

Abstract
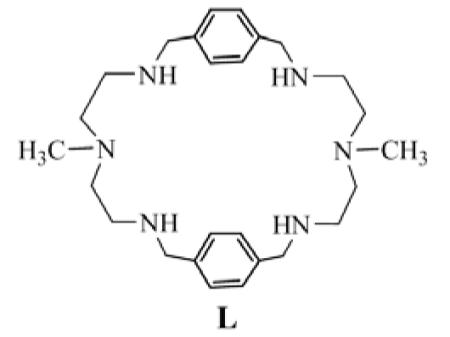
A hexaaminomacrocycle L, containing four secondary and two tertiary amines has been synthesized and crystallized with hydrobromic acid. Structural analysis of the bromide complex suggests that the ligand in its tetrtaprotonated form, is involved in coordinating two bromides from both sides via hydrogen bonding interactions with N…Br- distance of 3,351 Å, forming a ditopic complex. The other two bromides are outside the cavity, and singly bonded to the macrocycle. The molecules are packed showing layer structures in which the internal bromides are locked between the layers of macrocycles. The bromide anions are coordinated alternatively by one and two hydrogen bonds with the protonated amines from the two adjacent macrocycles.
Anion coordination chemistry is expanding rapidly with structurally and functionally diverse receptors capable of hosting a variety of anions in solution and solid states.1 Much interest is focused on aminomacrocyles as anion binding hosts,2 because they mimic many natural polyamines, e.g., spermine and spermidine binding to nucleic acids, predominantly through electrostatic interactions with the negatively charged phosphate groups.3 In general polyazamonocycles, as compared to their bicyclic analogues4, do not bind anions strongly in aqueous solutions, but in many cases they form complexes in solid state.5-9 For example [18]N65 and [24]N85 were shown to crystallize nitrates in water, while halide complexation occurred in [18]N6,6 or metacyclophane.7 On the other hand m-xylyl macrocycle with the expanded cavity and rigid spacers is suitable for binding larger anions like, sulfate,8 pyrophosphate,9 and triphosphate.9
The hexaazamacrocycles are conveniently synthesized in two steps from the condensation of diamine with aromatic dialdehyde (e.g. m-xylyl, p-xylyl, furan or pyridine) and diborane reduction of the corresponding Schiff base.10 In such cases, the diamine linker used in the cyclization process, is 2,2′-diaminodiethylamine which contributes the necessary binding sites for anions. Following the same protocol, the longer 3,3′-diaminodipropylamine was also successfully condensed with m-xylyl spacers to obtain a larger macrocyclic analogue, that was shown to interact with bromide11 and chloride12 in solution as well as in solid state. High affinity was observed in the case of p-xylyl analogue with tripolyphosphate and ATP in aqueous solution where ATP was assumed to interact with the macrocycle via both hydrogen bonding and π–π stacking interactions.13 Another potential diamine, N-methyl-2,2′-diaminodiethylamine, that contains a methyl group at the central nitrogen, has been identified as a potential linker for macrocylic amides,14 thioamides15 or quaternized amines.16 With the exception of a m-xylyl macrocycle demonstrating the ability to recognize pyruvate through H/D exchange at the CH3 position,17 the incorporation of N-methyl-2,2′-diaminodiethylamine as an amine linker, however, has not been explored in a macrocyclic amine.
In an effort to design selective receptors for hosting anionic guests, we incorporated methyl-2,2′-diaminodiethylamine as a linker in polyaza-based macrocycles. Herein we report the synthesis of a new macrocycle L, the crystal structure of bromide complex, and the results of 1H NMR titration studies for bromide anion in water.
The synthesis of ligand L is summarized in Scheme 1. It was prepared following the similar strategy employed for the related compounds.10 The reaction of an equimolar amount of N-methyl-2,2′-diaminodiethylamine and terephthaldehyde under high dilution condensation in CH3OH afforded the macrocyclic Schiff base that was reduced with NaBH4 to obtain the target compound L in 60% yield.18
Scheme 1.
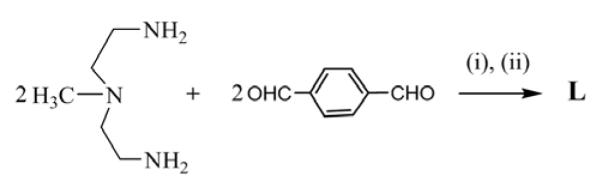
Synthesis of macrocycle L. (i) high dilution, r.t. 24 h, (ii) NaBH4 reduction, r.t., overnight.
The bromide complex of the ligand was obtained from the reaction of free L (45 mg, 0.10 mM) in methanol (2 mL) with 48% aqueous HBr. Crystals suitable for X-ray analysis were grown in three days from a H2O/CH3OH solution (5:1, v/v) at room temperature. X-ray analysis of the complex indicates that L crystallizes in the triclinic space group with four bromide anions.19 In the complex, the macrocycle is tetratprotonated, leaving two bridgehead nitrogens unprotonated. The cationic species and the anions sit on the inversion center.
As shown in Figure 1, all secondary amines act as hydrogen bond donors for bromide anions, contributing a total of six hydrogen bonds. Six out of eight ammonium protons are directed toward the cavity making the macrocycle an ideal host for chelating anions. Two symmetry related bromides (Br1), are bonded to N2 and N3 with N…Br- distances 3.3398(12) and 3.3622(12) Å, respectively, forming a ditopic complex. The N…Br- distances are comparable with those observed previously in the bromide complexes of [18]N6 (N…Br- = 3.343(8) − 3.416(8) Å)6 and [14]metacyclophane (N…Br- = 3.279(16) − 3.437(17) Å).7 Each of the two bromides lies 2.454 Å from the plane of four secondary nitrogen atoms, and is located close to the dien units sitting on the opposite faces of the macrocyle. On the other hand, the remaining two symmetry related bromides (Br2) are singly coordinated to protonated amines with N…Br- distance, 3.2614 (12) Å. Two methyl groups are positioned in anti-parallel and pointed to an internal anion possibly by a weak C…Br- interaction (3.611 Å). The intramolecular aromatic groups are paralleled to each other facing to the cavity center, and separated by a distance, Arcentroid… Arcentroid 5.560 Å, that is longer than that needed for π–π stacking.
Figure 1.
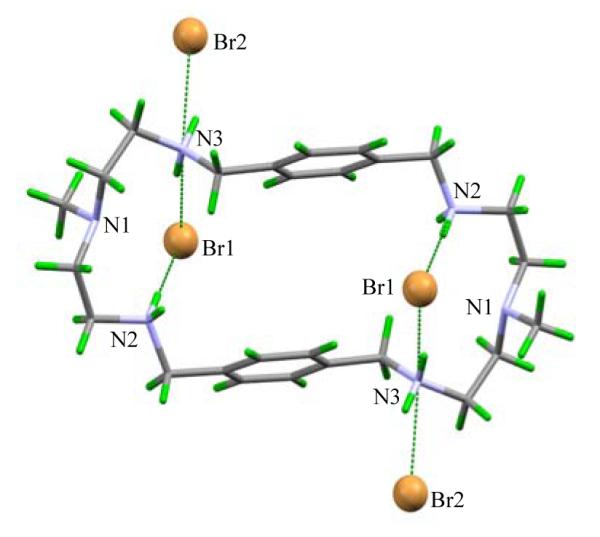
Crystal structure of [H4L(Br)2](Br)2 showing ditopic complex fromed by two bromides bonded to the macrocycle (N2…Br(1) = 3.3398(12) and N3…Br(1) = 3.3622(12) Å).
As shown in Figure 2, bromides are not only coordinated to the protonated amines of L, they are also involved in bridging the intermolecular macrocycles. In the packing diagram, each internal bromide is coordinated alternatively by two hydrogen bonds with a macrocycle, and one hydrogen bond with other macrocycle. Figure 3 illustrates two dimensional sheets formed by the macrocycles and all bromides, in which two bromides are locked between the layers of macrocycles. The anions within the layers are involved in bridging two macrocycles through hydrogen bonding networks. The internal bromide lies 7.062 Å from the plane formed by the other bromide and two ammonium coordinating nitrogens. In this structure, all four bromides form four different lines in which the repeating bromides (both internal and external) are separated by an equal distance of 5.799 Å.
Figure 2.
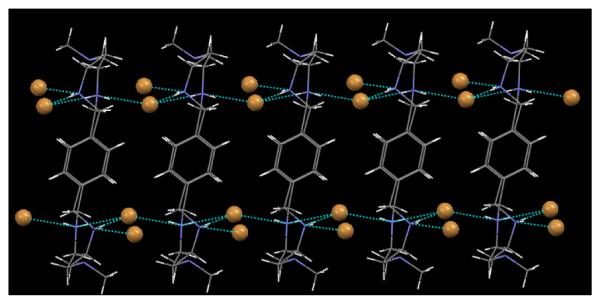
Packing diagram of [H4L1(Br)2](Br)2 viewed along b axis, showing intra- and inter-intermolecular hydrogen bonding interactions.
Figure 3.
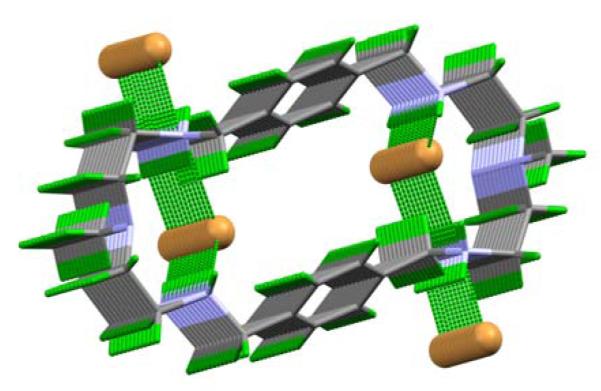
Packing diagram of [H4L(Br)2](Br)2 grown towards a and viewed along a* axis, showing 2D sheets.
In order to evaluate the binding affinity of L for bromide, the 1H NMR titration studies were performed in D2O at pH 3.3, and also in CDCl3. In the case of D2O, the solution pH was adjusted with a concentrated solution of TsOH and NaOH, and NaBr (50 mM in D2O) was used as a titrant. The addition of NaBr solution to the ligand (5 mM), resulted a downfield shift of the methylene protons (H1) adjacent to tertiary amines (Figure 4). Other protons did not shift significantly. The change in the chemical shift of H1 in the twelve independent 1H NMR spectra, as recorded with an increasing amount of anion solution at room temperature, yielded a binding constant K=90 M−1.20 The binding constant is, however, not strong in water as expected, due to the high solvation effect of the competitive polar solvent. The obtained value is in agreement with the results for halide binding in [18]N6,21 however much lower than that observed for bromide or chloride binding in p-xylyl cryptand.22 The presence of methyl group in macrocyclic moiety made the tosylate salt of L (prepared from the reaction of L with four equivalent of TsOH) fairly soluble in non-polar solvent, thus allowing us to measure the binding constant in CDCl3. Because of the solubility reason, n-(Bu)4NBr was used as a titrant, which resulted a significant downfield shift of H1 (Figure 4) The titration of H4L(OTs)4 (5 mM) with the anion (50 mM) gave the binding constant K = 550 M−1, that is about six fold higher than that obtained in D2O.
Figure 4.
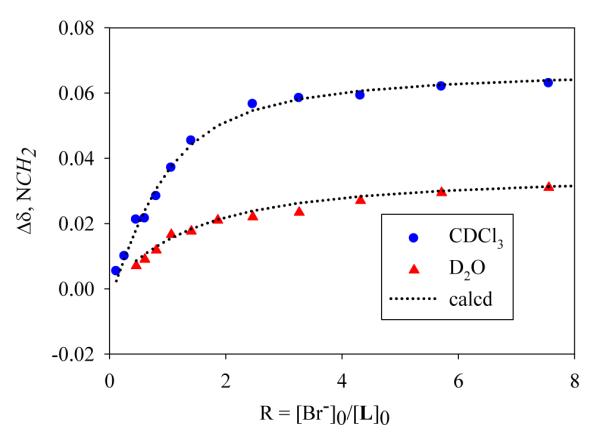
1H NMR titration curves of L with bromide following the chemical shifts of NCH2 in CDCl3(●) and D2O(▲).
In summary, the synthesis of an azamacrocycle, L has been described and a bromide complex of the new ligand has structurally been determined. In solid state, the macrocycle is capable of chelating two anions at the both faces with strong N…Br- interactions, forming a ditopic complex. The synthesized molecule does not form a strong complex with a singly charged bromide in water, however, the incorporation of methyl group to the macrocyclic moiety make the ligand lipophilic, thus allowing to study in non-polar solvent to give a moderate binding constant. The structural information of the bromide complex provides insight into the binding modes and recognition sites in the solid state. This and related ligands might be useful for binding polyatomic oxoanions2a.8,13,23 with multiple charges, as well as transition metal ions.24 Further research in this area is in progress.
Acknowledgments
This work was supported by National Institutes of Health, Division of National Center for Research Resources, under Grant Number G12RR013459. Purchase of the diffractometer was made possible by grant No. LEQSF (1999-2000)-ENH-TR-13, administered by the Louisiana Board of Regents.
References
- 1 (a).Bianchi A, García-España E, Bowman-James K, editors. Supramolecular Chemistry of Anions. Wiley-VCH; New York: 1997. [Google Scholar]; (b) Gale PA, editor. 35 years of Synthetic Anion Receptor Chemistry. Coord. Chem. Rev. 2003;2 [Google Scholar]; (c) Hossain MA. Curr. Org. Chem. 2008;12:1231–1256. [Google Scholar]; (d) Caltagirone C, Gale PA. Chem. Soc. Rev. 2009;38:520–563. doi: 10.1039/b806422a. [DOI] [PubMed] [Google Scholar]
- 2 (a).Llinares JM, Powell D, Bowman-James K. Coord. Chem. Rev. 2003;240:57–75. [Google Scholar]; (b) García-España E, Díaz P, Llinares JM, Bianchi A. Coord. Chem. Rev. 2006;250:2952–2986. [Google Scholar]; (c) Gale PA. Acc. Chem. Res. 2006;39:465–475. doi: 10.1021/ar040237q. [DOI] [PubMed] [Google Scholar]; (d) Bowman-James K. Acc. Chem. Res. 2005;38:671–678. doi: 10.1021/ar040071t. [DOI] [PubMed] [Google Scholar]
- 3.Nagarajan S, Ganem B. J. Org. Chem. 1987;52:5044–5046. [Google Scholar]
- 4.Wiórkiewicz-Kuczera J, Kuczera K, Bazzicalupi C, Bencini A, Valtancoli B, Bianchi A, Bowman-James K. New J. Chem. 1999;23:1007–1013. [Google Scholar]
- 5.Papoyan G, Gu K, Wiórkiewicz-Kuczera J, Kuczera K, Bowman-James K. J. Am. Chem. Soc. 1996;118:1354–1364. [Google Scholar]
- 6.Warden AC, Warren M, Hearn MTW, Spiccia L. New J. Chem. 2004;28:1160–1167. [Google Scholar]
- 7.Illioudis CA, Steed JW. Org. Biomol. Chem. 2005;3:2935–2945. doi: 10.1039/b506828b. [DOI] [PubMed] [Google Scholar]
- 8.Clifford T, Danby A, Llinares JM, Mason S, Alcock NW, Powell D, Aguilar JA, García-España E, Bowman-James K. Inorg. Chem. 2001;40:4710–4720. doi: 10.1021/ic010135l. [DOI] [PubMed] [Google Scholar]
- 9 (a).Lu Q, Motekaitis RJ, Reibenspies J, Martell AE. Inorg. Chem. 1995;34:4958–4964. [Google Scholar]; (b) Lu Q, Riebenspies JH, Caroll RI, Martell AE, Clearfield A. Inorg. Chim. Acta. 1998;270:207–215. [Google Scholar]
- 10.Chen D, Martell AE. Tetrahedron. 1991;47:6900–6901. [Google Scholar]
- 11.Llobet A, Reibenspies J, Martell AE. Inorg. Chem. 1994;33:5946–5951. [Google Scholar]
- 12.Liu H-Y, Wei G-H, Ma J-F. Acta Crystallogr. Sect. E. 2008;E64:o126. doi: 10.1107/S1600536807063064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anda C, Martinez MA, Llobet A. Supramol. Chem. 2005;17:257–266. [Google Scholar]
- 14.Hossain MA, Llinares JM, Powell D, Bowman-James K. Inorg. Chem. 2001;40:2936–2937. doi: 10.1021/ic015508x. [DOI] [PubMed] [Google Scholar]
- 15.Hossain MA, Kang SO, Llinares JM, Powell D, Bowman-James K. Inorg. Chem. 2003;42:5043–5045. doi: 10.1021/ic034735r. [DOI] [PubMed] [Google Scholar]
- 16.Hossain MA, Kang SO, Powell D, Bowman-James K. Inorg. Chem. 2003;42:1397–1399. doi: 10.1021/ic0263140. [DOI] [PubMed] [Google Scholar]
- 17.Fenniri H, Dallaire C, Funeriu DP, Lehn J-M. Perk. Trans. 1997;2:2073–2081. [Google Scholar]
- 18. The synthesis of L was accomplished from the condensation reaction under high dilution conditions. In a typical reaction, N-methyl-2,2′-diaminodiethylamine (1.00 g, 8.53 × 10−3 mol) in CH3OH (200 mL) and terephthaldehyde (1.15 g, 8.53 × 10−3 mol) in CH3OH (200 mL) were added simultaneously to 400 mL of CH3OH over 4 h. The resulting mixture was stirred at room temperature for another 24 h. After the evaporation of the solvent, the oily product was dissolved in CH3OH (100 mL), and reduced with NaBH4 overnight at room temperature. The solvent was removed in vacuo, and the resulting oily product was dissolved in water (100 mL). The aqueous phase was extracted by CH2Cl2 (3×100 mL). The organic layers were combined and dried by adding anhydrous MgSO4 (2 g). After the filtration, the solvent was evaporated under reduced pressure resulting a light yellowish powder. The crude product was purified by column chromatography (neutral alumina, 2% CH3OH in CH2Cl2).Yield: 0.67 g, 60%. 1H NMR (300 MHz, CDCl3, TMS): δ 2.16 (s, 6H, CH3), 2.54 (t, 8H, NCH2), 2.77 (t, 8H, NHCH2), 3.75 ((s, 8H, ArCH2), 7.19 (s, 8H, ArH). 13C NMR (100 MHz, CDCl3, TMS): 42.2 (NHCH2), 46.9 (CH3), 53.9 (NHCH2), 56.5 (NCH2), 128.2 (Ar-CH), 138.9 (Ar-C). Anal. Calcd. for (C26H42N6): C, 71.19; H, 9.65; N, 19.16. Found: C, 71.53; H, 9.77; N, 19.31.
- 19. [H4L(Br)2](Br)2: C26H46N6Br4 M = 762.33, crystal size 0.25 × 0.23 × 0.20 mm3, Triclinic, PĪ, a =5.7991 (5), b =9.5238 (10), c = 14.7954 (14) Å, α =74.360 (5)°, β = 84.001 (6)°, γ = 88.462 (6)°, V = 782.58 (13) Å3, Z = 1, dcalc = 1.618 g cm−3, T = 90 K, F(000) = 384, μ(Mο-Kα) = 5.17 mm−1, 7513 independent reflections (33284 measured), R = 0.028, Rint = 0.023, wR(F2) = 0.068. Intensity data for the complex were collected using Nonius KappaCCD diffractmeter, λ (MοKα) = 0.71073 Å..The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F. CCDC 742965.
- 20. Binding constants were obtained by 1H NMR (300 MHz Bruker) titrations of H4L·(TsO)4 with NaCl in D2O at pH 2, and with n-(Bu)4NBr in CDCl3. Initial concentrations were [ligand]0 = 5 mM, and [anion]0 = 50 mM. In the case of D2O, sodium salt of 3-(trimethylsilyl)propionic-2,2,3,3,-d4 acid (TSP) in D2O was used as an external reference in a capillary tube. The pH was adjusted with a concentrated solution of TsOH and NaOH in D2O. Each titration was performed by twelve measurements at room temperature. The association constant K was calculated using Sigma Plot software, from the following equations: Δδ = ([A]0 + [L]0 + 1/K − (([A]0 + [L] + 1/K)2 − 4[L]0[A]0)1/2) Δδmax / 2[L]0 (where L = ligand and A = chloride). Error limit in K was less that 15%.
- 21.Cullinane J, Gelb RI, Margulis TN, Zompa LJ. J. Am.Chem. Soc. 1982;104:3048–3053. [Google Scholar]
- 22.Hossain MA, Morehouse P, Powell PD, Bowman-James K. Inorg. Chem. 2005;44:2143–2149. doi: 10.1021/ic048937e. [DOI] [PubMed] [Google Scholar]
- 23 (a).Anda C, Llobet A, Salvado V, Reibenspies J, Motekaitis RJ, Martell AE. Inorg. Chem. 2000;39:2986–2999. doi: 10.1021/ic990818p. [DOI] [PubMed] [Google Scholar]; (b) Gerasimchuk OA, Mason S, Llinares AM, Song M, Alcock NW, Bowman-James K. Inorg. Chem. 2000;39:1371–1375. doi: 10.1021/ic9911116. [DOI] [PubMed] [Google Scholar]
- 24 (a).Gao J, Reibenspies JH, Martell AE. Inorg. Chim. Acta. 2002;335:125–129. [Google Scholar]; (b) Graham B, Spiccia L, Batten SR, Skelton BW, White AH. Inorg. Chim. Acta. 2002;338:3983–3994. [Google Scholar]; (c) Costas M, Anda C, Llobet A, Parella T, Evans HS, Pinilla E. Eur. J. Inorg. Chem. 2004:857–865. [Google Scholar]