based on studies involving in vivo induction of multiple DSBs in mtDNA, it was suggested that infrequent intermolecular recombination is a potential consequence of DSBs in mtDNA (Bacman et al., 2009). Nevertheless, the biological significance of the low frequency of recombination observed in mammalian cells is still unclear. Key proteins known to be involved in nuclear repair, such as RAD51, RAD52, M/R/N, ATM and others have not been detected in mammalian mitochondria. It may be speculated that mitochondrial recombinational DNA repair in mammalian cells may depend on enzymes involved in other DNA repair pathways.
4. Mitochondrial Mismatch Repair
Mismatch repair (MMR) is a post-replicative DNA repair system which corrects base mismatches and small loops. It increases the fidelity of nuclear replication 1000-fold and is conserved from bacteria to humans (Schofield and Hsieh, 2003). The MMR pathway in humans is initiated by heterodimers consisting of homologs of Escherichia coli Mut S, which bind directly to single base mismatches and insertion/deletion loops (IDL). In both humans and yeast, MSH2 and MSH6 combine to form MutSα while MSH2 and MSH3 can form MutSβ, which are the two mismatch recognizing complexes. MutSα recognizes and binds to base-base mismatches and small IDLs, while MutSβ binds to larger IDLs (Larsen et al., 2005). The downstream assembly of additional proteins involved in MMR include E.coli MutL homolog heterodimers, PCNA, hEXO1, and RPA (Li, 2003).
Mismatch repair proteins as we know them from the nucleus have not yet been identified in mammalian mitochondria, except for MSH2, which was detected in rat mitochondrial lysates by one laboratory but not another (Chen et al., 2001; Mason et al., 2003). One of the six MutS isoforms called MSH1 localizes to yeast mitochondria, and when disrupted causes a severe mtDNA instability phenotype (Renan and Kolodner, 1992; Chi and Kolodner, 1994a; Chi and Kolodner, 1994b). However, a homologue of MSH1 has not been found in mammals. Mammalian mitochondria repair cisplatin crosslinks, which in the nucleus are recognized by MMR as well as by NER proteins – suggesting that a functional MMR pathway may exist in mammalian mitochondria (LeDoux et al., 1992). When Lightowlers and coworkers used a DNA repair assay, which measures the capacity of mitochondrial lysates to repair nicked heteroduplex substrates, they found that GG and GT mismatches are repaired efficiently by rat liver mitochondria (Mason et al., 2003). The repair did, however, not seem to be directed to the nicked strand like its nuclear counterpart – but it can not be excluded that this observation can be ascribed to a methodological problem. Mitochondria from rat liver cells deficient in the essential MMR protein MSH2 were still displaying efficient MMR, which suggests that mitochondrial MMR activity is different from the classical MMR found in the nucleus. In a recent study we characterized a mismatch-binding activity in human mitochondria, which was not dependent on classical nuclear MMR factors (Souza Pinto et al., 2009). The mismatch-binding activity co-purified with YB-1, a multifunctional protein, known to be implicated in the nuclear BER pathway, and shown to melt mismatch- and cisplatin containing DNA duplexes in vitro and interact with numerous proteins including MSH2 and Werner syndrome protein (WRN) (Gaudreault et al., 2004). Cells depleted of YB-1 showed decreased cellular respiration and increased mtDNA mutagenesis, implicating this protein in mitochondrial function and mutation avoidance. Together our results show that human mitochondrial extracts do support mismatch repair and that the pathway is independent of nuclear MMR factors but involves the activity of polγ and the multifunctional YB-1 protein. YB-1 has previously been reported to play a role in other nuclear DNA repair pathways including BER by interaction with various BER proteins (Das et al., 2007). In the human mtMMR pathway it is likely to act as a mismatch recognizing protein.
5. Mitochondrial base excision repair
Base excision repair is the main DNA repair pathway coping with most alkylation and oxidative DNA lesions. The latter are mainly induced by free radicals, which are generated endogenously in mitochondria. Investigations indicate that in eukaryotic cells, mtDNA is not floating in the mitochondrial matrix. Instead, it is associated with the inner mitochondrial membrane and arranged in nucleoprotein complexes, or nucleoids (Albring et al., 1977; Holt et al., 2007). Knowing that superoxide radicals are mainly released towards the mitochondrial matrix from the main ROS generation-sites (Barja, 1999; Muller et al., 2004), the location of mtDNA at the inner mitochondrial membrane makes it especially vulnerable to oxidative damage. Hence, BER becomes a crucial mechanism to avoid mtDNA damage accumulation and mtDNA instability.
BER takes place both in the nucleus and in mitochondria, and although the mechanisms are similar, mitochondria possess an independent BER machinery, the components of which are coded by nuclear genes (Bohr 2002b). Similarly to nuclear BER (nBER), mtBER involves a cascade of reactions starting with the recognition of the damage followed by enzymatic processing steps that aim to remove the lesion and restore genomic integrity (for a recent review see Wilson and Bohr, 2007). Briefly, mtBER includes four distinct steps (Figure 1); the pathway is initiated by a specific DNA glycosylase, which recognizes the modified or inappropriate base and cleaves the N-glycosidic bond, creating an abasic site. The glycosylases can be divided into mono-functional or bi-functional glycosylases. In addition to the cleavage activity towards the N-glycosylic bond, the bi-functional glycosylases also possess apurinic/apyrimidinic (AP) lyase activity, which allows the cleavage of the DNA backbone and generation of a 3’ -deoxyribose phosphate (dRP) at the AP site. The resulting AP site is then processed by an AP endonuclease, which creates a strand break with a 3’-hydroxyl end and a 5’- (dRP) residue. Repair then proceeds with the filling of the single nucleotide gap by the mitochondrial DNA polymerase, pol γ. Besides the polymerase activity, pol γ possesses 3’-5’ exonuclease and 5’dRP lyase activities. Thus, when mtBER is initiated by a monofunctional DNA glycosylase, the 5’-dRP moiety generated when the AP endonuclease cleaves the strand break can be removed by the 5’dRP lyase function of pol γ resulting nick is sealed by a DNA ligase.
Figure 1.

Mitochondrial handling of oxidative DNA damage by BER. For details, please see text.
The sequence of events described above illustrates the short-patch (SP) BER, which involves the removal of the DNA lesion and the incorporation of a single nucleotide. Nuclear BER can also proceed via long-patch (LP) BER, which involves the incorporation of two to twelve nucleotides during repair synthesis (Frosina et al., 1996). LP-BER processing of the DNA damage results in the exposure of the ancient DNA strand as part of a single strand overhang or a flap structure. These flap structures are recognized and cleaved by flap endonuclease 1 (FEN1), which is an essential enzyme for nuclear LP-BER (Klungland and Lindahl, 1997) previous to ligation by DNA ligase. Besides, certain BER intermediates, such as oxidized AP sites, cannot be properly handled by the SP-BER (Demple and DeMott, 2002). In the nucleus, the process of these intermediates requires the 5’exo/endonuclease activity of FEN1 and LP-BER (Sung et al., 2005).
Until recently, it was believed that only SP-BER occurred in mitochondria. However, considering that the exposure of mtDNA to ROS generation is continuous, it is likely that oxidized AP sites are generated in mtDNA at a significant rate (Demple and DeMott, 2002). There is now consensus that, similarly to nBER, mtBER can also take place in a long-patch manner (Akbari et al., 2008; Kalifa et al., 2009; Liu et al., 2008; Szczesny et al., 2008). Although initially FEN-1 was not detected in mammalian mitochondrial extracts (Akbari et al., 2008), other investigations have detected FEN-1 in human (Liu et al., 2008; Szczesny et al., 2008) and mouse (Kalifa et al., 2009; Szczesny et al., 2008) mitochondria. Furthermore, the investigation by Liu and coworkers reported that FEN-1 was recovered in the mitochondrial nucleoid fraction and showed a significant dependency of mitochondrial LP-BER on FEN-1 using immunodepletion assays in human lymphoblasts. In addition, it has been reported that Dna2, a helicase/ nuclease protein, interacts with FEN-1 in human HeLa cell mitochondria in the processing of the 5’ flap structure (Zheng et al., 2008). On the other hand, Szczesny et al., (2008) observed that LP-BER was only marginally affected when FEN-1 was depleted in human HCT116 cells (Szczesny et al., 2008), suggesting that mitochondrial LP-BER is mediated primarily by an unidentified 5’-exo/endonuclease. New data show that depletion of the Saccharomyces cerevisiae FEN1 ortholog, Rad27p, increases the frequency of mtDNA mutations, supporting a role of FEN-1 in the mitochondrial LP-BER (Kalifa et al., 2009).
As mentioned above, all the proteins involved in the DNA repair mechanisms in mitochondria are coded in the nuclear DNA, most of them being splice-variants, alternative translation-initiation products, or post-translationally modified versions of the nuclear-encoded protein forms (Nakabeppu, 2001; Nilsen et al., 1997; Nishioka et al., 1999). Interestingly, Stuart et al. (2005) reported that most of the mitochondrial enzymes involved in SP-BER were not freely soluble in the mitochondrial matrix. Instead, they constitute a multiprotein complex, which is strongly associated with an inner membrane-containing particulate fraction, and independent of mtDNA binding (Stuart et al., 2005). The association of the proteins involved in the mtBER to the inner mitochondrial membrane may be crucial for the efficiency of the whole mtDNA repair process, since mtDNA is associated to the inner mitochondrial membrane (Albring et al., 1977) and the main sites of mitochondrial ROS generation are located in the inner mitochondrial membrane as well (Barja, 1999). A model illustrating the location of some of the described mitochondrial components is shown in Figure 2.
Figure 2.

Mitochondrial generation of ROS, mtDNA damage and mtDNA repair occur in close proximity at the mitochondrial inner membrane. Schematic representation of the mitochondrial inner membrane with the electron transport chain (ETC) complexes, nucleoids, mtDNA and BER proteins. Electron leak occurs from complexes I and III (Barja, 1999), leading to the formation of superoxide radical towards the mitochondrial matrix. The mitochondrial DNA is associated in nucleoprotein complexes (nucleoids) to the mitochondrial inner membrane in close proximity to the ETC and the sites of ROS generation, increasing the rate of oxidative attack to mtDNA. In order to avoid mtDNA mutation accumulation due to oxidative damage, mtDNA repair mechanisms are required. Some mitochondrial proteins involved in BER have been located to the nucleoids such as polymerase γ (Garrido et al., 2003) and FEN1 (Liu et al., 2009). Moreover, mitochondrial BER activity has been shown to localize to an inner membrane-associated particulate fraction, and electrostatic interactions have been suggested to mediate the association of mitochondrial BER proteins with the mitochondrial inner membrane (Stuart et al., 2005). Black arrows: electron flux; White/ Red arrows: Electron leak and ROS production; Cx: Complex; UQ: Ubiquinone.
5.1. DNA glycosylases
The first step of the BER is catalyzed by DNA glycosylases. These enzymes recognize the modified or inappropriate base and cleave the N-glycosidic bond, creating an abasic site. Up to eleven different DNA glycosylases have been characterized in mammals (Robertson et al., 2009), but only a few of them have been detected in mitochondria (Slupphaug et al., 2003).
One of the most extensively investigated mitochondrial DNA glycosylases is Ogg1. Ogg1 is a DNA glycosylase with AP lyase activity that recognizes and cleaves 8-hydroxy-guanine (8-oxoG) from DNA (Bjorås et al., 1997; Zharkov et al., 2000). 8-oxoG is a mutagenic lesion when present in double-stranded DNA, because it can mispair with adenine, leading to GC to TA transversion mutations (Grollman and Moriya, 1993). Ring-opened formamidopyrimidine lesions (4,6- diamino-5- formamidopyrimidine (FapyA) and 2,6- diamino-4- hydroxy-5- formamidopyrimidine (FapyG)) have been detected in mouse liver genomic DNA at similar or higher levels than 8-oxoG (Hu et al., 2005), suggesting that the endogenous accumulation with age of the formamidopyrimidines may be higher than that of 8-oxoG. However, 8-oxoG is the DNA lesion that has been most broadly used as a DNA damage marker, both in nuclear and mitochondrial DNA (Richter et al., 1988; Shigenaga et al., 1994; Barja and Herrero, 2000). This is because 8-oxoG is relatively easy to detect and measure, but not because it is established as the most frequent or most biologically important lesion.
The human OGG1 gene produces two major distinct transcripts via alternative splicing (Nishioka et al., 1999). While only one isoform of Ogg1 has been described in rodents, two different isoforms of Ogg1 proteins have been identified in humans: α- Ogg1, and β- Ogg1. β- Ogg1 appears to be localized exclusively in mitochondria (Nishioka et al 1999; Takao et al., 1998) and, although the α-isoform is mainly present in the nucleus, it has been described to be present to a lesser extent in the mitochondria as well (Hashiguchi, et al., 2004). Furthermore, Hashiguchi et al., reported that the β-form is a non-active splice variant with unknown function and that the α- Ogg1 isoform is the one accounting for the 8-oxoG repair activity both in nuclei and mitochondria. It is unclear what the role of βOGG1 is in mitochondrial DNA metabolism. Our own studies have indicated that there is no evident role for βOGG1 in mitochondrial BER, but other studies have indicated a role for it in apoptosis (Oka et al., 2008)). In contrast to the situation for αOGG1 very little is known about protein interactions for βOGG1, but it would be of great future interest to explore these further. Mammalian Ogg1 has been shown to posses glycosylase activity not only towards 8-oxoG, but also towards other oxidative lesions such as 8-oxoA and FapyG (Jensen et al., 2003; Zharkov et al., 2000).
The first DNA glycosylase activity identified was the uracil-DNA glycosylase (UNG) in E. coli (Lindahl, 1974). Uracil in DNA emerges by deamination of cytosine or by misincorporation of dUMP (Hagen et al 2006). The removal of uracil from DNA is crucial due to its ability to pair with adenine, causing GC to AT transition mutations upon replication (Duncan and Miller, 1980).
In mammals, two different forms of UNG, nuclear (UNG2) and mitochondrial (UNG1), are encoded by the UNG gene and are generated by alternative transcription initiation sites and alternative splicing (Nilsen et al., 1997). They share a common catalytic domain, and hence substrate specificity for uracil. However, they differ in the N-terminal sequence that determines the subcellular sorting as well as the interaction with other proteins. Although other DNA glycosylases have been described to catalyze the excision of uracil residues from DNA (Haushalter et al., 1999), UNG1 and UNG2 are the most efficient DNA glycosylases removing uracil, catalyzing excision of uracil from both single- and double-stranded DNA (Slupphaug et al., 1995). Moreover, UNG1 is the only uracil-DNA glycosylase known to be present in mammalian mitochondria (Akbari et al., 2008).
A new group of DNA glycosylases, named NEIL, have recently been identified in mammalian tissues (Hazra et al., 2002a, b). They are homologous of the E. coli DNA glycosylases Fpg/Nei, and primarily excise oxidized purines such as FapyG and FapyA. NEIL1, NEIL2 and NEIL3 have been identified in mammalian genomes, and characterization of the specific activities indicate that, in addition to excision of fapy intermediates, NEIL1 and NEIL2 are also involved in repair mechanisms of oxidative lesions in transcribed or replicating DNA and are bi-functional enzymes (Dou et al., 2003). Unlike other bi-functional DNA glycosylases, NEIL1 and NEIL2 generate DNA strand breaks with 3' phosphate termini, which are then removed in an APE endonuclease independent mechanism mediated by the polynucleotide kinase (PNK) (Wiederhold et al., 2004). Although NEILs were initially localized in the nucleus, Hu et al. (2005) have reported the presence of NEIL1 in mouse liver mitochondria, and NEIL activity has recently been observed in mouse brain mitochondria as well (Gredilla et al., 2008). Whether PNK, or another enzyme possessing 3’ phosphate activity, is involved in the processing of the 3’ phosphate termini in mitochondria is unclear. However, PNK has been located primarily in nuclei (Karimi-Busheri et al., 1999) and described to be involved mainly in nuclear DNA repair events (Bernstein et al., 2005), thus it is possible that the processing of the 3’ phosphate termini in mitochondrial DNA is mediated by a different enzyme than in the nucleus. The importance of this new set of DNA glycosylases in the maintenance of mtDNA has been confirmed by NEIL1 knockout mice. These mice accumulate mtDNA deletions to a higher extent than wild type mice in hepatic tissue and in some cases develop symptoms associated with the metabolic syndrome (Vartanian et al., 2006). NEIL1 and NEIL2 share part of the substrate specificity with another DNA glycosylase, NTH1. The mammalian NTH1 is an ortholog of E. coli endonuclease III (Nth). It is a DNA glycosylase with AP lyase activity that excises a wide range of pyrimidine lesions, including thymine glycol and 5-hydroxy cytosine. Moreover, it has been described to remove Fapy residues in human cells (Luna et al., 2000). NTH1 appears to localize mostly in mitochondria in mice (Ikeda et al., 2002). However, human NTH1 has been shown to localize almost exclusively to the nucleus (Ikeda et al., 2002; Takao et al., 1998), even though it contains a putative site sensitive to mitochondrial matrix proteases (Takao et al., 1998).
5.2. AP endonuclease
After a DNA glycosylase has removed the modified or erroneous base, the resulting abasic site is further processed by an AP endonuclease. APE1 has been described to be the main AP endonuclease in mammalian cells (Demple and Harrison, 1994). A second mammalian AP endonuclease protein (APE2) has been described in human cells (Hadi and Wilson 3rd, 2000; Tsuchimoto et al., 2001), however, with no detectable APE activity (Ribar et al., 2004; Wiederhold et al., 2004). APE1 has two major enzymatic functions: (a) cleavage of the phosphodiester backbone 5’ to AP sites, generating 3’-OH substrates for DNA polymerases; (b) 3’-diesterase activity specific for the 3’-dRP products generated by β-elimination by bi-functional glycosylases, again creating 3’-OH substrates for DNA polymerases (Demple and Harrison, 1994). In addition, APE1 also has a redox activity (Tell et al., 2005) that is involved in signal transduction, and not very well understood.
Unlike most of the enzymes taking part in the BER process in mitochondria, the nature of mtAPE still remains unclear. It has been suggested that the mitochondrial APE is an N-terminal truncation product of APE1 (Chattopadhyay et al., 2006). Moreover, the deletion of the N-terminal residues, which contain the nuclear localization signal, induced a 3-fold increase in the specific activity of mtAPE. However, Chattopadhyay and coworkers reported that the presence of the truncated APE1 protein at a significant level was restricted to bovine liver, while only full-length APE1 was detected in mitochondrial extracts of various human cell lines. Unlike knock-out (KO) mice for most of the individual DNA glycosylases, which do not exhibit a significant phenotype due to the partial redundancy among these enzymes, APE1 KO mice show embryonic lethality (Ludwig et al., 1998), confirming the critical role that APE1 plays in cell survival. However, in addition to its AP site processing activity, APE1 functions as a transcriptional co-activator for different transcription factors, thereby participating in the control of several different important cellular mechanisms such as apoptosis, proliferation, and differentiation (Tell et al., 2005; Xanthoudakis et al., 1992). Thus, it remains to be established if the embryonic lethality observed in APE1 KO mice is due to its role in BER or in transcription.
In the nucleus, the processing of AP sites is not always APE1-dependent. The description of the new NEILs in mammalian cells and the characterization of the mechanisms of action of these glycosylases questioned the absolute requirement of APE1 in BER. NEIL2-initiated repair of oxidized bases in nuclear DNA has been reported to occur in an APE-independent manner (Hazra et al., 2007). Instead, the 3’-phosphate at the strand break left after β-δ-elimination by the bi-functional NEIL1 and NEIL2 is processed by the mammalian polynucleotide kinase (PNK) (Das et al., 2006; Wiederhold et al., 2004). Further investigation is needed to determine whether the NEIL-initiated BER takes place in mitochondria in an APE-independent manner as well.
5.3 DNA polymerase and DNA ligase
Independent of whether the AP site is processed by the short- or long-patch subpathway, the next step in the BER pathway is catalyzed by a DNA polymerase, which will insert the correct nucleotide(s) in the generated gap. Mammalian mitochondria posses a single DNA polymerase, polymerase γ (pol γ), which functions in all replication, recombination and repair processes involving mtDNA. Together with the DNA synthesis activity, the mtDNA polymerase in mammalian cells has an inherent 3’- 5’ exonuclease activity necessary for proof-reading newly synthesized mtDNA together with a 5’dRP lyase function, both of which are important for accurate gap-filling and termini processing during BER (Graziewicz et al., 2006). Similarly to APE1 KO mice, Polγ KO mice show embryonic lethality (Hance et al., 2005). Again, since polγ is involved not only in DNA repair but also in replicative events, it is not possible to discriminate the repair capacity from the KO lethality. Interestingly, the creation by two independent groups of a knock-in mice expressing proof-reading deficient pol γ, but conserving its replicative function has provided the first direct evidence that accelerating the mtDNA mutation rate results in premature aging (Trifunovic et al., 2004; Kujoth et al., 2005).
The final step of mitochondrial BER, the ligation of the remaining single-strand nick is catalyzed by a DNA ligase. The human mitochondrial ligase is DNA ligase III, a splice variant from the LIG3 gene which encodes both the nuclear and mitochondrial enzymes (Lakshmipathy and Campbell, 1999a). Nuclear and mitochondrial variants appear to differentially interact with other proteins involved in DNA repair, such as XRCC1. XRCC1 interacts with the nuclear variant of LIG3 and stabilizes it (Thompson and West, 2000), while mitochondrial DNA ligase III has been described to be independent of XRCC1 (Lakshmipathy and Campbell, 2000).
Single strand breaks (SSBs) are some of the most common lesions found in chromosomal DNA. In SSB repair, PARP1 is believed to function as a SSB sensor that binds to the break and attracts ligase 3. The repair of SSBs then follows essentially the same procedure as BER. Interestingly, it was very recently demonstrated that the predominantly nuclear enzyme, PARP-1, localizes to mitochondria, associates to mitochondrial DNA, and is involved in maintenance of mitochondrial DNA integrity (Rossi et al., 2009).
Increasing knowledge on the mitochondrial DNA repair regulation as well as in the proteins that are actually present in mitochondria suggest that other still unknown proteins and pathways may play important roles in the mtDNA repair mechanisms. Figure 3 gives an overview of the current knowledge regarding DNA repair pathways shown to function in mitochondria.
Figure 3.
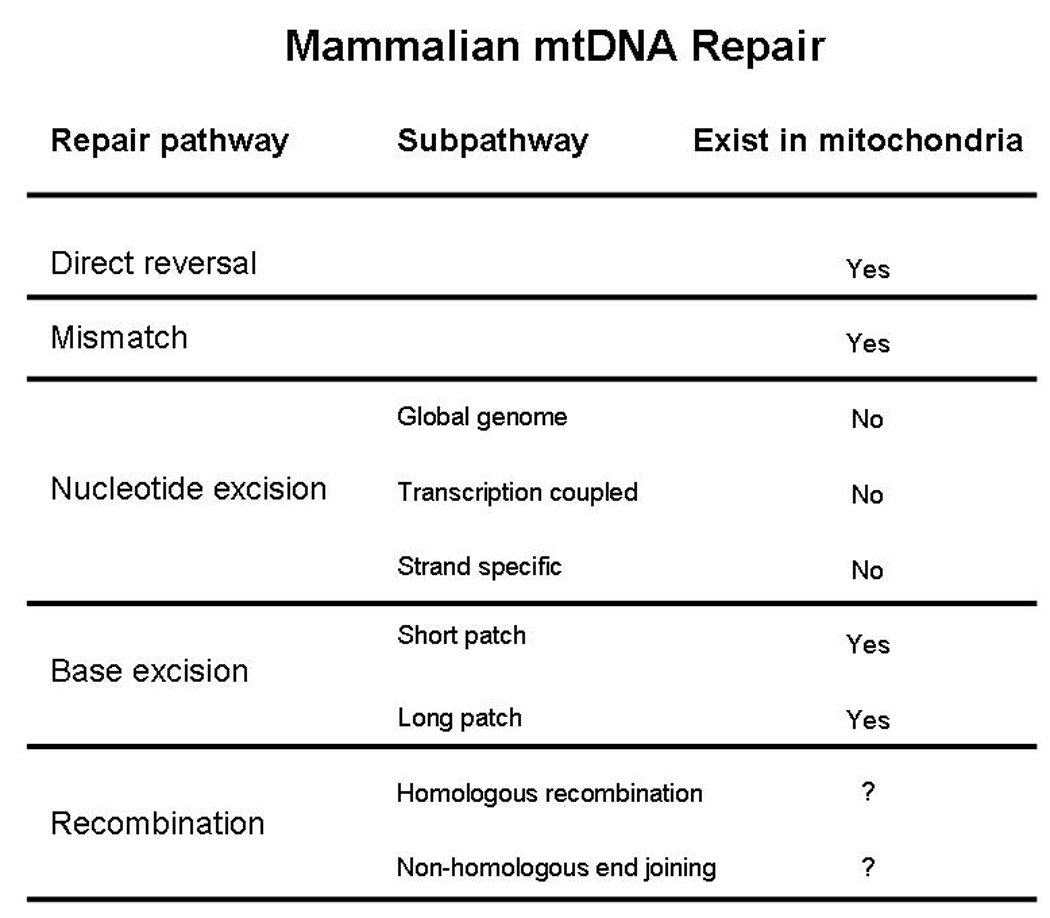
Overview of DNA repair pathways shown to function in mammalian mitochondria
6. Base excision repair: a conserved mechanism involved in the aging process
All living organisms are exposed to DNA damaging agents, and DNA repair pathways have evolved as important systems for maintenance of DNA stability and cell survival. E. coli and the yeast S. cerevisiae are the two model organisms in which DNA repair has been described in most detail. Homologues between prokaryotes and eukaryotes have been reported in different repair pathways (Aravind et al., 1999; Gros et al., 2002). Proteins in the main DNA repair pathways are highly conserved and several of them contain highly conserved sequences. Besides, most of the genes involved in DNA repair are also present in higher plants (Kimura and Sacaguchi, 2006). Such high degree of conservation indicates that DNA repair pathways are fundamental mechanisms in cell survival. BER is not an exception, and many of the genes involved in this repair pathway are highly conserved from bacteria to humans. Examples are those coding for UNG and NTH1 (Krokan et al., 1997; Sarker et al., 1998; Soerensen et al., 2009). Alignment of these BER protein sequences has revealed high levels of sequence conservation. The human and the bacterial UNG proteins possess 55.7% identical residues (Olsen et al., 1989), while the protein sequence of the human NTH1 showed showed 81.0% identity to the mouse homologue (Sarker et al., 1998) and 47% identity to the Podospora anserina homologue (Soerensen et al., 2009). The similarity significantly increases when conservative amino acids are included in the comparison, reaching 73.3% when comparing the human and bacterial UNG (Olsen et al., 1989).
Model organisms other than mammalian species are excellent model systems for the study of the mechanisms underlying the aging process (Strehler, 1962). The advantage being that they have relatively short life span and genetic modification is easier than in mammals. Along with the studies performed in unicellular organisms, multicellular eukaryotes such as Caenorhabditis elegans (Hyun et al., 2008; Meyer et al., 2007; Morinaga et al 2009), or filamentous fungi like P. anserina (Goldman et al., 2002; Soerensen et al 2009) may become important models in DNA repair and aging research. Together with the studies carried out in human cells and in other mammals, these investigations may notably contribute to the understanding of the role of DNA repair mechanisms in the aging process, particularly those taking place in mitochondria.
Although the DNA repair mechanisms in mitochondria are not as well known as those taking place in the nucleus, the knowledge on mtDNA repair functions is increasing. Currently, it is of great interest to understand whether the mtDNA repair mechanisms, particularly BER, play an important role in the aging process and age-associated diseases. Increased oxidative damage and mutations in mtDNA have been observed in different tissues during aging, and the role of such an increase has been linked to aging in mammals (Barja, 1999; Beckman and Ames, 1999; Bender et al., 2006; Kujoth et al., 2007; Melov, 2004). The effects of mtDNA damage and mutations are believed to be particularly important in tissues containing postmitotic cells, since these cells can not be replaced by intact ones. Together with the accumulation of mtDNA damage with aging, it has been reported that oxidative damage to mtDNA in cardiac and neuronal tissue is inversely related to the maximum life span potential (MLSP) in mammals (Barja and Herrero, 2000) suggesting that such accumulation may play a causative role in the determination of lifespan. Different investigations have analyzed whether DNA repair mechanisms are also related to MLSP determination. When the UV-light induced DNA damage was investigated, DNA repair activities were inversely correlated to MLSP in mammals (Hart and Setlow, 1974). Similarly, in a recent investigation on C. elegans strains showing different longevities, long-lived strains showed higher rates of UV-induced DNA damage repair than the wild type nematodes. These studies provide evidence that NER repair capacity correlates with MLSP (Hyun et al., 2008). However, when the relation of BER and MLSP was investigated, the results were different. Brown and Stuart (2007) reported that BER enzyme activities in fibroblast whole cell extracts showed either no correlation (APE1), or correlated negatively (polymerase β) with MLSP in mammals. Similar results have been observed when mitochondrial BER was investigated in relation to MLSP. Caloric restriction is the only experimental manipulation that increases maximum life span in several species (Gredilla and Barja, 2005). Although caloric restriction has been reported to enhance BER in the nucleus (Cabelof et al., 2003), Stuart et al. (2004) showed that the mtBER capacity did not change significantly in liver and even decreased in the brain and kidney of calorically restricted rats. Likewise, Soerensen et al. (2009) have recently observed that the activity of BER enzymes in mitochondrial extracts of two different long-lived strains of P. anserina is significantly lower than in the wild-type mates. The results obtained for mtBER may reflect an internal balance between DNA damage and DNA repair. Similarly to mtDNA oxidative damage, mtROS generation is inversely correlated to maximum life span in mammals (Ku et al., 1993; Lambert et al., 2007). Moreover, mitochondria from caloric restricted rodents have been shown to generate ROS at lower rates than ad libitum fed animals (Sohal et al., 1994a; Gredilla et al., 2001), and the levels of oxidative damage to DNA have been reported to be lower in restricted animals as well (Sohal et al., 1994b; Gredilla et al 2001; Sanz et al 2005). Similarly, long-lived strains of P. anserina generate lower mitochondrial ROS (Gredilla et al., 2006) and show lower mtDNA instability (Borghouts et al., 1997) than in the wild-type strain. Hence, when the mtDNA damage is significantly reduced, it is possible for the organism to invest less energy in DNA repair capacity without negative consequences. Since BER activities and the overall BER capacity are known to be up-regulated in vivo, in part by oxidative stress (Cabelof et al., 2002; Mitra et al., 2007; Akbari et al., 2006), it is likely that reduced oxidative stress has the opposite effect, leading to down-regulation of the mtBER, so it is proportional to the rate of mtDNA damage. In order to completely understand the role of DNA repair mechanisms in MLSP determination, it would be necessary to investigate whether the mechanisms of mtDNA repair correlate with life span in mammals.
On the other hand, compiling evidence supports that BER is altered in aging, and that deficiencies in mtBER, particularly in postmitotic tissues, play a role in cell function and survival. The recent creation of the knock-in mice expressing proof-reading deficient pol γ appears as a promising good mouse model in which to investigate the causative link between mtDNA mutation accumulation and aging, and the role that mtBER may play in the process (Kujoth et al., 2007; Kukat and Trifunovic, 2009). In various tissues of this mouse model, mtDNA point mutations as well as mtDNA deletions accumulate at a much higher rate than in the wild type mice (Trifunovic et al., 2004; Kujoth et al., 2005). However, how the accumulation of mtDNA mutations lead to the loss of mitochondrial function and aging is still discussed. Investigations on brain and heart samples have suggested that it is the accumulation of large mtDNA deletions and clonal expansion, instead of mtDNA point mutations, that drive the premature aging phenotype (Vermulst et al., 2008; Vermulst et al., 2007). However, after performing research on hepatic and cardiac tissues, Edgar et al. (2009) recently reported that circular mtDNA molecules with large deletions represent only a minor proportion of the total mtDNA in this mouse model, and suggested that random point mutations occurring in mtDNA are the driving force behind the premature aging phenotype. The mechanisms by which mtDNA mutations are generated in postmitotic and mitotic tissues have been suggested to be different (Reeve et al., 2009). It has been proposed that in postmitotic cells, such as neurons, mtDNA deletions occur primary during repair of damaged DNA rather than during DNA replication, whereas in mitotic tissues mtDNA point mutations are more likely to be caused during replication (Krishnan et al., 2008). This could lead to significant differences in the type of mtDNA mutations being detected in postmitotic and mitotic cells during normal aging. Thus, for substantia nigra neurons from aged individuals and Parkinson’s disease patients direct evidence exists of mitochondrial dysfunction due to accumulation of mtDNA deletions (Bender et al., 2006; Kraytsberg et al., 2006), while mtDNA point mutations has been reported to occur rarely in these neurons during aging (Reeve et al., 2009). Changes in mitochondrial BER fidelity with age play a role in mtDNA damage accumulation, contributing to age-related decline. Mitochondrial BER capacity has been described to be organ-specific, with the brain being one of the tissues with the lowest capacity (Karahalil et al., 2002). Various studies have investigated the role of changes in mitochondrial BER in age-related functional decline, showing tissue specific age-related changes. While an increase in APE1 and increases or no changes in DNA glycosylase activities have been observed to occur during aging in mitochondria from liver and heart of rodents (Mitra et al., 2007; Souza-Pinto et al., 1999; Souza-Pinto et al., 2001), a general decline in DNA glycosylases has been observed in brain cortical mitochondria in rats (Chen et al., 2003) and mice (Imam et al., 2006; Gredilla et al., 2008). Moreover, important differences in mtBER have been observed among various brain regions during aging (Gredilla et al., 2008). The cortical region and the cerebellum have been described to accumulate less mtDNA lesions with aging and to be more resistant to oxidative stress conditions (Brown et al., 2004; Filburn et al., 1996; Hota et al., 2007). Interestingly, those regions showed higher BER capacity than hippocampus, which has been described to be a much more vulnerable region in the brain (Brown et al., 2004; Filburn et al., 1996; Abd El Mohsen et al., 2005). These results support the idea that mtBER plays a critical role in the development and maintenance of the central nervous system during aging (Weissman et al., 2007). Recent investigations in our laboratory support the relevance of mtBER in brain aging and the importance of the central nervous system heterogeneity in these processes (Gredilla et al. in preparation). In addition to these studies, increasing Ogg1 activity in mitochondria from oligodendrocytes after targeting hOGG1 into mitochondria increases cell survival and protects against induced-oxidative stress (Druzhyna et al., 2003). Recently, studies in the aging model P. anserina have shown that in this model, aging is also associated with a decrease in mitochondrial BER (Soerensen et al., 2009), contributing to the observed mtDNA instability in the aged fungi (Borghouts et al., 1997). Taken together all these data suggest that there is a relationship between DNA damage, increased DNA damage repair and aging. Further investigations will clarify whether other DNA repair mechanisms are involved in the aging process as well. Investigation of the role of BER on aging that have been performed in human samples have mainly focused on nuclear BER, with a limited number of studies addressing the role of mtBER. Several reports have associated mtDNA mutations and deletions with Alzheimer’s disease (AD) (Chang et al., 2000; Hutchin et al., 1995; Davis et al., 1997; Coskun et al., 2004; Lin and Beal, 2006) and recent data show that a significant decline in total BER takes place in the cortical area of AD patients (Weissman et al., 2007). Moreover, it has been reported that targeting hOGG1 into mitochondria increases survival and protects against induced-oxidative stress in various cell types including oligodendrocytes and INS-1 cells (Druzhyna et al., 2003; Rachek et al., 2006), and that mitochondrial BER activity decreases in cultured human fibroblast with higher passage number (Shen et al., 2003).
In order to understand the normal aging process in humans, genetically inherited premature aging syndromes are valuable tools that have been widely used (Bohr et al., 2005). Among them, the DNA repair defective disease Cockayne syndrome (CS) shows deficiency in transcription coupled nucleotide excision repair. CS is a multisystemic disease characterized by developmental and neurological abnormalities and premature aging. Although NER is the most important repair pathway affected in this syndrome, recent investigations indicate that BER is affected as well. In the CS complementation group B (CSB), a deficiency in the repair of 8-oxoG has been reported both in whole cell extracts (Dianov et al., 1999) and in mitochondria from human CSB-deficient cells (Stevnsner et al., 2002). However, the precise mechanism by which CSB participates in the repair of 8-oxoG in the nuclear or in the mitochondrial DNA is not yet known. Nevertheless, it is likely that impairment in mtBER contributes to the neurological phenotype and progression of the disease in CSB patients (Stevnsner et al., 2008). In Xeroderma Pigmentosum (XP) complementation group C (XPC), another rare autosomal recessive disorder with impaired NER activity, a significant impairment in 8-oxoG repair has recently been described in the nucleus as well (D’Errico et al., 2006). Similarly to CS, it has been suggested that such decline in the repair of oxidative lesions may significantly contribute to the neurological symptoms observed in XPC patients (D’Errico et al., 2006).
In conclusion, several investigations strongly support the hypothesis that mtBER impairment and mtDNA instability contribute to aging. Further investigation is required in order to confirm the role of mtBER in the determination of MLSP in mammals as well as whether other mitochondrial DNA repair pathways that are now known to take place in mitochondria play a role in the aging process. Finally, certain DNA adducts, such as those derived from lipid peroxidation, are processed via NER.
Such adducts are likely to be generated in mtDNA, since mitochondrial phospholipids are main targets of mitochondrial free radicals and mtDNA is associated with the inner mitochondrial membrane. Thus, it is tempting to speculate about the potential presence of NER-like mechanisms in mitochondria, even though UV induced DNA damage is not repaired in these organelles. Further knowledge of how DNA repair takes place in mitochondria will provide help to find strategies to potentially retard mtDNA mutations and hence cellular dysfunction observed during aging and with age-related diseases.
Acknowledgements
This work was supported by a grant from the European Commission (LSHM-CT-2004-512020), The Danish Aging Research Center (VELUX 95-103-11419), the Danish Cancer Society (DP05118 and DP03131), The Danish Research Council (271-08-0697) and the Lundbeck Foundation. Some of this work was also supported by funds from the Intramural Program of the National Institute on Aging, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abd El Mohsen MM, Iravani MM, Spencer JP, Rose S, Fahim AT, Motawi TM, Ismail NA, Jenner P. Age-associated changes in protein oxidation and proteasome activities in rat brain: modulation by antioxidants. Biochem Biophys Res Commun. 2005;336:386–391. doi: 10.1016/j.bbrc.2005.07.201. [DOI] [PubMed] [Google Scholar]
- Akbari M, Otterlei M, Pena-Diaz J, Krokan HE. Different organization of base excision repair of uracil in DNA in nuclei and mitochondria and selective upregulation of mitochondrial uracil-DNA glycosylase after oxidative stress. Neuroscience. 2006;145:1201–1212. doi: 10.1016/j.neuroscience.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Akbari M, Visnes T, Krokan HE, Otterlei M. Mitochondrial base excision repair of uracil and AP sites takes place by single-nucleotide insertion and long-patch DNA synthesis. DNA Repair. 2008;7:605–616. doi: 10.1016/j.dnarep.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Albring M, Griffith J, Attardi G. Association of a protein structure of probable membrane derivation with HeLa cell mitochondrial DNA near its origin of replication. Proc. Natl. Acad. Sci. USA. 1977;74:1348–1352. doi: 10.1073/pnas.74.4.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Moraes CT. Intra- and inter-molecular recombination of mitochondrial DNA after in vivo induction of multiple double-strand breaks. Nucleic Acids Res. 2009;37:4218–4226. doi: 10.1093/nar/gkp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barja G. Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 1999;31:347–366. doi: 10.1023/a:1005427919188. [DOI] [PubMed] [Google Scholar]
- Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000;14:312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN. Endogenous oxidative damage of mtDNA. Mutat Res. 1999;424:51–58. doi: 10.1016/s0027-5107(99)00007-x. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Bernstein NK, Williams RS, Rakovszky ML, Cui D, Green R, Karimi-Busheri F, Mani RS, Galicia S, Koch CA, Cass CE, Durocher D, Weinfeld M, Glover JN. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol. Cell. 2005;17:657–670. doi: 10.1016/j.molcel.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Bjorås M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T, Seeberg E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997;16:6314–6322. doi: 10.1093/emboj/16.20.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr VA. DNA damage and its processing. Relation to human disease. J. Inherit. Metab. Dis. 2002a;25:215–222. doi: 10.1023/a:1015681929316. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Repair of oxidative DNA damage in nuclear and mitochondrial DNA, and some changes with aging in mammalian cells. Free Radic. Biol. Med. 2002b;32:804–812. doi: 10.1016/s0891-5849(02)00787-6. [DOI] [PubMed] [Google Scholar]
- Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–620. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr VA, Ottersen OP, Tønjum T. Genome instability and DNA repair in brain, ageing and neurological disease. Neuroscience. 2007;145:1183–1186. doi: 10.1016/j.neuroscience.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Borghouts C, Kimpel E, Osiewacz HD. Mitochondrial DNA rearrangements of Podospora anserina are under the control of the nuclear gene grisea. Proc. Natl. Acad. Sci. USA. 1997;94:10768–10773. doi: 10.1073/pnas.94.20.10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MF, Stuart JA. Correlation of mitochondrial superoxide dismutase and DNA polymerase beta in mammalian dermal fibroblasts with species maximal lifespan. Mech. Ageing Dev. 2007;128:696–705. doi: 10.1016/j.mad.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Brown MR, Geddes JW, Sullivan PG. Brain region-specific, age-related, alterations in mitochondrial responses to elevated calcium. J. Bioenerg. Biomembr. 2004;36:401–406. doi: 10.1023/B:JOBB.0000041775.10388.23. [DOI] [PubMed] [Google Scholar]
- Cabelof DC, Raffoul JJ, Yanamadala S, Guo Z, Heydari AR. Induction of DNA polymerase beta-dependent base excision repair in response to oxidative stress in vivo. Carcinogenesis. 2002;23:1419–1425. doi: 10.1093/carcin/23.9.1419. [DOI] [PubMed] [Google Scholar]
- Cabelof DC, Yanamadala S, Raffoul JJ, Guo Z, Soofi A, Heydari AR. Caloric restriction promotes genomic stability by induction of base excision repair and reversal of its age-related decline. DNA Repair. 2003;2:295–307. doi: 10.1016/s1568-7864(02)00219-7. [DOI] [PubMed] [Google Scholar]
- Chang SW, Zhang D, Chung HD, Zassenhaus HP. The frequency of point mutations in mitochondrial DNA is elevated in the Alzheimer’s brain. Biochem. Biophys. Res. Commun. 2000;273:203–208. doi: 10.1006/bbrc.2000.2885. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay R, Wiederhold L, Szczesny B, Boldogh I, Hazra TK, Izumi T, Mitra S. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006;34:2067–2076. doi: 10.1093/nar/gkl177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Cao G, Hastings T, Feng Y, Pei W, O'Horo C, Chen J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J. Neurochem. 2002;81:1273–1284. doi: 10.1046/j.1471-4159.2002.00916.x. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Felsheim R, Wong P, Augustin LB, Metz R, Kren BT, Steer CJ. Mitochondria isolated from liver contain the essential factors required for RNA/DNA oligonucleotide-targeted gene repair. Biochem. Biophys. Res. Commun. 2001;285:188–194. doi: 10.1006/bbrc.2001.5156. [DOI] [PubMed] [Google Scholar]
- Chi NW, Kolodner RD. Purification and characterization of MSH1, a yeast mitochondrial protein that binds to DNA mismatches. J. Biol. Chem. 1994a;269:29984–29992. [PubMed] [Google Scholar]
- Chi NW, Kolodner RD. The effect of DNA mismatches on the ATPase activity of MSH1, a protein in yeast mito chondria that recognizes DNA mismatches. J. Biol. Chem. 1994b;269:29993–29997. [PubMed] [Google Scholar]
- Clayton DA, Doda JM, Friedberg EC. The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc Natl Acad Sci USA. 1974;71:2777–2781. doi: 10.1073/pnas.71.7.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun PE, Beal MF, Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinane C, Bohr VA. DNA interstrand cross links induced by psoralen are not repaired in mammalian mitochondria. Cancer Res. 1998;58:1400–1404. [PubMed] [Google Scholar]
- Das A, Wiederhold L, Leppard JB, Kedar P, Prasad R, Wang H, Boldogh I, Karimi-Busheri F, Weinfeld M, Tomkinson AE, Wilson SH, Mitra S, Hazra TK. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells DNA Repair. 2006;5:1439–1448. doi: 10.1016/j.dnarep.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Chattopadhyay R, Bhakat KK, Boldogh I, Kohno K, Prasad R, Wilson SH, Hazra TK. Stimulation of Neil2-mediated oxidized base excision repair via YB-1 interaction during oxidative stress. J. Biol. Chem. 2007;282:28474–28484. doi: 10.1074/jbc.M704672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE, Miller S, Herrnstadt C, Ghosh SS, Fahy E, Shinobu LA, Galasko D, Thal LJ, Beal MF, Howell N, Parker WD., Jr Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1997;94:4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Demple B, DeMott MS. Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene. 2002;21:8926–8934. doi: 10.1038/sj.onc.1206178. [DOI] [PubMed] [Google Scholar]
- Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- D'Errico M, Parlanti E, Teson M, de Jesus BM, Degan P, Calcagnile A, Jaruga P, Bjørås M, Crescenzi M, Pedrini AM, Egly JM, Zambruno G, Stefanini M, Dizdaroglu M, Dogliotti E. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 2006;25:4305–4315. doi: 10.1038/sj.emboj.7601277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Sunesen M, Bohr VA. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res. 1999;27:1365–1368. doi: 10.1093/nar/27.5.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- Druzhyna NM, Hollensworth SB, Kelley MR, Wilson GL, Ledoux SP. Targeting human 8-oxoguanine glycosylase to mitochondria of oligodendrocytes protects against menadione-induced oxidative stress. Glia. 2003;42:370–378. doi: 10.1002/glia.10230. [DOI] [PubMed] [Google Scholar]
- Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560–561. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, Nedergaard J, Cannon B, Larsson NG, Trifunovic A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab. 2009;10:131–138. doi: 10.1016/j.cmet.2009.06.010. [DOI] [PubMed] [Google Scholar]
- Filburn CR, Edris W, Tamatani M, Hogue B, Kudryashova I, Hansford RG. Mitochondrial electron transport chain activities and DNA deletions in regions of the rat brain. Mech. Ageing Dev. 1996;87:35–46. doi: 10.1016/0047-6374(96)01696-x. [DOI] [PubMed] [Google Scholar]
- Frosina G, Fortini P, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandolo A, Dogliotti E. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996;271:9573–9578. doi: 10.1074/jbc.271.16.9573. [DOI] [PubMed] [Google Scholar]
- Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM, Spelbrink JN. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell. 2003;14:1583–1596. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreault I, Guay D, Lebel M. YB-1 promotes strand separation in vitro of duplex DNA containing either mispaired bases or cis-platin modifications, exhibits endonucleolytic activities and binds several DNA repair proteins. Nucleic Acids Res. 2004;32:316–327. doi: 10.1093/nar/gkh170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase gamma in mitochondrial DNA replication and repair. Chem. Rev. 2006;106:383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- Gredilla R, Barja G. Minireview: the role of oxidative stress in relation to caloric restriction and longevity. Endocrinology. 2005;146:3713–3717. doi: 10.1210/en.2005-0378. [DOI] [PubMed] [Google Scholar]
- Gredilla R, Garm C, Holm R, Bohr VA, Stevnsner T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol. 2008 doi: 10.1016/j.neurobiolaging.2008.07.004. Aging doi:10.1016/j.neurobiolaging.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredilla R, Grief J, Osiewacz HD. Mitochondrial free radical generation and lifespan control in the fungal aging model Podospora anserina. Exp. Gerontol. 2006;41:439–447. doi: 10.1016/j.exger.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Gredilla R, Sanz A, Lopez-Torres M, Barja G. Caloric restriction decreases mitochondrial free radical generation at complex I and lowers oxidative damage to mitochondrial DNA in the rat heart. FASEB J. 2001;15:1589–1591. doi: 10.1096/fj.00-0764fje. [DOI] [PubMed] [Google Scholar]
- Greenberg MM, Dizdaroglu M, Bohr VA. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1 and NEIL1 enzymes. J. Biol. Chem. 2005;280:40544–40551. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- Grollman AP, Moriya M. Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- Gros L, Saparbaev MK, Laval J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene. 2002;21:8905–8925. doi: 10.1038/sj.onc.1206005. [DOI] [PubMed] [Google Scholar]
- Hadi MZ, Wilson DM., 3rd Second human protein with homology to the Escherichia coli abasic endonuclease exonuclease III. Environ. Mol. Mutagen. 2000;36:312–324. [PubMed] [Google Scholar]
- Hagen L, Peña-Diaz J, KAvli B, Otterlei M, Sluphaug G, Krokan HE. Genomic uracil and human disease. Exp. Cell Res. 2006;312:2666–2672. doi: 10.1016/j.yexcr.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Hamilton ML, Guo Z, Fuller CD, Van Remmen H, Ward WF, Austad SN, Troyer DA, Thompson I, Richardson A. A reliable assessment of 8-oxo-2-deoxyguanosine levels in nuclear and mitochondrial DNA using the sodium iodide method to isolate DNA. Nucleic Acids Res. 2001;29:2117–2126. doi: 10.1093/nar/29.10.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hance N, Ekstrand MI, Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum. Mol. Genet. 2005;14:1775–1783. doi: 10.1093/hmg/ddi184. [DOI] [PubMed] [Google Scholar]
- Hart RW, Setlow RB. Correlation between deoxyribonucleic acid excision-repair and life-span in a number of mammalian species. Proc. Natl. Acad. Sci. USA. 1974;71:2169–2173. doi: 10.1073/pnas.71.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi K, Stuart JA, Souza-Pinto NC, Bohr VA. The C-terminal alphaO helix of human Ogg1 is essential for 8-oxoguanine DNA glycosylase activity: the mitochondrial beta-Ogg1 lacks this domain and does not have glycosylase activity. Nucleic Acids Res. 2004;32:5596–5608. doi: 10.1093/nar/gkh863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haushalter KA, Todd Stukenberg MW, Kirschner MW, Verdine GL. Identification of a new uracil-DNA glycosylase family by expression cloning using synthetic inhibitors. Curr. Biol. 1999;9:174–185. doi: 10.1016/s0960-9822(99)80087-6. [DOI] [PubMed] [Google Scholar]
- Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair. 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra M. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2002a;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, Mitra M, Izumi T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002b;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- Holt IJ, He J, Mao C, Boyd-Kirkup JD, Martinsson P, Sembongi H, Reyes A, Spelbrnk JN. Mammalian mitochondrial nucleoids: organizing an independently minded genome. Mitochondrion. 2007;7:311–321. doi: 10.1016/j.mito.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Hota SK, Barhwal K, Singh SB, Ilavazhagan G. Differential temporal response of hippocampus, cortex and cerebellum to hypobaric hypoxia: A biochemical approach. Neurochem. Int. 2007;51:384–390. doi: 10.1016/j.neuint.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Hu J, de Souza-Pinto NC, Haraguchi K, Hogue BA, Jaruga P, Greenberg MM, Dizdaroglu M, Bohr VA. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005;280:40544–40551. doi: 10.1074/jbc.M508772200. [DOI] [PubMed] [Google Scholar]
- Hutchin T, Cortopassi G. A mitochondrial DNA clone is associated with increased risk for Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1995;92:6892–6895. doi: 10.1073/pnas.92.15.6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun M, Bohr VA, Ahn B. Biochemical characterization of the WRN-1 RecQ helicase of Caenorhabditis elegans. Biochemistry. 2008;47:7583–7593. doi: 10.1021/bi800197m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun M, Lee J, Lee K, May A, Bohr VA, Ahn B. Longevity and resistance to stress correlate with DNA repair capacity in Caenorhabditis elegans. Nucleic Acids Res. 2008;36:1380–1389. doi: 10.1093/nar/gkm1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Kohmoto T, Tabata R, Seki Y. Differential intracellular localization of the human and mouse endonuclease III homologs and analysis of the sorting signals. DNA Repair. 2002;1:847–854. doi: 10.1016/s1568-7864(02)00145-3. [DOI] [PubMed] [Google Scholar]
- Imam S, Karahalil B, Hogue B, Souza-Pinto N, Bohr V. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol. Aging. 2006;27:1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Jensen A, Calvayrac G, Karahalil B, Bohr VA, Stevnsner T. Mammalian 8-oxoguanine DNA glycosylase 1 incises 8-oxoadenine opposite cytosine in nuclei and mitochondria, while a different glycosylase incises 8-oxoadenine opposite guanine in nuclei. J. Biol. Chem. 2003;278:19541–19548. doi: 10.1074/jbc.M301504200. [DOI] [PubMed] [Google Scholar]
- Kalifa L, Beutner G, Phadnis N, Sheu SS, Sia EA. Evidence for a role of FEN1 in maintaining mitochondrial DNA integrity. DNA repair. 2009;8:1242–1249. doi: 10.1016/j.dnarep.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karahalil B, Hogue BA, de Souza-Pinto NC, Bohr VA. Base excision repair capacity in mitochondria and nuclei: tissue-specific variations. FASEB J. 2002;16:1895–1902. doi: 10.1096/fj.02-0463com. [DOI] [PubMed] [Google Scholar]
- Karimi-Busheri F, Daly G, Robins P, Canas B, Pappin DJ, Sgouros J, Miller GG, Fakhrai H, Davis EM, Le Beau MM, Weinfeld M. Molecular characterization of a human DNA kinase. J. Biol. Chem. 1999;274:24187–24194. doi: 10.1074/jbc.274.34.24187. [DOI] [PubMed] [Google Scholar]
- Kavli B, Otterlei M, Slupphaug G, Krokan HE. Uracil in DNA-General mutagen, but normal intermediate in acquired immunity. DNA repair. 2007;6:505–516. doi: 10.1016/j.dnarep.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Kimura S, Sakaguchi K. DNA repair in plants. Chem. Rev. 1997;106:753–766. doi: 10.1021/cr040482n. [DOI] [PubMed] [Google Scholar]
- Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) Embo J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DG, Chinnery PF, Blackwood JK, Taylor RW, Wandooij S, Spelbrink JN, Lighttowlers RN, Turnbull DM. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku HH, Brunk UT, Sohal RS. Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species. Free Rad. Biol. Med. 1993;15:621–627. doi: 10.1016/0891-5849(93)90165-q. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. Plos genetics. 2007;3:161–173. doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Kukat A, Trifunovic A. Somatic mtDNA mutations and aging--facts and fancies. Exp. Gerontol. 2009;44:101–105. doi: 10.1016/j.exger.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Lakshmipathy U, Campbell C. Double strand break rejoining by mammalian mitochondrial extracts. Nucl. Acids Res. 1999a;27:1198–1204. doi: 10.1093/nar/27.4.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmipathy U, Campbell C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell Biol. 1999b;19:3869–3876. doi: 10.1128/mcb.19.5.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmipathy U, Campbell C. Mitochondrial DNA ligase III function is independent of Xrcc1. Nucleic Acids Res. 2000;28:3880–3886. doi: 10.1093/nar/28.20.3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AJ, Boysen HM, Buckingham JA, Yang T, Podlutsky A, Austad SN, Kunz TH, Buffenstein R, Brand MD. Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell. 2007;6:607–618. doi: 10.1111/j.1474-9726.2007.00312.x. [DOI] [PubMed] [Google Scholar]
- Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- LeDoux SP, Wilson GL, Beecham EJ, Stevnsner T, Wassermann K, Bohr VA. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis. 1992;13:1967–1973. doi: 10.1093/carcin/13.11.1967. [DOI] [PubMed] [Google Scholar]
- Li GM. DNA mismatch repair and cancer. Front. Biosci. 2003;8:997–1017. doi: 10.2741/1121. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Alzheimer's APP mangles mitochondria. Nat, Med. 2006;12:1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- Lindahl T. An N-glycosidase from Escherichia coli that releases free uracil fromDNAcontaining deaminated cytosine residues. Proc. Natl. Acad. Sci. USA. 1974;71:3649–3653. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Qian L, Sung J, Souza-Pinto NC, Zheng L, Bogenhagen DF, Bohr VA, Wilson DM, III, Shen B, Demple B. Removal of oxidative DNA damage via FEN1- dependent long-patch base excision repair in human cell mitochondria. Mol Cel Biol. 2008;28:4975–4987. doi: 10.1128/MCB.00457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, Meneses J, Pedersen RA, Chen DJ. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res. 2009;409:17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- Luna L, Bjørås M, Hoff E, Rognes T, Seeberg E. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res. 2000;460:95–104. doi: 10.1016/s0921-8777(00)00015-x. [DOI] [PubMed] [Google Scholar]
- Mason PA, Matheson EC, Hall AG, Lightowlers RN. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003;31:1052–1058. doi: 10.1093/nar/gkg167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S. Modeling mitochondrial function in aging neurons. Trends Neurosci. 2004;27:601–606. doi: 10.1016/j.tins.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Meyer JN, Boyd WA, Azzam GA, Haugen AC, Freedman JH, Van Houten B. Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biol. 2007;8:R70. doi: 10.1186/gb-2007-8-5-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S, Izumi T, Boldogh I, Bhakat KK, Chattopadhyay R, Szczesny B. Intracellular trafficking and regulation of mammalian AP-endonuclease 1 (APE1), an essential DNA repair protein. DNA Repair. 2007;6:461–946. doi: 10.1016/j.dnarep.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Morel F, Renoux M, Lachaume P, Alziari S. Bleomycin-induced double-strand breaks in mitochondrial DNA of Drosophila cells are repaired. Mut.Res. 2008;637:111–117. doi: 10.1016/j.mrfmmm.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Morinaga H, Yonekura S, Nakamura N, Sugiyama H, Yonei S, Zhang-Akiyama QM. Purification and characterization of Caenorhabditis elegans NTH, a homolog of human endonuclease III: essential role of N-terminal region. DNA Repair. 2009;8:844–851. doi: 10.1016/j.dnarep.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- Myers KA, Saffhill R, O’Connor PJ. Repair of alkylated purines in the hepatic DNA of mitochondria and nuclei in the rat. Carcinogenesis. 1998;9:285–292. doi: 10.1093/carcin/9.2.285. [DOI] [PubMed] [Google Scholar]
- Nakabeppu Y. Regulation of intracellular localization of human MTH1, OGG1 and MYH proteins for repair of oxidative DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2001;68:75–94. doi: 10.1016/s0079-6603(01)68091-7. [DOI] [PubMed] [Google Scholar]
- Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan FE. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997;25:750–755. doi: 10.1093/nar/25.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K, Nakabeppu Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell. 1999;10:1637–1652. doi: 10.1091/mbc.10.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka S, Ohno M, Tsuchimoto D, Sakumi K, Furuichi M, Nakabeppu Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008;27:421–432. doi: 10.1038/sj.emboj.7601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen LC, Aasland R, Wittwer CU, Krokan HE, Helland DE. Molecular cloning of human uracil DNA glycosylase, a highly conserved DNA repair enzyme. EMBO J. 1989;8:3121–3125. doi: 10.1002/j.1460-2075.1989.tb08464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachek LI, Thornley NP, Grishko VI, LeDoux SP, Wilson GL. Protection of INS-1 cells from free fatty acid-induced apoptosis by targeting hOGG1 to mitochondria. Diabetes. 2006;55:1022–1028. doi: 10.2337/diabetes.55.04.06.db05-0865. [DOI] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Taylor G, Elson JL, Bender A, Taylor RW, Morris CM, Turnbull DM. The low abundance of clonally expanded mitochondrial DNA point mutations in aged substantia nigra neurons. Aging Cell. 2009;8:496–498. doi: 10.1111/j.1474-9726.2009.00492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renan RA, Kolodner RD. Characterizationof insertion mutations in the saccharomyces cerevisiae MSH1 and MSH2 genes: evidence for separate mitochondrial and nuclear functions. Genetics. 1992;132:975–985. doi: 10.1093/genetics/132.4.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribar B, Izumi T, Mitra S. The major role of human AP-endonuclease homolog Apn2 in repair of abasic sites in Schizosaccharomyces pombe. Nucleic Acids Res. 2004;32:115–126. doi: 10.1093/nar/gkh151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter C, Park JW, Ames BN. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci U S A. 1988;85:6465–6467. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AB, Klungland A, Rognes T, Leiros I. Base excision repair: the long and short of it. Cell. Mol. Life Sci. 2009;66:981–993. doi: 10.1007/s00018-009-8736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi MN, Carbone M, Mostocotto C, Mancone C, Tripodi M, Maione R, Amati P. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 2009;284:31616–31624. doi: 10.1074/jbc.M109.025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz A, Caro P, Ibañez J, Gómez J, Gredilla R, Barja G. Dietary restriction at old age lowers mitochondrial oxygen radical production and leak at complex I and oxidative DNA damage in rat brain. J Bioenerg Biomembr. 2005;37:83–90. doi: 10.1007/s10863-005-4131-0. [DOI] [PubMed] [Google Scholar]
- Sarker AH, Ikeda S, Nakano H, Terato H, Ide H, Imai K, Akiyama K, Tsutsui K, Bo Z, Kubo K, Yamamoto K, Yasui A, Yoshida MC, Seki S. Cloning and characterization of a mouse homologue (mNthl1) of Escherichia coli endonuclease III. J. Mol. Biol. 1998;282:761–774. doi: 10.1006/jmbi.1998.2042. [DOI] [PubMed] [Google Scholar]
- Sastre J, Pallardo FV, Garcia de la Asuncion J, Vina J. Mitochondria, oxidative stress and aging. Free Radic Res. 2000;32:189–198. doi: 10.1080/10715760000300201. [DOI] [PubMed] [Google Scholar]
- Satoh MS, Huh N, Rajewski, Rajewsky MF, Kuroki T. Enzymatic removal of O6-ethylguanine from mitochondrial DNA in rat tissues exposed to N-ethyl-N-nitrosurea in vivo. J. Biol. Chem. 1998;263:6854–6856. [PubMed] [Google Scholar]
- Seeberg E, Eide L, Bjorås M. The base excision repair pathway. Trends Biochem. Sci. 1995;20:391–397. doi: 10.1016/s0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- Shen GP, Galick H, Inoue M, Wallace SS. Decline of nuclear and mitochondrial oxidative base excision repair activity in late passage human diploid fibroblasts. DNA Repair. 2003;2:673–693. doi: 10.1016/s1568-7864(03)00006-5. [DOI] [PubMed] [Google Scholar]
- Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. U.S.A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu.Rev.Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- Sickmann A, Reinders J, Wagner Y, Joppich C, Zahedi R, Meyer HE, Schonfisch B, Perschil I, Chacinska A, Guiard B, Rehling P, Pfanner N, Meisinger C. The proteome of Saccharomyces cerevisiae mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13207–13012. doi: 10.1073/pnas.2135385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slupphaug G, Eftedal I, Kavli B, Bharati S, Helle NM, Haug T, Levine DW, Krokan HE. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry. 1995;34:128–138. doi: 10.1021/bi00001a016. [DOI] [PubMed] [Google Scholar]
- Slupphaug G, Kavli B, Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res. 2003;531:231–251. doi: 10.1016/j.mrfmmm.2003.06.002. [DOI] [PubMed] [Google Scholar]
- Soerensen M, Gredilla R, Müller-Ohldach M, Werner A, Bohr VA, Osiewacz HD, Stevnsner T. A potential impact of DNA repair on ageing and lifespan in the ageing model organism Podospora anserina: decrease in mitochondrial DNA repair activity during ageing. Mech. Ageing Dev. 2009;130:487–496. doi: 10.1016/j.mad.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Agarwal S, Candas M, Forster MJ, Lal H. Effect of age and caloric restriction on DNA oxidative damage in different tissues of C57BL/6 mice. Mech. Ageing Dev. 1994b;76:215–224. doi: 10.1016/0047-6374(94)91595-4. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H. Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse. Mech. Ageing Dev. 1994a;74:121–133. doi: 10.1016/0047-6374(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Souza-Pinto N, Hogue BA, Bohr VA. DNA repair and aging in mouse liver: 8-oxodG glycosylase activity increases in mitochondrial but not in nuclear extracts. Free Rad. Biol. Med. 2001;30:916–923. doi: 10.1016/s0891-5849(01)00483-x. [DOI] [PubMed] [Google Scholar]
- Souza-Pinto NC, Croteau DL, Hudson EK, Hansford RG, Bohr VA. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/AP lyase activity in rat mitochondria. Nucleic Acids Res. 1999;27:1935–1942. doi: 10.1093/nar/27.8.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza-Pinto NC, Mason PA, Hashiguchi K, Weissman L, Tian J, Guay D, Lebel M, Stevnsner TV, Rasmussen LJ, Bohr VA. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair. 2009;8:704–719. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza-Pinto NC, Wilson DM, 3rd, Stevnsner TV, Bohr VA. Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair. 2008;7:1098–1109. doi: 10.1016/j.dnarep.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevnsner T, Nyaga S, de Souza-Pinto NC, van der Horst GT, Gorgels TG, Hogue BA, Thorslund T, Bohr VA. Mitochondrial repair of 8- oxoguanine is deficient in Cockayne syndrome group B. Oncogene. 2002;21:8675–8682. doi: 10.1038/sj.onc.1205994. [DOI] [PubMed] [Google Scholar]
- Strehler BL. Time, cells, and aging. New York: Academic Press; 1962. [Google Scholar]
- Stuart JA, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA base excision repair are affected differently by caloric restriction. FASEB J. 2004;18:595–597. doi: 10.1096/fj.03-0890fje. [DOI] [PubMed] [Google Scholar]
- Stuart JA, Mayard S, Hashiguchi K, Souza-Pinto NC, Bohr VA. Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulate fraction. Nucleic Acids Res. 2005;33:3722–3732. doi: 10.1093/nar/gki683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JS, DeMott MS, Demple B. Long-patch base excision DNA repair of 2-deoxyribonolactone prevents the formation of DNA-protein cross-links with DNA polymerase beta. J. Biol. Chem. 2005;280:39095–39103. doi: 10.1074/jbc.M506480200. [DOI] [PubMed] [Google Scholar]
- Szczesny B, Tann AW, Longley MJ, Copeland WC, Mitra S. Long patch base excision repair in mammalian mitochondrial genomes. J. Biol. Chem. 2008;283:26349–26356. doi: 10.1074/jbc.M803491200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao M, Aburatani H, Kobayashi K, Yasui A. Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res. 1998;26:2917–2922. doi: 10.1093/nar/26.12.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid. Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- Thompson LH, West MG. XRCC1 keeps DNA from getting stranded. Mutat. Res. 2000;459:1–18. doi: 10.1016/s0921-8777(99)00058-0. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Törnell J, Jacobs JT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Tsuchimoto D, Sakai Y, Sakumi K, Nishioka K, Sasaki M, Fujiwara T, Nakabeppu Y. Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res. 2001;29:2349–2360. doi: 10.1093/nar/29.11.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK, Lloyd RS. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. PNAS. 2006;103:1864–1869. doi: 10.1073/pnas.0507444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Weissman L, de Souza-Pinto NC, Stevnsner T, Bohr VA. DNA repair, mitochondria, and neurodegeneration. Neuroscience. 2007a;145:1318–1329. doi: 10.1016/j.neuroscience.2006.08.061. [DOI] [PubMed] [Google Scholar]
- Weissman L, Jo DG, Sørensen MM, de Souza-Pinto NC, Markesbery WR, Mattson MP, Bohr VA. Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Res. 2007b;35:5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, Hazra TK. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell. 2004;15:209–220. doi: 10.1016/j.molcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Wilson DM, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair. 2007;6:544–559. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. Embo J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharkov DO, Rosenquist TA, Gerchman SE, Grollman AP. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem. 2000;275:28607–28617. doi: 10.1074/jbc.M002441200. [DOI] [PubMed] [Google Scholar]
- Zheng L, Zhou M, Gou Z, Lu H, Qian L, Dai H, Qiu J, Yakubovxkaya E, Bogenhagen DF, Demple B, Shen B. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol. Cell. 2008;32:325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]