

Abstract
Acute lung injury (ALI) is associated with severe alterations in lung structure and function and is characterized by hypoxemia, pulmonary edema, low lung compliance and widespread capillary leakage. Asymmetric dimethylarginine (ADMA), a known cardiovascular risk factor, has been linked to endothelial dysfunction and the pathogenesis of a number of cardiovascular diseases. However, the role of ADMA in the pathogenesis of ALI is less clear. ADMA is metabolized via hydrolytic degradation to L-citrulline and dimethylamine by the enzyme, dimethylarginine dimethylaminohydrolase (DDAH). Recent studies suggest that lipopolysaccharide (LPS) markedly increases the level of ADMA and decreases DDAH activity in endothelial cells. Thus, the purpose of this study was to determine if alterations in the ADMA/DDAH pathway contribute to the development of ALI initiated by LPS-exposure in mice. Our data demonstrate that LPS exposure significantly increases ADMA levels and this correlates with a decrease in DDAH activity but not protein levels of either DDAH I or DDAH II isoforms. Further, we found that the increase in ADMA levels cause an early decrease in nitric oxide (NOx) and a significant increase in both NO synthase (NOS)-derived superoxide and total nitrated lung proteins. Finally, we found that decreasing peroxynitrite levels with either uric acid or Manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin (MnTymPyp) significantly attenuated the lung leak associated with LPS-exposure in mice suggesting a key role for protein nitration in the progression of ALI. In conclusion, this is the first study that suggests a role of the ADMA/DDAH pathway during the development of ALI in mice and that ADMA may be a novel therapeutic biomarker to ascertain the risk for development of ALI.
Keywords: Nitration, Superoxide, Arginine metabolism
1. Introduction
Acute lung injury (ALI) and Acute respiratory distress syndrome (ARDS) are acute inflammatory states which are characterized by an onset of dyspnea, severe hypoxemia, neutrophil pulmonary sequestration, and pulmonary edema secondary to disruption of pulmonary capillary integrity thus leading to significant morbidity and mortality (Martinez et al., 2009). In ALI/ARDS, the integrity of the separation between the alveolus and the pulmonary circulation is compromised either by endothelial and/or epithelial injury. This damage leads to increased vascular permeability, alveolar flooding, and surfactant abnormalities (due to damage of type II pneumocytes). ALI can occur in response to a number of insults that either directly or indirectly induce lung injury. The most common indirect pulmonary insult leading to ALI is the release of lipopolysaccharide (LPS; endotoxin) from the outer cell wall of most gram-negative bacteria producing sepsis (Erickson et al., 2009). Despite great advances in understanding the pathophysiology of ALI/ARDS, the available therapies have not led to a significant reduction in mortality or an increased quality of life in survivors. Thus, a greater understanding of the mechanisms by which the pathways leading to ALI are disrupted could lead to the development of more effective therapies.
ADMA is an endogenously produced competitive inhibitor of NO synthases (Vallance et al., 1992) and has been shown to be a cardiovascular risk factor for numerous diseases. ADMA is constantly produced in the course of normal protein turnover in many tissues, including vascular endothelial cells, and is derived from the hydrolysis of methylated proteins (Kakimoto and Akazawa, 1970). ADMA is metabolized via hydrolytic degradation to citrulline and dimethylamine by the enzyme dimethylarginine dimethylaminohydrolase (DDAH) (Kimoto et al., 1995). Elevated ADMA levels have been shown to attenuate endothelium-dependent vasodilation in humans (Boger, 2003a; Boger and Bode-Boger, 2000). In addition, inhibition of DDAH results in vasoconstriction of vascular segments that can be reversed by L-arginine (MacAllister et al., 1996). Earlier studies have shown that ADMA can disrupt NO signaling and induce endothelial dysfunction (Boger, 2003a,b, 2004; Boger and Bode-Boger, 2000; Tran et al., 2003). There is also increasing evidence that ADMA causes NOS uncoupling in endothelial cells leading to increased superoxide generation (Sud et al., 2008; Antoniades et al., 2009). Superoxide free radicals can react with NO to form peroxynitrite (ONOO–), which is a potent reactive nitrogen species (RNS) that causes the irreversible nitration of tyrosine residues within proteins that can in turn lead to cellular damage and cytotoxicity. Nitrotyrosine (3-NT) is a major product formed by peroxynitrite mediated nitration of proteins (Szabo, 2003). Our previous studies have shown that ADMA uncouples eNOS leading to an increase in superoxide production resulting in increased peroxynitrite generation and nitrotyrosine protein levels in endothelial cells (Sud et al., 2008).
In a recent study, LPS was found to increase the levels of ADMA and decrease DDAH activity in human endothelial cells. LPS also increased intracellular reactive oxygen species production in these cells (Xin et al., 2007). Another study has shown that ADMA levels were elevated in patients with septic shock (O'Dwyer et al., 2006). Peroxynitrite has been shown to play a role in the pathogenesis of endotoxin-induced homodynamic instability and organ dysfunction (Zingarelli et al., 1997). Previous studies in animal models of ALI have shown the elevated levels of 3-NT levels in the pulmonary tissue and BAL fluid (Laffey et al., 2004; Chen et al., 2003; Tsuji et al., 2000; Shang et al., 2008) while increases in 3-NT levels in ALI have previously been shown to be iNOS-dependent (Tsuji et al., 2000; Chen et al., 2003; Razavi et al., 2005). However, at present there have been no studies that evaluate the early effects on ADMA levels and NOS signaling in the murine model of ALI induced by LPS. Thus, in this study we utilized the LPS-induced mouse model of ALI to investigate whether alterations in the ADMA/DDAH pathway may contribute to the pathogenesis of this disease. Furthermore, we explored the role played by increased ADMA levels in increasing nitrosative stress and subsequent nitration of proteins as a possible mechanism leading to LPS induced ALI. Finally, we evaluated whether the scavenging of ONOO– could exert a protective effect on the lung leak associated with ALI.
2. Materials and methods
2.1. In vivo experiments
2.1.1. LPS treatment
Adult male C57BL/6NHsd mice (7–8 weeks; Harlan Indianapolis, IN) were used in all experiments. All animal care and experimental procedures were approved by the Committee on Animal Use in Research and Education of the Medical College of Georgia (Augusta, GA). Stock solutions of lipopolysaccharide (LPS), purified from Escherichia coli (serotype 0111:B4) was prepared in 0.9% saline. Mice received vehicle (10% DMSO in saline) or LPS (6.75×104 EU/gm body wt) intraperitoneally. Mice were then euthanized at 0, 2, 4, and 12 h after LPS injection and the lungs were flushed with 1 ml of ice-cold EDTA-PBS excised, snap-frozen in liquid nitrogen, and stored at −80 °C until used.
2.1.2. Peroxynitrite scavenger treatments
Manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin (MnTymPyp, A.G. Scientific, Inc. San Diego, CA), was prepared in distilled water, 0 h control mice received an intraperitoneal injection (IP) of water. Uric acid was dissolved in 25% glycerol and 75%, of 0.9% saline, 0 h control mice received an intraperitoneal injection (IP) of 25% glycerol and 75% of 0.9% saline. In the experiments to determine lung leak (Evans Blue), MnTymPyp (5 mg/kg body weight), uric acid (5 mg/kg body weight) or corresponding vehicle was injected I.P. 30 min prior to LPS injections. Subsequent doses of uric acid were injected 3 and 6 h post LPS injection (Hooper et al., 1998). After 12 h of LPS exposure, animals were anesthetized and Evans Blue surgery was performed. To determine total nitration levels, MnTymPyp, uric acid, or vehicle was injected I.P. 30 min prior to LPS injections. A subsequent dose of uric acid was injected 3 h post LPS injection. Animals were then euthanized, blood was collected by ventricular puncture and the lungs were flushed with ice-cold phospho-buffered saline and EDTA. The lungs for total nitration were then excised, snap frozen in liquid nitrogen and stored at −80 °C until used.
2.2. Lung tissue homogenates
Lung protein extracts were prepared by homogenizing mouse lung tissues in Triton lysis buffer (50 mM Tris–HCL, pH 7.6, 0.5%Triton X-100, 20% glycerol) containing a protease inhibitor cocktail (Sigma). Extracts were then clarified by centrifugation (15,000 g×10 min at 4 °C). Supernatant fractions were then assayed for protein concentration using the Bradford reagent (Bio-Rad, Richmond, CA).
2.3. Western blot analyses
Western blot analysis was performed as previously described (Sharma et al., 2008, 2007; Sud et al., 2008)). Briefly, protein extracts (25–50 μg) were separated on 4–20% denaturing polyacrylamide gels and transferred to Immunoblot-PVDF membranes (Biorad Lab, Hercules, CA). The membranes were blocked with 5% nonfat dry milk in TBS containing 0.1% Tween. After blocking, the membranes were incubated overnight at 4 °C with eNOS (1:1000, BD Transduction), nNOS (1:1000, BD Transduction), iNOS (1:1000, Upstate), DDAH I (1:500, Biosynthesis Inc., Louisville, TX) and DDAH II (Biosynthesis Inc., Louisville, TX), 3-nitrotyrosine (3-NT) antibody (1:1000, Calbiochem, San Diego, CA), mouse β-actin (1:10,000, Sigma), washed with TBS containing 0.1% Tween, and then incubated with a goat anti-mouse IgG-horseradish peroxidase. After washing, the protein bands were visualized with chemiluminescence (West Femto kit, Pierce) using a Kodak Digital Science Image Station. All protein bands were densitometrically analyzed using Kodak Imaging software. To normalize for protein loading, blots were re-probed with β-actin, the housekeeping protein.
2.4. Measurement of ADMA levels
ADMA levels were analyzed by high-performance liquid chromatography (HPLC) as we have previously published (Sud et al., 2008). The crude fraction of cell lysate was isolated using a solid phase extraction column and subsequently, ADMA was separated using pre-column derivatization with ortho-phthaldialdehyde (OPA) reagent (4.5 mg/mL in borate buffer, pH 8.5, containing 3.3 μl/mL β-mercaptoethanol) prior to injection. HPLC was performed using a Shimadzu UFLC system with a Nucleosil phenyl reverse phase column (4.6 × 250 mm; Supelco, Bellefonte, PA), equipped with an RF-10AXL fluorescence detector (Shimadzu USA Manufacturing Corporation). ADMA levels were quantified by fluorescence detection at 450 nm (emission) and 340 nm (excitation). Mobile phase A was composed of 95% potassium phosphate (50 mM, pH 6.6), 5% methanol and mobile phase B was composed of 100% methanol. ADMA was separated using a pre-gradient wash of 25% mobile phase B(flow rate 0.8 mL/min), followed by a linear increase in mobile phase B concentration from 20% to 25% over 7 min followed by a constant flow at 25% for 10 min and another linear increase from 25% to 27% mobile phase B over 5 min followed by constant flow at 27% mobile phase B for another 7 min. Retention time for ADMA was approximately 28 min. ADMA concentrations were calculated using standards and an internal homoarginine standard. The detection limit of the assay was 0.1 μmol/L.
2.5. Measurement of DDAH activity
DDAH activity in LPS treated mouse lungs was assessed directly by measuring the amount of ADMA metabolized by this enzyme as previously described (Lin et al., 2002). DDAH activity is defined as the amount of ADMA degraded per mg protein.
2.6. Measurement of BH4 levels
BH4 levels were determined using the differential iodine oxidation method as we have previously described (Kumar et al., 2009; Wainwright et al., 2005). Lung tissue was homogenized in an extraction buffer (50 mM pH 7.4 Tris-HCl, 1 mM EDTA, 1 mM DTT) and divided into equal volumes between two centrifuge tubes containing either 1 M NaOH or 1 M H3PO4. A solution of 1% I2 in 2% KI was added to each tube and samples were then incubated in the dark at RT for 90 min. 1 M H3PO4 was then added to the tubes containing NaOH. Excess I2 was removed from the samples by adding 2% ascorbic acid and samples were centrifuged at 15,000 ×g for 10 min to remove the precipitated protein. Eac supernatant was then analyzed for BH4 content by HPLC using a Spherisorb ODS-1 column (Waters, Franklin MA). BH4 levels were calculated by subtracting the area of the biopterin peak resulting from the oxidation of BH2 in the base solution from the peak resulting from the oxidation of both BH2 and BH4 in the acidic solution. Levels were normalized for protein concentration by Bradford assay.
2.7. Assessment of lung capillary leakage
Mice were anesthetized 12 h after LPS administration, with ketamine (80 mg/kg) and xylazine-HCl (8 mg/kg). Evans blue dye (EB) dissolved in saline was injected (100 mg/kg) through the left jugular vein, using a 30-gauge needle inserted to PE-10 tubing. After 30 min, blood was withdrawn via cardiac puncture and stored at 4 °C. The lungs were flushed with 1 ml of EDTA–PBS (pH 7.4, 4 °C), excised, snap-frozen in liquid nitrogen, and stored at −80 °C. Frozen lungs were homogenized in ice-cold PBS (1 ml/100 mg tissue), incubated with 2 volumes of formamide (60 °C, 18 h), and centrifuged (5,000 ×g for 30 min), and supernatant absorbance at 620 nm (A620) and 740 nm (A740) was recorded. Tissue EB content was calculated by correcting the A620 optical density for the presence of heme pigments: A620 (corrected) = A620 − (1.426 × A740 + 0.030) and by then comparing this value with a standard curve of EB in formamide–PBS. Total EB leak was expressed as lungEB content divided by serumEB content.
2.8. Superoxide quantitation in lung tissue
Superoxide levels in mouse lung tissue taken from 0, 2, 4, and 12 h post LPS treatments, were estimated by electronic paramagnetic resonance (EPR) assay using the spin-trap compound 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine HCl (CMH) as we have previously described (Lakshminrusimha et al., 2007). Briefly, 0.1 g of tissue was sectioned from fresh-frozen lung tissue and immediately immersed, while still frozen, in 200 μl of EPR Buffer (PBS supplemented with 5 μM diethydithiocarbamate [DETC, Sigma-Aldrich], and 25 μM desferrioxamine [Def MOS, Sigma-Aldrich]). All samples were then incubated for 30 min on ice then homogenized for 30 s with a VWR PowerMAX AHS 200 tissue homogenizer. Following incubation, samples were analyzed for protein content using Bradford analysis. Sample volumes were then adjusted with EPR buffer and 25 mg/ml CMH-hydrochloride in order to achieve equal protein content and a final CMH concentration of 5 mg/ml. Samples were further incubated for 60 min on ice and centrifuged at 14,000×g for 15 min at room temperature. 35 μl of supernatant from each sample was loaded into a 50 μl capillary tube and analyzed with a MiniScope MS200 ESR (Magnettech, Berlin, Germany) at a microwave power of 40 mW, modulation amplitude of 3000 mG, and modulation frequency of 100 kHz, with a magnetic strength of 333.95–3339.94 mT. Resulting EPR spectra were analyzed using ANALYSIS v.2.02 software (Magnettech), whereby the EPR maximum and minimum spectral amplitudes for the CM superoxide spin-trap product waveform were quantified. Experimental groups were normalized to fold vs. untreated control samples, then compared for differences in O2−. concentration using statistical analysis. The specificity for superoxide and the level of NOS-derived superoxide were determined by incubating duplicate samples with either PEG-SOD (100 U) or the NOS inhibitor NG-monomethyl L-arginine (L-NMMA; 100 μM) respectively.
2.9. Measurement of NOx levels
In order to quantify bioavailable NO, NO and its metabolites were determined in mouse lung tissue. In solution, NO reacts with molecular oxygen to form nitrite, and with oxyhemoglobin and superoxide anion to form nitrate. Nitrite and nitrate are reduced using vanadium (III) and hydrochloric acid at 90 °C. NO is purged from solution resulting in a peak of NO for subsequent detection by chemiluminescence (NOA 280, Sievers Instruments Inc. Boulder CO), as we have previously described (Black et al., 1999; McMullan et al., 2000). The sensitivity is 1 × 10−12 mol, with a concentration range of 1×10−9 to 1×10−3 mol of nitrate.
2.10. Human lung microvascular endothelial cell isolation and culture
Isolation and culture of human lung microvascular endothelial cell (HLMVEC) was performed by a modification of the method of Hewett and Murray (Hewett and Murray, 1993) and Burg et al (Burg et al., 2002). Normal human lung tissue was obtained from lobectomy specimens resected due to lung disease. Briefly, isolation of HPMEC was performed as follows: subpleural lung tissue was cut into small fragments with scissors. After removal of debris and erythrocytes by filtering through a 40 μm nylon net, the tissue was treated with dispase (1 U/ml at 4 °C for 18 h). After filtration through a 100 μm nylon net, the tissue was treated in a volume of 15 ml M199, 15% FBS, 1 mg dispase/ml at 37 °C for 1 h followed by a further flitration through a 100 μm nylon net. The cell clumps within the filtrate were repeatedly resuspended in M199 and filtered through a 40 μm net, followed by centrifugation for 10 min and resuspension in M199 containing 20% serum. Undigested tissue was washed from the 100 μm net, collected and digested again in 1 mg dispase/ml as above. The positive selection of HLMVEC was achieved by interacting the cell suspension with magnetic beads (Tosyl activated Dynabeads: Invitrogen) coated with Ulex europaeus I according to the method of Jackson et al (Jackson et al., 1990). After purification, cells were cultured in M199, 20% FBS, 100U Heparin/ml, 150 μg ECGF/ml, 1 μg hydrocortisone/ml, 292mg L-glutamine/l, and 110 mg sodium pyruvate/l. EC identity was confirmed by uptake of 1,1_-dioctadecyl-1,3,3_,3_-tetramethyl-indocarbocyanineacetylated low-density lipoprotein (Dil-Ac-LDL) and used between passages 1–3.
2.11. Measurement of transendothelial cell electrical impedance
Transendothelial impedance was measured using an electric cell impedance sensing (ECIS) apparatus (Applied Biophysics, Troy, NY). Equal number of HLMVEC were seeded on L-cysteine coated gold electrode arrays (8W10E) and allowed to grow to confluence then serum starved for 4 h. Transendothelial impedance was monitored for 30 min to establish baseline. The cells were treated or not with ADMA (5 μM) in the presence or absence of vascular endothelial growth factor (VEGF,100 ng) and the effect on endothelial permeability measured over 2 h.
2.12. Statistical analysis
Statistical analysis was performed using GraphPad Prism version 4.01 for Windows (GraphPad Software, San Diego, CA). The mean±SEM were calculated for all samples and significance was determined either by the unpaired t-test (for 2 groups) and ANOVA (for≥3 groups) followed by Newman-Keuls multiple comparisons test. A value of P< 0.05 was considered significant.
3. Results
3.1. Superoxide levels in LPS treated mouse lungs
Relative superoxide levels were determined by EPR in lung tissues harvested from 0- (Control), 2-, 4-, and 12-h after LP exposure (Fig. 1). Our data indicate that lung superoxide levels were significantly increased early after LPS-exposure: 2 h (~2-fold) and 4 h (~1.5 fold) and that this was a transient event as there was no change in superoxide levels 12 h post-LPS (Fig. 1). To determine the contribution of uncoupled eNOS in the increased superoxide generation, duplicate samples were incubated with the NOS inhibitor, NG-monomethyl L-arginine (L-NMMA; 100 μM, 30 min) on ice prior to the addition of CMH. The LPS-mediated increase in superoxide generation was blocked in the presence of L-NMMA suggesting that NOS uncoupling is a significant contributor to superoxide generation after LPS exposure (Fig. 1). Specificity of the EPR assay for superoxide was confirmed by a significant reduction in the waveform amplitude with the addition of superoxide scavenger, polyethylene glycolconjugated superoxide dismutase (PEG-SOD) to the samples (Fig. 1).
Fig. 1.
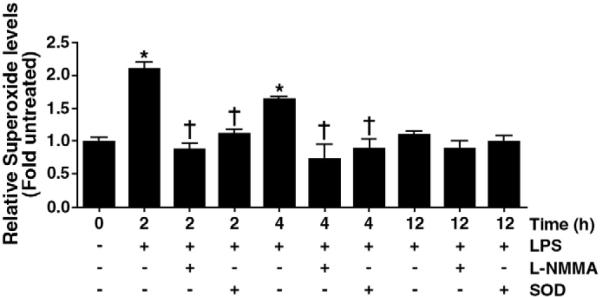
Superoxide levels in the mouse lung after LPS exposure. Relative superoxide levels were determined by electron paramagnetic resonance (EPR) in LPS-treated mouse lungs. There is a significant increase in superoxide radical generation 2- and 4 h after LPS exposure that is blocked by the addition of the NOS inhibitor, NG-monomethyl L-arginine (L-NMMA; 100 μM) or polyethylene glycol-superoxide dismutase (PEGSOD; 100 U/ml). There is no change in superoxide generation 12 h after LPS exposure. Values are means ± SE, N=6. *P<0.05 vs. no LPS, †P<0.05 vs. LPS alone.
3.2. NOx and BH4 levels after LPS exposure
NOx levels were determined in mouse lung tissue 0-, 2-, 4-, and 12-h after LPS treatment. Correlating with the increase in NOS uncoupling, we found there was a significant decrease in NOx levels 2 h (−40%) after LPS exposure whereas we found an increase in NOx levels 4 h (+60%) and 12 h (+160%) after LPS administration (Fig. 2 A). In addition, we measured lung BH4 levels after LPS exposure. BH4 levels were unaltered 2 h after LPS exposure but were significantly elevated at 4 h (~2 fold) and 12 h (~3 fold) post LPS treatment (Fig. 2 B).
Fig. 2.

Lung tissue NOx and tetrahydrobiopterin levels after LPS exposure. Tissue NOx levels are significantly decreased 2 h after LPS exposure. However, NOx levels were significantly increased 4- and 12-h after LPS exposure (A). Lung BH4 levels were measured by HPLC. There was no change in BH4 levels 2 h after LPS exposure, but BH4 levels were significantly increased 4- and 12-h post LPS treatment (B). Values are mean±SE, N=5 for each group. *P<0.05 compared to no LPS, †P<0.05 compared to previous time point.
3.3. NOS expression in control and ALI mouse lungs
To attempt to correlate the changes in NOx levels with NOS isoform protein levels, whole lung homogenates were assessed for changes in the expression of NOS isoforms (eNOS, nNOS and iNOS) 2- and 4-h after LPS exposure. Western blot analyses showed that there was no difference in eNOS (Fig. 3 A) or nNOS protein levels (Fig. 3 B). However, iNOS protein levels although unchanged 2 h post-LPS treatment were significantly increased (~6-fold) 4 h after LPS treatment (Fig. 3C) suggesting the increase in NOx levels 4 h post-LPS is likely due to increases in iNOS protein levels.
Fig. 3.
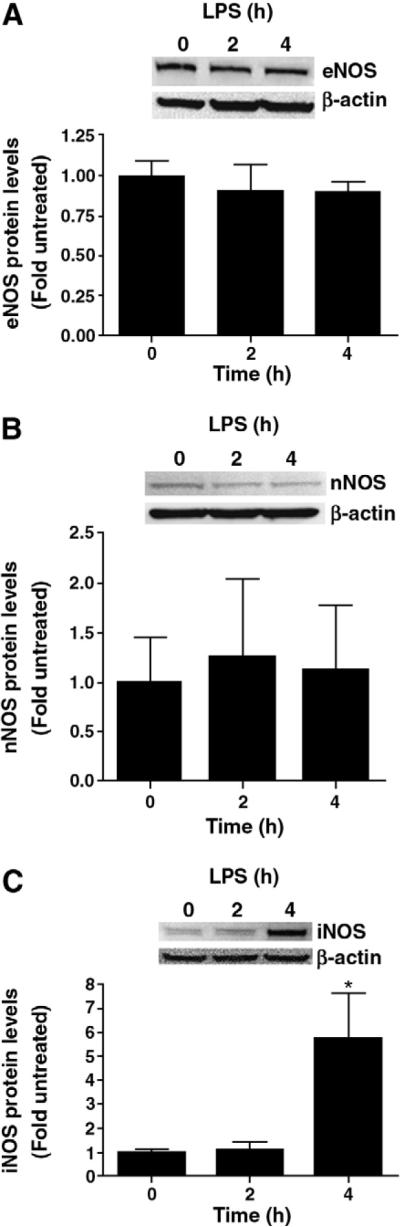
NOS isoform protein levels in the mouse lung after LPS exposure. Protein levels for eNOS, nNOS, and iNOS were determined in lung tissues 2- and 4-h after LPS exposure by Western blot analysis using specific antisera raised against eNOS, nNOS, or iNOS respectively and re-probed with β-actin to normalize for loading. Representative Western blots are shown for eNOS (panel A), nNOS (panel B), and iNOS (panel C). There are no changes in eNOS or nNOS protein levels but iNOS protein levels are significantly increased 4 h after LPS exposure. Values are mean ± SE, N=5 for each group *P<0.05 compared to no LPS.
3.4. Elevated ADMA levels and decreased DDAH activity in LPS-treated mouse lungs
In an attempt to evaluate the mechanism responsible for the increase in NOS uncoupling early after LPS exposure, we next determined if LPS altered the levels of ADMA in the mouse lung. We found significantly increased ADMA levels in LPS treated mouse lungs at 2 h (12.13 ± 0.84 vs. 7.53 ± 0.57 nmol/gww; Fig. 4 A), 4 h (13.40 ± 2.10 vs. 7.53 ± 0.57 nmol/gww; Fig. 4 A) and 12 h (19.10 ± 1.90 vs. 7.53 ± 0.57 nmol/gww; Fig. 4 A) after LPS exposure. To determine whether the increased ADMA levels in the LPS treated mouse lungs were a result of decreased DDAH I and/or DDAH II protein expression, we measured protein levels of the two isoforms by Western blot analyses. DDAH I and DDAH II anti-serum detected ~37-kDA and ~33-kDA bands, respectively in lung tissue homogenates. No significant differences were detected between protein levels of either DDAH I (Fig. 4 B) or DDAH II (Fig. 4 C) in mouse lung tissue post-LPS. However, we found that DDAH enzyme activity was significantly decreased (~2-fold) in mouse lung tissue homogenates both 2- and 4-h after LPS exposure (Fig. 4 D).
Fig. 4.

LPS exposure leads to elevated ADMA and decreased DDAH activity in the mouse lung. ADMA levels and DDAH activity were analyzed by high-performance liquid chromatography (HPLC) in the mice lungs after LPS exposure. There was a progressive increase in ADMA levels after 2-, 4- and 12-h LPS exposure (A). Although there were no changes in either DDAH I (B) or DDAH II (C) protein levels there was a significant decrease in DDAH activity 2- and 4-h after LPS exposure (panel D). Values are mean ± SE, N=5 for each group, *P<0.05 compared to no LPS.
3.5. Elevated nitrotyrosine levels after LPS exposure
Superoxides react with NO to form peroxynitrite that can modify proteins by interacting with and nitrating tyrosine residues to form 3-NT. To determine the presence of tyrosine-nitrated proteins in the mouse lungs after LPS exposure, the levels of 3-NT were assessed by Western blotting to detect nitrated proteins. The 3-NT levels were quantified by obtaining the densitometric units of all nitrated proteins (Fig. 5 A). Our data indicate that LPS exposure significantly increases 3-NT levels 4 h after LPS-treatment in the mouse lung (Fig. 5 B).
Fig. 5.
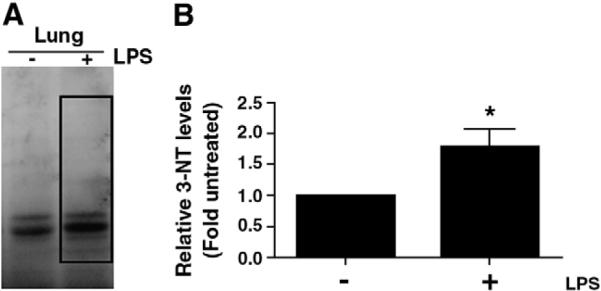
Nitrated protein levels are increased in the mouse lung after LPS exposure. Protein levels for total nitrated proteins were determined in the mouse lung 4 h after LPS exposure. Homogenates (25 μg) were separated on a 4–20% denaturing polyacrylamide gel, electrophoretically transferred to PVDF-nitrocellulose membranes, and analyzed using specific antiserum raised against 3-NT residues. A representative image for the Western blot analysis for 3-NT protein levels is shown (A). The boxed area shows the region of total nitrated proteins that was used to quantify 3-NT levels. Densitometric analysis indicates that LPS exposure increases total nitrated proteins in the mouse lung (B). Values are mean ± SE, N=5. *P<0.05 vs. no LPS.
3.6. Peroxynitrite scavengers cause a reduction in nitrated proteins after LPS exposure
To determine the effect of peroxynitrite scavenging on LPS-mediated 3-NT levels, we used two potent peroxynitrite scavengers, MnTymPyp and uric acid. The animals were treated with peroxynitrite scavengers prior to LPS exposure and 3-NT levels were determined 4 h post-LPS. Both, MnTymPyp and uric acid significantly attenuated the LPS-induced increase in 3-NT levels (Fig. 6 A).
Fig. 6.

Peroxynitrite scavenging decreases the LPS-mediated hyperpermeability in the mouse lung. Protein levels for total nitrated proteins were determined in the mouse lung 4 h after LPS exposure in the presence and absence of the ONOO– scavengers, uric acid and MnTymPyp. The presence of either uric acid and MnTymPyp significantly decreased total nitrated protein levels compared to that obtained with LPS alone (A) and this correlated with a significant decrease in Evans blue dye levels in the lungs indicating a decrease in lung permeability (B). Values are mean ± SE, N=5. *P<0.05 vs. no LPS, †P<0.05 vs. LPS alone.
3.7. Peroxynitrite scavengers attenuate the LPS mediated increase in lung permeability
The increase in lung permeability in response to LPS was determined by measuring the Evan Blue dye leak, 12 h after LPS challenge. Our data indicate that there was a significant increase in the lung leak in the LPS treated mice (~1.7 fold) while this increase in lung leak was significantly reduced in the animals pre-treated with the peroxynitrite scavengers (MnTymPyp and uric acid) (Fig. 6 B).
3.8. ADMA potentiates VEGF-induced decrease in endothelial barrier function
To determine if increases in ADMA alone are sufficient to induce endothelial cell barrier disruption, HLMVEC were exposed or not to ADMA (5 μM) in the presence or absence of VEGF (100 ng) and the effect on endothelial barrier function estimated by measuring changes in transendothelial resistance (TER) using an ECIS apparatus. Our data indicate that ADMA alone is not sufficient to induce barrier disruption but it does potentiate the VEGF-mediated reduction in TER (Fig. 7).
Fig. 7.
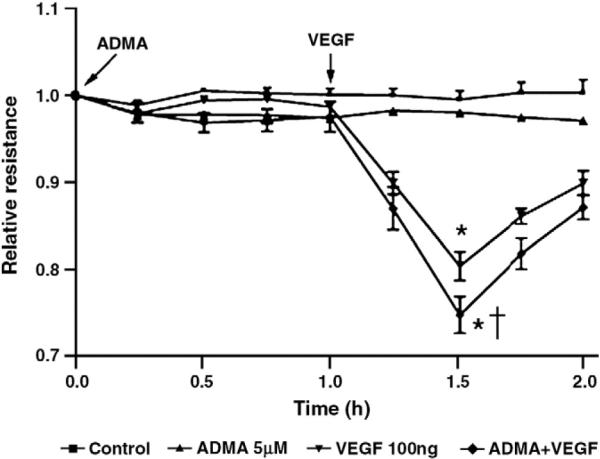
ADMA potentiates the decrease in transendothelial electrical resistance (TER) associated with VEGF exposure in human lung microvascualr endothelial cells. Confluent were exposed or not to ADMA (5 μM, 1 h). Cells were then exposed or not to VEGF (100 ng) and changes in TER across the endothelial cell monolayer were measured by ECIS. Although ADMA alone does not induce a change in TER there is a potentiation in the decrease in TER associated with VEGF exposure. Values are mean ± SE, N=5. *P<0.05 vs. untreated, †P<0.05 vs. VEGF alone.
4. Discussion
This study provides insight into a novel mechanism by which ADMA mediated nitrosative damage may cause a loss of lung function in an LPS-induced mouse model of ALI. We found: (1) decreased DDAH activity, which was correlated with increased ADMA levels; (2) increased NOS uncoupling at early stages and increased iNOS expression at later stages of the pathogenesis; (3) peroxynitrite mediated increase in 3-NT levels and subsequent increase in lung permeability; (4) reduced 3-NT levels and decreased lung leak in mice receiving peroxynitrite scavengers before endotoxin exposure. Interestingly our data also indicate that alone increases in ADMA levels do not cause endothelial barrier disruption in vitro using HLMVEC. However, ADMA does potentiate VEGF-induced barrier disruption suggesting ADMA may be necessary but not sufficient to induce ALI. Thus, ADMA may act in concert with other proteins to produce endothelial barrier dysfunction. Indeed a recent study has shown that the ADMA/DDAH pathway regulates pulmonary endothelial barrier function through the modulation of Rac1 signaling (Wojciak-Stothard et al., 2009). Further studies will be required to elucidate the mechanism and key targets involved in ADMA mediated EC barrier disruption.
We, and others, have previously shown that increased levels of the endogenous NOS inhibitor, ADMA can cause uncoupling of NOS and increased production of both reactive oxygen species (ROS) and reactive nitrogen species (RNS) (Sud et al., 2008; Boger et al., 2000), which results in oxidative and nitrosative stress in the cell. There is growing evidence that increased ADMA levels are involved in the pathogenesis of a number of cardiovascular diseases (Miyazaki et al., 1999; Takiuchi et al., 2004; Boger, 2003c; Bae et al., 2005). Further, ADMA has been shown to cause increased peroxynitrite generation, and increased nitration events leading to pathological conditions (Sud et al., 2008). However, the role of ADMA in ALI has not been clarified. In this study, we demonstrate that lung tissue ADMA levels were significantly increased within 2 h of LPS exposure suggesting that this is an early event in the pathogenesis of the disease. ADMA is degraded through active metabolism by the enzyme, DDAH. DDAH has two isoforms and it has been previously shown that mRNA (Tran et al., 2000) and protein (Arrigoni et al., 2003) for both DDAH isoforms are expressed in the lung. We observed a decrease in DDAH activity in ALI lung tissue, however our Western blot analyses did not detect alterations in either DDAH I or DDAH II protein levels. This suggests that the decrease in DDAH activity is not due to altered protein levels but is perhaps due to a post-translational modification. However, further studies will be necessary to elucidate the mechanism by which LPS regulates DDAH activity. Consistent with our results other studies have shown that reduced DDAH activity, but not expression, is responsible for the plasma ADMA elevation in hypercholesterolemia and hyperhomocysteinemia (Boger et al., 1998; Lin et al., 2002; Stuhlinger et al., 2001). It has also been shown that the treatment of mice with a DDAH inhibitor can cause higher plasma and blood vessel concentrations of ADMA (Leiper et al., 2007). Our results are also consistent with a recent study in which LPS significantly increased the levels of ADMA, decreased DDAH activity, and increased intracellular ROS production in human endothelial cells (Xin et al., 2007). While prior studies have reported elevated ADMA levels in patients with septic shock (O'Dwyer et al., 2006) and found that serum ADMA was associated with increased vascular superoxide generation and eNOS uncoupling in human atherosclerosis (Antoniades et al., 2009). Our data also indicate that the NOS uncoupling is a transient phenomenon after LPS exposure and as all three NOS isoforms are present our data cannot determine if one isoform predominates. NOS uncoupling is a complex process that can be induced by a variety of conditions including increases in ADMA (Sud et al., 2008), decreases in the substrate L-arginine (Settergren et al., 2009), and decreases in the NOS cofactor, BH4 (Bevers et al., 2006; Cai et al., 2005). We found that there was no change in lung BH4 levels after 2 h LPS exposure but there was a substantial increase after 4- and 12-h. This suggests that the increase in BH4 at the later time points may be able to overcome the uncoupling effect of the increases in ADMA. Although this is speculative our BH4 data are in agreement with a prior study that reported increases in BH4 levels in the rat in both the plasma and tissues 3 h after LPS exposure (Hattori et al., 1996).
Tissue NOx levels, a stable metabolite of NO, were significantly decreased 2 h after LPS exposure but were markedly elevated 4- and 12-h post-LPS treatment. The decreased NOx levels 2 h post-LPS were not due to decreased NOS protein levels but rather were due to increased NOS uncoupling. NO is formed from arginine by the enzyme NO synthase with three known isoforms: eNOS, nNOS, and iNOS which contribute to total NOS activity. eNOS and nNOS are the two constitutive forms, whereas iNOS is induced by cytokines and bacterial products (Szabo et al., 1995; Minc-Golomb et al., 1994). Previously, LPS treatment has been shown to reduce eNOS protein and mRNA expression in bovine endothelial cells (Lu et al., 1996) and a decrease in eNOS protein expression 12 h post-LPS in the mouse lung has been suggested (Chatterjee et al., 2008). Studies have also found increased lung NO production associated with increased iNOS expression and/or iNOS activity in various models of ALI (Webert et al., 2000; Farley et al., 2006; Razavi et al., 2004; Scumpia et al., 2002; Okamoto et al., 2000). Interestingly, in our studies, although increased NO production assessed indirectly by measuring the levels of the oxidative metabolites of NO (NOx levels) at 4- and 12-h post-LPS exposure, is associated with increased iNOS expression, we did not find any change in eNOS and nNOS protein levels, suggesting that the increased bioavailable NO may be iNOS derived. It is worth noting that some studies have indicated that nNOS also may be induced in pathological conditions (Gocan et al., 2000) and that nNOS-derived NO may be involved in the pathogenesis of ALI in sheep with sepsis (Enkhbaatar et al., 2003). These conflicting reports suggest that studies using selective NOS inhibitors and investigating the individual roles of eNOS, nNOS and iNOS on NO generation may be warranted.
NO and superoxide radicals can combine to form the toxic product, ONOO–, a potent RNS which causes the irreversible nitration of tyrosine residues within proteins leading to cellular damage and cytotoxicity. Increasing evidence suggests that peroxynitrite is involved in the pathogenesis of endotoxin-induced endothelial injury, multiple organ dysfunction, and hemodynamic instability (Salvemini et al., 2006; Cuzzocrea et al., 2006; Zingarelli et al., 1997; Beckman, 2002). The levels of nitrated proteins have been shown to be significantly elevated during inflammatory diseases in humans, including ALI (Haddad et al., 1994; Zhu et al., 2001; Lamb et al., 1999; Salvemini et al., 2006; Cuzzocrea et al., 2006). In addition, a recent study has shown evidence that immunoglobulins against tyrosine-nitrated proteins are present in plasma of patients with ALI (Thomson et al., 2007) while increased iNOS expression has been previously associated with increased 3-NT concentrations in the lungs of animal with ALI (Peng et al., 2005; Mehta, 2005). Our previous in vitro studies have also shown that ADMA uncouples eNOS leading to an increased peroxynitrite generation and subsequent elevation in 3-NT protein levels (Sud et al., 2008). In the present study, we found an almost 2-fold increase in the 3-NT levels 4 h after LPS exposure and while the increases in 3-NT levels were blocked by the addition of the ONOO– scavenger, uric acid and the superoxide scavenger, MnTymPyp. Both scavenging ONOO– directly, by uric acid, or indirectly by decreasing superoxide levels, with MnTymPyp, decreased total nitrated proteins and the lung permeability associated with LPS exposure. Thus, our data along with the findings of previous studies suggest that that blocking protein nitration events may be a potential therapeutic target for ALI. However, to allow more specific therapies to be developed further studies will be required to identify the relevant nitrated proteins and determine how nitration modulates their activity leading to endothelial barrier disruption and ALI.
In conclusion, in this study we found a significant role for the ADMA/DDAH pathway in the development of ALI. Increased ADMA levels lead to peroxynitrite mediated nitrosative damage of proteins as indicated by increased 3-NT levels. Peroxynitrite scavengers reduced the nitrated protein levels and decreased permeability events associated with ALI. Given increasing evidences of the role of ADMA in the pathogenesis of ALI, it may be a target for pharmacotherapeutic interventions in patients with ALI.
Acknowledgments
This research was supported in part by grants HL60190 (SMB), HL67841 (SMB), HL084739 (to SMB), R21HD057406 (to SMB), and HL61284 (to JRF) all from the National Institutes of Health, 0550133Z from the American Heart Association, Pacific Mountain Affiliates (SMB), 09BGIA2310050 from the Southeast Affiliates (to SS), and by a Transatlantic Network Development Grant from the Fondation Leducq (to SMB & JRF). This work was also supported by a Programmatic Development award (to SMB, JC, and DF) and Seed Awards (to SS and SJ) from the Cardiovascular Discovery Institute of the Medical College of Georgia. Anita Smith was supported in part by NIH training Grant 5T32HL06699.
Abbreviations
- ALI
acute lung injury
- LPS
lipopolysaccharide
- ADMA
asymmetric dimethylarginine
- DDAH
dimethylarginine dimethylaminohydrolase
- 3-NT
3-nitrotyrosine
- NO
nitric oxide
- NOS
nitric oxide synthase
References
- Antoniades C, Shirodaria C, Leeson P, Antonopoulos A, Warrick N, Van-Assche T, Cunnington C, Tousoulis D, Pillai R, Ratnatunga C, Stefanadis C, Channon KM. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur. Heart J. 2009;30:1142–1150. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]
- Arrigoni FI, Vallance P, Haworth SG, Leiper JM. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation. 2003;107:1195–1201. doi: 10.1161/01.cir.0000051466.00227.13. [DOI] [PubMed] [Google Scholar]
- Bae SW, Stuhlinger MC, Yoo HS, Yu KH, Park HK, Choi BY, Lee YS, Pachinger O, Choi YH, Lee SH, Park JE. Plasma asymmetric dimethylarginine concentrations in newly diagnosed patients with acute myocardial infarction or unstable angina pectoris during two weeks of medical treatment. Am. J. Cardiol. 2005;95:729–733. doi: 10.1016/j.amjcard.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Beckman JS. Protein tyrosine nitration and peroxynitrite. Faseb. J. 2002;16:1144. doi: 10.1096/fj.02-0133lte. [DOI] [PubMed] [Google Scholar]
- Bevers LM, Braam B, Post JA, van Zonneveld AJ, Rabelink TJ, Koomans HA, Verhaar MC, Joles JA. Tetrahydrobiopterin, but not L-arginine, decreases NO synthase uncoupling in cells expressing high levels of endothelial NO synthase. Hypertension. 2006;47:87–94. doi: 10.1161/01.HYP.0000196735.85398.0e. [DOI] [PubMed] [Google Scholar]
- Black SM, Heidersbach RS, McMullan DM, Bekker JM, Johengen MJ, Fineman JR. Inhaled nitric oxide inhibits NOS activity in lambs: potential mechanism for rebound pulmonary hypertension. Am. J. Physiol. 1999;277:H1849–H1856. doi: 10.1152/ajpheart.1999.277.5.H1849. [DOI] [PubMed] [Google Scholar]
- Boger RH. Association of asymmetric dimethylarginine and endothelial dysfunction. Clin. Chem. Lab. Med. 2003a;41:1467–1472. doi: 10.1515/CCLM.2003.225. [DOI] [PubMed] [Google Scholar]
- Boger RH. The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc. Res. 2003b;59:824–833. doi: 10.1016/s0008-6363(03)00500-5. [DOI] [PubMed] [Google Scholar]
- Boger RH. When the endothelium cannot say `NO' anymore. ADMA, an endogenous inhibitor of NO synthase, promotes cardiovascular disease. Eur. Heart J. 2003c;24:1901–1902. doi: 10.1016/j.ehj.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Boger RH. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J. Nutr. 2004;134:2842S–2847S. doi: 10.1093/jn/134.10.2842S. discussion 2853S. [DOI] [PubMed] [Google Scholar]
- Boger RH, Bode-Boger SM. Asymmetric dimethylarginine, derangements of the endothelial nitric oxide synthase pathway, and cardiovascular diseases. Semin. Thromb. Hemost. 2000;26:539–545. doi: 10.1055/s-2000-13210. [DOI] [PubMed] [Google Scholar]
- Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998;98:1842–1847. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- Boger RH, Bode-Boger SM, Tsao PS, Lin PS, Chan JR, Cooke JP. An endogenous inhibitor of nitric oxide synthase regulates endothelial adhesiveness for monocytes. J. Am. Coll. Cardiol. 2000;36:2287–2295. doi: 10.1016/s0735-1097(00)01013-5. [DOI] [PubMed] [Google Scholar]
- Burg J, Krump-Konvalinkova V, Bittinger F, Kirkpatrick CJ. GM-CSF expression by human lung microvascular endothelial cells: in vitro and in vivo findings. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;283:L460–L467. doi: 10.1152/ajplung.00249.2001. [DOI] [PubMed] [Google Scholar]
- Cai S, Khoo J, Channon KM. Augmented BH4 by gene transfer restores nitric oxide synthase function in hyperglycemic human endothelial cells. Cardiovasc. Res. 2005;65:823–831. doi: 10.1016/j.cardiores.2004.10.040. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, Catravas JD. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;294:L755–L763. doi: 10.1152/ajplung.00350.2007. [DOI] [PubMed] [Google Scholar]
- Chen LW, Wang JS, Chen HL, Chen JS, Hsu CM. Peroxynitrite is an important mediator in thermal injury-induced lung damage. Crit. Care Med. 2003;31:2170–2177. doi: 10.1097/01.CCM.0000079605.28852.D0. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Di Paola R, Esposito E, Macarthur H, Matuschak GM, Salvemini D. A role for nitric oxide-mediated peroxynitrite formation in a model of endotoxin-induced shock. J. Pharmacol. Exp. Ther. 2006;319:73–81. doi: 10.1124/jpet.106.108100. [DOI] [PubMed] [Google Scholar]
- Enkhbaatar P, Murakami K, Shimoda K, Mizutani A, McGuire R, Schmalstieg F, Cox R, Hawkins H, Jodoin J, Lee S, Traber L, Herndon D, Traber D. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole attenuates acute lung injury in an ovine model. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003;285:R366–R372. doi: 10.1152/ajpregu.00148.2003. [DOI] [PubMed] [Google Scholar]
- Erickson SE, Martin GS, Davis JL, Matthay MA, Eisner MD. Recent trends in acute lung injury mortality: 1996–2005. Crit. Care Med. 2009;37:1574–1579. doi: 10.1097/CCM.0b013e31819fefdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley KS, Wang LF, Razavi HM, Law C, Rohan M, McCormack DG, Mehta S. Effects of macrophage inducible nitric oxide synthase in murine septic lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006;290:L1164–L1172. doi: 10.1152/ajplung.00248.2005. [DOI] [PubMed] [Google Scholar]
- Gocan NC, Scott JA, Tyml K. Nitric oxide produced via neuronal NOS may impair vasodilatation in septic rat skeletal muscle. Am. J. Physiol. Heart Circ. Physiol. 2000;278:H1480–H1489. doi: 10.1152/ajpheart.2000.278.5.H1480. [DOI] [PubMed] [Google Scholar]
- Haddad IY, Pataki G, Hu P, Galliani C, Beckman JS, Matalon S. Quantitation of nitrotyrosine levels in lung sections of patients and animals with acute lung injury. J. Clin. Invest. 1994;94:2407–2413. doi: 10.1172/JCI117607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, Nakanishi N, Kasai K, Murakami Y, Shimoda S. Tetrahydrobiopterin and GTP cyclohydrolase I in a rat model of endotoxic shock: relation to nitric oxide synthesis. Exp. Physiol. 1996;81:665–671. doi: 10.1113/expphysiol.1996.sp003967. [DOI] [PubMed] [Google Scholar]
- Hewett PW, Murray JC. Human microvessel endothelial cells: isolation, culture and characterization. In Vitro Cell Dev. Biol. Anim. 1993;29A:823–830. doi: 10.1007/BF02631356. [DOI] [PubMed] [Google Scholar]
- Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, Chaudhry I, Koprowski H. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 1998;95:675–680. doi: 10.1073/pnas.95.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CJ, Garbett PK, Nissen B, Schrieber L. Binding of human endothelium to Ulex europaeus I-coated dynabeads: application to the isolation of microvascular endothelium. J. Cell Sci. 1990;96(Pt 2):257–262. doi: 10.1242/jcs.96.2.257. [DOI] [PubMed] [Google Scholar]
- Kakimoto Y, Akazawa S. Isolation and identification of N-G, N-G- and N-G, N′-G-dimethyl-arginine, N-epsilon-mono-, di-, and trimethyllysine, and glucosylgalactosyl- and galactosyl-delta-hydroxylysine from human urine. J. Biol. Chem. 1970;245:5751–5758. [PubMed] [Google Scholar]
- Kimoto M, Whitley GS, Tsuji H, Ogawa T. Detection of NG, NG-dimethylarginine dimethylaminohydrolase in human tissues using a monoclonal antibody. J. Biochem. (Tokyo) 1995;117:237–238. doi: 10.1093/jb/117.2.237. [DOI] [PubMed] [Google Scholar]
- Kumar S, Sun X, Sharma S, Aggarwal S, Ravi K, Fineman JR, Black SM. GTP cyclohydrolase I expression is regulated by nitric oxide: role of cyclic AMP. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;297:L309–L317. doi: 10.1152/ajplung.90538.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffey JG, Honan D, Hopkins N, Hyvelin JM, Boylan JF, McLoughlin P. Hypercapnic acidosis attenuates endotoxin-induced acute lung injury. Am. J. Respir. Crit. Care Med. 2004;169:46–56. doi: 10.1164/rccm.200205-394OC. [DOI] [PubMed] [Google Scholar]
- Lakshminrusimha S, Wiseman D, Black SM, Russell JA, Gugino SF, Oishi P, Steinhorn RH, Fineman JR. The role of nitric oxide synthase-derived reactive oxygen species in the altered relaxation of pulmonary arteries from lambs with increased pulmonary blood flow. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H1491–H1497. doi: 10.1152/ajpheart.00185.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb NJ, Quinlan GJ, Westerman ST, Gutteridge JM, Evans TW. Nitration of proteins in bronchoalveolar lavage fluid from patients with acute respiratory distress syndrome receiving inhaled nitric oxide. Am. J. Respir. Crit. Care Med. 1999;160:1031–1034. doi: 10.1164/ajrccm.160.3.9810048. [DOI] [PubMed] [Google Scholar]
- Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O'Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat. Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- Lin KY, Ito A, Asagami T, Tsao PS, Adimoolam S, Kimoto M, Tsuji H, Reaven GM, Cooke JP. Impaired nitric oxide synthase pathway in diabetes mellitus: role of asymmetric dimethylarginine and dimethylarginine dimethylaminohydrolase. Circulation. 2002;106:987–992. doi: 10.1161/01.cir.0000027109.14149.67. [DOI] [PubMed] [Google Scholar]
- Lu JL, Schmiege LM, III, Kuo L, Liao JC. Downregulation of endothelial constitutive nitric oxide synthase expression by lipopolysaccharide. Biochem. Biophys. Res. Commun. 1996;225:1–5. doi: 10.1006/bbrc.1996.1121. [DOI] [PubMed] [Google Scholar]
- MacAllister RJ, Parry H, Kimoto M, Ogawa T, Russell RJ, Hodson H, Whitley GS, Vallance P. Regulation of nitric oxide synthesis by dimethylarginine dimethylaminohydrolase. Br. J. Pharmacol. 1996;119:1533–1540. doi: 10.1111/j.1476-5381.1996.tb16069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez O, Nin N, Esteban A. Prone position for the treatment of acute respiratory distress syndrome: a review of current literature. Arch. Bronconeumol. 2009;45:291–296. doi: 10.1016/j.arbres.2008.05.010. [DOI] [PubMed] [Google Scholar]
- McMullan DM, Bekker JM, Parry AJ, Johengen MJ, Kon A, Heidersbach RS, Black SM, Fineman JR. Alterations in endogenous nitric oxide production after cardiopulmonary bypass in lambs with normal and increased pulmonary blood flow. Circulation. 2000;102:III172–III178. doi: 10.1161/01.cir.102.suppl_3.iii-172. [DOI] [PubMed] [Google Scholar]
- Mehta S. The effects of nitric oxide in acute lung injury. Vascul. Pharmacol. 2005;43:390–403. doi: 10.1016/j.vph.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Minc-Golomb D, Tsarfaty I, Schwartz JP. Expression of inducible nitric oxide synthase by neurones following exposure to endotoxin and cytokine. Br. J. Pharmacol. 1994;112:720–722. doi: 10.1111/j.1476-5381.1994.tb13136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki H, Matsuoka H, Cooke JP, Usui M, Ueda S, Okuda S, Imaizumi T. Endogenous nitric oxide synthase inhibitor: a novel marker of atherosclerosis. Circulation. 1999;99:1141–1146. doi: 10.1161/01.cir.99.9.1141. [DOI] [PubMed] [Google Scholar]
- O'Dwyer MJ, Dempsey F, Crowley V, Kelleher DP, McManus R, Ryan T. Septic shock is correlated with asymmetrical dimethyl arginine levels, which may be influenced by a polymorphism in the dimethylarginine dimethylaminohydrolase II gene: a prospective observational study. Crit. Care. 2006;10:R139. doi: 10.1186/cc5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, Abe M, Shibata K, Shimizu N, Sakata N, Katsuragi T, Tanaka K. Evaluating the role of inducible nitric oxide synthase using a novel and selective inducible nitric oxide synthase inhibitor in septic lung injury produced by cecal ligation and puncture. Am. J. Respir. Crit. Care Med. 2000;162:716–722. doi: 10.1164/ajrccm.162.2.9907039. [DOI] [PubMed] [Google Scholar]
- Peng X, Abdulnour RE, Sammani S, Ma SF, Han EJ, Hasan EJ, Tuder R, Garcia JG, Hassoun PM. Inducible nitric oxide synthase contributes to ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 2005;172:470–479. doi: 10.1164/rccm.200411-1547OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razavi HM, Wang le F, Weicker S, Rohan M, Law C, McCormack DG, Mehta S. Pulmonary neutrophil infiltration in murine sepsis: role of inducible nitric oxide synthase. Am. J. Respir. Crit. Care Med. 2004;170:227–233. doi: 10.1164/rccm.200306-846OC. [DOI] [PubMed] [Google Scholar]
- Razavi HM, Wang L, Weicker S, Quinlan GJ, Mumby S, McCormack DG, Mehta S. Pulmonary oxidant stress in murine sepsis is due to inflammatory cell nitric oxide. Crit. Care Med. 2005;33:1333–1339. doi: 10.1097/01.ccm.0000165445.48350.4f. [DOI] [PubMed] [Google Scholar]
- Salvemini D, Doyle TM, Cuzzocrea S. Superoxide, peroxynitrite and oxidative/nitrative stress in inflammation. Biochem. Soc. Trans. 2006;34:965–970. doi: 10.1042/BST0340965. [DOI] [PubMed] [Google Scholar]
- Scumpia PO, Sarcia PJ, DeMarco VG, Stevens BR, Skimming JW. Hypothermia attenuates iNOS, CAT-1, CAT-2, and nitric oxide expression in lungs of endotoxemic rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;283:L1231–L1238. doi: 10.1152/ajplung.00102.2002. [DOI] [PubMed] [Google Scholar]
- Settergren M, Bohm F, Malmstrom RE, Channon KM, Pernow J. L-arginine and tetrahydrobiopterin protects against ischemia/reperfusion-induced endothelial dysfunction in patients with type 2 diabetes mellitus and coronary artery disease. Atherosclerosis. 2009;204:73–78. doi: 10.1016/j.atherosclerosis.2008.08.034. [DOI] [PubMed] [Google Scholar]
- Shang Y, Li X, Prasad PV, Xu S, Yao S, Liu D, Yuan S, Feng D. Erythropoietin attenuates lung injury in lipopolysaccharide treated rats. J. Surg. Res. 2008 doi: 10.1016/j.jss.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Sharma S, Grobe AC, Wiseman DA, Kumar S, Englaish M, Najwer I, Benavidez E, Oishi P, Azakie A, Fineman JR, Black SM. Lung antioxidant enzymes are regulated by development and increased pulmonary blood flow. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;293:L960–L971. doi: 10.1152/ajplung.00449.2006. [DOI] [PubMed] [Google Scholar]
- Sharma S, Sud N, Wiseman DA, Carter AL, Kumar S, Hou Y, Rau T, Wilham J, Harmon C, Oishi P, Fineman JR, Black SM. Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008;294:L46–L56. doi: 10.1152/ajplung.00247.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhlinger MC, Tsao PS, Her JH, Kimoto M, Balint RF, Cooke JP. Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation. 2001;104:2569–2575. doi: 10.1161/hc4601.098514. [DOI] [PubMed] [Google Scholar]
- Sud N, Wells SM, Sharma S, Wiseman DA, Wilham J, Black SM. Asymmetric dimethylarginine inhibits HSP90 activity in pulmonary arterial endothelial cells: role of mitochondrial dysfunction. Am. J. Physiol. Cell Physiol. 2008;294:C1407–C1418. doi: 10.1152/ajpcell.00384.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol. Lett. 2003;140–141:105–112. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- Szabo C, Salzman AL, Ischiropoulos H. Endotoxin triggers the expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitrite in the rat aorta in vivo. FEBS Lett. 1995;363:235–238. doi: 10.1016/0014-5793(95)00322-z. [DOI] [PubMed] [Google Scholar]
- Takiuchi S, Fujii H, Kamide K, Horio T, Nakatani S, Hiuge A, Rakugi H, Ogihara T, Kawano Y. Plasma asymmetric dimethylarginine and coronary and peripheral endothelial dysfunction in hypertensive patients. Am. J. Hypertens. 2004;17:802–808. doi: 10.1016/j.amjhyper.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Thomson L, Christie J, Vadseth C, Lanken PN, Fu X, Hazen SL, Ischiropoulos H. Identification of immunoglobulins that recognize 3-nitrotyrosine in patients with acute lung injury after major trauma. Am. J. Respir. Cell Mol. Biol. 2007;36:152–157. doi: 10.1165/rcmb.2006-0288SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CT, Fox MF, Vallance P, Leiper JM. Chromosomal localization, gene structure, and expression pattern of DDAH1: comparison with DDAH2 and implications for evolutionary origins. Genomics. 2000;68:101–105. doi: 10.1006/geno.2000.6262. [DOI] [PubMed] [Google Scholar]
- Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler. Suppl. 2003;4:33–40. doi: 10.1016/s1567-5688(03)00032-1. [DOI] [PubMed] [Google Scholar]
- Tsuji C, Shioya S, Hirota Y, Fukuyama N, Kurita D, Tanigaki T, Ohta Y, Nakazawa H. Increased production of nitrotyrosine in lung tissue of rats with radiation-induced acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000;278:L719–L725. doi: 10.1152/ajplung.2000.278.4.L719. [DOI] [PubMed] [Google Scholar]
- Vallance P, Leone A, Calver A, Collier J, Moncada S. Endogenous dimethylarginine as an inhibitor of nitric oxide synthesis. J. Cardiovasc. Pharmacol. 1992;20(Suppl 12):S60–S62. doi: 10.1097/00005344-199204002-00018. [DOI] [PubMed] [Google Scholar]
- Wainwright MS, Arteaga E, Fink R, Ravi K, Chace DH, Black SM. Tetrahydrobiopterin and nitric oxide synthase dimer levels are not changed following hypoxia-ischemia in the newborn rat. Brain Res. Dev. Brain Res. 2005;156:183–192. doi: 10.1016/j.devbrainres.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Webert KE, Vanderzwan J, Duggan M, Scott JA, McCormack DG, Lewis JF, Mehta S. Effects of inhaled nitric oxide in a rat model of Pseudomonas aeruginosa pneumonia. Crit. Care Med. 2000;28:2397–2405. doi: 10.1097/00003246-200007000-00035. [DOI] [PubMed] [Google Scholar]
- Wojciak-Stothard B, Torondel B, Zhao L, Renne T, Leiper JM. Modulation of Rac1 activity by ADMA/DDAH regulates pulmonary endothelial barrier function. Mol. Biol. Cell. 2009;20:33–42. doi: 10.1091/mbc.E08-04-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin HY, Jiang DJ, Jia SJ, Song K, Wang GP, Li YJ, Chen FP. Regulation by DDAH/ADMA pathway of lipopolysaccharide-induced tissue factor expression in endothelial cells. Thromb. Haemost. 2007;97:830–838. [PubMed] [Google Scholar]
- Zhu S, Ware LB, Geiser T, Matthay MA, Matalon S. Increased levels of nitrate and surfactant protein a nitration in the pulmonary edema fluid of patients with acute lung injury. Am. J. Respir. Crit. Care Med. 2001;163:166–172. doi: 10.1164/ajrccm.163.1.2005068. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Day BJ, Crapo JD, Salzman AL, Szabo C. The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. Br. J. Pharmacol. 1997;120:259–267. doi: 10.1038/sj.bjp.0700872. [DOI] [PMC free article] [PubMed] [Google Scholar]