

Abstract
We report the copper(II)-catalyzed conversion of organoboron compounds into the corresponding azide derivatives. A systematic series of phenylboronic acid derivatives is evaluated to examine the importance of steric and electronic effects of the substituents on reaction yield as well as functional group compatibility. Heterocyclic substrates are also shown to participate in this mild reaction while compounds incorporating B–C(sp3) bonds are unreactive under the reaction conditions. The copper(II)-catalyzed boronic acid–azide coupling reaction is further extended to both boronate esters and potassium organotrifluoroborate salts. The method described herein complements existing procedures for the preparation of aryl azides from the respective amino, triazene, and halide derivatives and we expect that it will greatly facilitate copper- and ruthenium-catalyzed azide–alkyne cycloaddition reactions for the preparation of diversely functionalized 1-aryl- or 1-heteroaryl-1,2,3-triazoles derivatives.
Keywords: azides; organoboron compounds; copper catalysis; 1,2,3-triazoles
Aryl and heteroaryl azides have emerged as valuable building blocks for the construction of 1,2,3-triazoles in combinatorial drug discovery.1,2 Traditionally, aryl azides have been prepared from the corresponding amines via diazotization. Moses and co-workers reported an improved diazotization procedure utilizing tert-butyl nitrite and azidotrimethylsilane under mild conditions while Bräse and co-workers described a mild synthesis of aryl azides from aryltriazenes.3,4 Ma and Zhu demonstrated the synthesis of both aryl and vinyl azides from the respective aryl halides by proline–copper(I)-catalyzed coupling with sodium azide.5 Pinhey and co-workers first reported the conversion of arylboronic acids into aryl azides using stoichiometric lead(IV) acetate and mercury(II) acetate to afford intermediate aryl–lead species, which were then reacted with sodium azide in dimethyl sulfoxide.6,7 In a significant advance, Guo, Liu, and co-workers recently showed that simple arylboronic acids can be elaborated to aryl azides using copper salts at room temperature.8 Herein, we disclose our results that examine in much greater detail the substrate scope of this powerful transformation including the influence of ligands, impact of electronics of the substrate on coupling efficiency, functional group compatibility, utilization of boronate esters and organotrifluoroborate salts, and examination of various heterocyclic substrates. Our results demonstrate the broad applicability and identify limitations of the copper(II)-catalyzed boronic acid–azide coupling reaction.
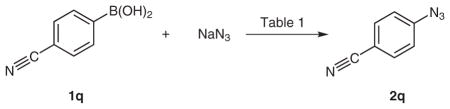
The copper(II)-catalyzed synthesis of aryl azides from arylboronic acids is based on the Chan–Lam coupling of arylboronic acids with N–H containing heteroarenes, anilines, phenols, and amides that employs stoichiometric copper(II) acetate in aprotic solvents and requires a base such as pyridine or triethylamine.9,10 Since sodium azide is sparingly soluble in most aprotic organic solvents, we examined protic solvents and discovered that in contrast to the Chan–Lam coupling conditions, the azidation of 4-cyanophenylboronic acid (1q) proceeds efficiently in water, methanol, and ethanol (Table 1). Additionally, we found removal of the obligate base (pyridine or triethylamine) in the Chan–Lam coupling improved the yield (Table 1, entries 4 and 7). The ligands 2,2′-bipyridine (Bipy) and 1,10-phenanthroline (Phen) were also evaluated as these have been shown to enhance copper(II)-catalyzed cross-coupling reactions; however, these provided no major benefit in terms of isolated yield or enhancement in the reaction rate (Table 1, entries 5 and 6). If the reaction was conducted under an aerobic atmosphere to reoxidize Cu(0) back to Cu(II), then copper could be used catalytically (Table 1, entry 8). Our standardized reaction conditions involved vigorously stirring a 0.2 M solution of arylboronic acid in methanol with sodium azide (1.5 equiv) and copper(II) acetate (0.1 equiv) at 55 °C for 1–3 hours under air (Table 1, entry 9). The higher temperature used herein allow the reactions to reach completion in 1–3 hours, but the reaction can also be performed at room temperature if the reaction time is extended to 24 hours, which is preferred when working on scales larger than several millimoles.8 The reaction was conveniently followed colorimetrically as the dark brown solution turned light green as the reaction reached completion.
Table 1.
Survey of Reaction Conditionsa
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Cu(OAc)2 Solvent (equiv) | Base/ligand (equiv) | Temp (°C) | Time (h) | Yieldb (%) | |
| 1 | 1.0 | CH2Cl2 | Py (1.0) | 25 | 24 | 83 ± 0 |
| 2 | 1.0 | H2O | Py (1.0) | 25 | 24 | 66 ± 3 |
| 3 | 1.0 | EtOH | Py (1.0) | 25 | 24 | 92 ± 4 |
| 4 | 1.0 | MeOH | Py (1.0) | 25 | 24 | 80 ± 4 |
| 5 | 1.0 | MeOH | Bipy (1.0) | 25 | 24 | 83 ± 1 |
| 6 | 1.0 | MeOH | Phen (1.0) | 25 | 24 | 95 ± 3 |
| 7 | 1.0 | MeOH | none | 25 | 24 | 94 ± 4 |
| 8 | 0.1 | MeOH | none | 25 | 24 | 77 ± 1 |
| 9 | 0.1 | MeOH | none | 55 | 3 | 98 ± 0.1 |
All reactions were carried out under air atmosphere using NaN3 (1.5 equiv), 4-cyanophenylboronic acid (1q, 0.2 M, 1.0 equiv).
Isolated yield ± standard deviation from 2–3 independent experiments.
A systematic series of para-substituted phenylboronic acids was initially examined to explore the influence of electron-donating and -withdrawing substituents on coupling efficiency (Table 2). Remarkably, all compounds underwent coupling providing 40–98% yields of the corresponding aryl azides. While coupling efficiency did not correlate with electron-donating ability as measured by the Hammett substituent constant σp, 11 we observed that arylboronic acids with strongly electron-withdrawing groups reacted more slowly, requiring three hours to reach completion and compounds with electron-donating groups, such as methoxy, were complete within one hour. In the case of volatile azides, such as phenyl azide (2e), 1-azido-4-fluorobenzene (2g), and 1-azido-4-(trifluoromethyl)benzene (2p), low isolated yields were obtained, therefore these were isolated as the corresponding triazole derivatives 3e, 3g, and 3p, respectively, by reacting the azides with methyl propiolate at room temperature. Overall, the results highlight the functional group compatibility of this reaction. The impact of sterics was examined with 2-(pivaloylamino)phenylboronic acid (1v) that provided 2v in 94% yield while biphenyl-2- (1u), -3- (1t), and -4-boronic acid (1c) provided 2u, 2t, and 2c, respectively, in yields ranging from 40–50% illustrating the relative lack of sensitivity to sterics (Table 2).
Table 2.
Scope of the Copper(II)-Catalyzed Azidation of Arylboronic Acids
 | ||||
|---|---|---|---|---|
| Entry | R | σpa | Product | Yieldb (%) |
| 1 | 4-OMe | −0.27 | 2a | 78 ± 5 |
| 2 | 4-OPh | −0.03 | 2b | 92 ± 8 |
| 3 | 4-Ph | −0.01 | 2c | 50 ± 1 |
| 4 | 4-CH2OH | 0 | 2d | 98 ± 1 |
| 5 | H | 0 | 3e | 64c |
| 6 | 4-SMe | 0 | 2f | 97 ± 1 |
| 7 | 4-F | +0.06 | 3g | 68c |
| 8 | 4-I | +0.18 | 2h | 63 ± 2 |
| 9 | 4-Cl | +0.23 | 2i | 73 ± 4 |
| 10 | 4-Br | +0.23 | 2j | 68 ± 6 |
| 11 | 4-CONH2 | +0.36 | 2k | 84 ± 3 |
| 12 | 4-CHO | +0.42 | 2l | 81 ± 4 |
| 13 | 4-CO2H | +0.45 | 2m | 89 ± 3 |
| 14 | 4-CO2Me | +0.45 | 2n | 99 ± 0 |
| 15 | 4-COMe | +0.50 | 2o | 81 ± 3 |
| 16 | 4-CF3 | +0.54 | 3p | 68c |
| 17 | 4-CN | +0.66 | 2q | 98 ± 1 |
| 18 | 4-SO2Me | +0.72 | 2r | 86 ± 3 |
| 19 | 4-NO2 | +0.78 | 2s | 85 ± 1 |
| 20 | 3-Ph | –d | 2t | 46 ± 1 |
| 21 | 2-Ph | –d | 2u | 40 ± 2 |
| 22 | 2-NHCOt-Bu | –d | 2v | 94 ± 2 |
Hammett substituent constants.11
Isolated yield ± standard deviation from 2–3 independent experiments.
Due to volatility, the crude azides were not isolated, but reacted in situ with methyl propiolate to afford the corresponding triazoles 3. The isolated yields are for the corresponding triazole products. See experimental section for details.
Not applicable.
Next, arylboronic acid pinacol esters were evaluated. 4-Cyanophenylboronic acid pinacol ester (4q) furnished azide 2q in 81% yield (Table 3, entry 1), while 2q was obtained in 98% yield from the corresponding boronic acid derivative 1q (Table 2, entry 17). The successful coupling of amino- and dimethylamino-substituted phenylboronic acid pinacol esters 4w and 4x to afford azides 2w and 2x further highlights the functional group compatibility of this reaction (Table 3). Examination of heterocyclic substrates revealed some limitations to this reaction as 4-azidopyrazole 2y and 5-azidoindole 2z were isolated in only modest yields.
Table 3.
Scope of the Copper(II)-Catalyzed Azidation of Boronic Acid Pinacol Esters
 | |||
|---|---|---|---|
| Entry | R | Product | Yielda (%) |
| 1 |  |
2q | 81 ± 3 |
| 2 | 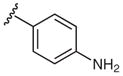 |
2w | 94 ± 1 |
| 3 |  |
2x | 83 ± 1 |
| 4 | 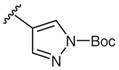 |
2y | 29 ± 5 |
| 5 | 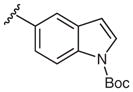 |
2z | 51 ± 1 |
Isolated yield ± standard deviation from 2–3 independent experiments.
Heterocyclic boronic acids were also examined including pyridine, pyrimidine, quinoline, and isoquinoline 5a–d and the corresponding azides 6a–d were isolated in yields ranging from 34–42% (Table 4). Finally, several substrates containing B–C(sp3) bonds were examined including cyclopropyl 5e, cyclohexyl 5f, and benzyl 5g; however, no coupling was observed with any of these substrates demonstrating the limitations of the current method, which is restricted to substrates with B–C(sp2) bonds.
Table 4.
Scope of the Copper(II)-Catalyzed Azidation of Heteroaryl- and Alkylboronic Acids
 | |||
|---|---|---|---|
| Entry | R | Product | Yielda (%) |
| 1 | 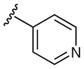 |
6a | 42 ± 1 |
| 2 | 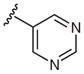 |
6b | 40 ± 8 |
| 3 | 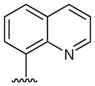 |
6c | 34 ± 4 |
| 4 |  |
6d | 40 ± 6 |
| 5 | 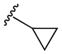 |
6e | 0 |
| 6 | 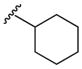 |
6f | 0 |
| 7 | 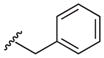 |
6g | 0 |
Isolated yield ± standard deviation from 2–3 independent experiments.
Potassium organotrifluoroborate salts have emerged as attractive reagents in organic synthesis due to their increased chemical stability, excellent reactivity, and improved physicochemical properties.12 Therefore we examined whether this valuable class of organoboron reagents would participate in the copper(II)-catalyzed cross-coupling reaction with sodium azide. A subset of the aryltrifluoroborates (Table 5) was selected in order to directly compare the results with the corresponding boronic derivatives from Table 1. The aryltrifluoroborates were competent substrates, but provided lower yields of the azides in all cases and were slightly less reactive than the respective arylboronic acids.
Table 5.
Scope of the Copper(II)-Catalyzed Azidation of Organofluoroborate Salts
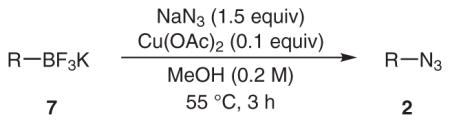 | |||
|---|---|---|---|
| Entry | R | Product | Yield (%) |
| 1 | 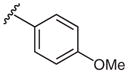 |
2a | 70 |
| 2 | 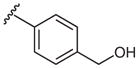 |
2d | 40 |
| 3 | 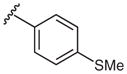 |
2f | 67 |
| 4 | 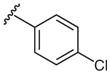 |
2i | 46 |
| 5 |  |
2m | 75 |
| 6 | 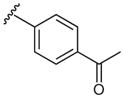 |
2o | 73 |
The Chan–Lam coupling of arylboronic acids with azides as first disclosed by Guo, Liu, and co-workers represents a powerful and more environmentally friendly method for the preparation of various aryl and heteroaryl azides.8 Another major advantage of this method is the compatibility with the conditions for the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction, thus enabling one to sequentially perform both reactions in the same vessel.13,14 Notably, the Chan–Lam coupling is a copper(II) catalytic cycle while the CuAAC reaction requires a copper(I) cycle. At the completion of the copper(II)–boronic acid–azide coupling, the addition of an alkyne and a reducing agent (aqueous solution of sodium ascorbate) results in a facile CuAAC reaction. The product azides in general are nonpolar and can be easily purified by filtration through a short silica gel column to remove all inorganic salts and potentially unreacted organoboron substrates. In almost all cases, except for heterocyclic compounds, the reaction as monitored by thin-layer chromatography resulted in complete consumption of the polar organoboranes followed by the formation of the more nonpolar aryl azide with no observed byproducts. The mass balance for compounds that resulted in less than 90% yield is likely due to losses obtained during purification of these volatile low molecular weight (MW < 150) azide derivatives. While we primarily investigated the coupling of arylboronic acids with sodium azide using catalytic copper(II) salts, the stoichiometric version may be preferred in common parallel synthesis reactors due to lack of turnover of the air headspace. Additionally, stoichiometric copper is more desirable for volatile organic azides as reaction can be done in a sealed flask.
The proposed catalytic cycle for the Chan–Lam coupling of organoboranes with the azide ion is shown in Scheme 1 and consists of an initial transmetalation to afford a R–Cu(II) intermediate. In the second step, the azide anion coordinates to the R–Cu(II) complex providing R–Cu(II)–N3 that subsequently undergoes reductive elimination to afford the organic azide and Cu(0). Oxidation of Cu(0) to Cu(II) by molecular oxygen completes the catalytic cycle. The successful copper-catalyzed coupling of aryl iodides with sodium azide as reported by Zhu and Ma, demonstrated that aryl–Cu(II)–azido complexes could undergo reductive elimination and immediately suggested the viability of the Chan–Lam coupling with the azide anion.5 Reoxidation of Cu(0) to Cu(II) is slow and can be rate-limiting if the reaction vessel is sealed. Alternatively, it is plausible that oxidation of the aryl–Cu(II)–azido complex to a Cu(III) species would serve to promote reductive elimination to afford the product aryl azide and a Cu(I) species, which could be rapidly oxidized to Cu(II). This mechanism has been proposed for the Chan–Lam coupling.15 Furthermore, the first Cu(III) intermediate, which was identified from conjugate addition to an enone, has been recently spectroscopically characterized.16
Scheme 1.

Proposed catalytic cycle for Chan–Lam coupling of boronic acids with the azide anion
In closing, it is important to note the excellent functional group tolerance of aryl azides and boronic acids. Thus, 1-azido-2-bromobenzene has been reported to successfully participate in Suzuki cross-coupling with arylboronic acids.17 In an even more impressive example, Ham, Molander, and co-workers demonstrated the chemoselective azidation of bromo- and iodoaryltrifluoroborates to form (azidoaryl)trifluoroborates employing catalytic copper(I) bromide followed by Suzuki cross-coupling of these bifunctional molecules.18 The use of a copper(I) salt by these authors led to azidation of the halogen while the use of copper(II) salts reported herein results in the azidation of the trifluoroborate moiety. These complimentary reaction outcomes highlight the critical role of the copper oxidation state.
All commercial reagents (Sigma-Aldrich, Acros, Boron Molecular) were used as provided. Flash chromatography was performed with an ISCO Combiflash Companion® purification system with pre-packed 4 g silica gel cartridges with the indicated solvent system. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Chemical shifts are reported in ppm from an internal standard of residual methanol (δ = 3.31 ppm for 1H NMR and δ = 49.1 ppm for 13C NMR). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant, integration. FT-IR spectra were obtained on a Nicolet Protégé 460 ESP spectrometer. The presence of an azido group was confirmed by a characteristic strong infrared absorption at 2100–2150 cm−1. High-resolution mass spectra were acquired on an Agilent TOF II TOF/MS instrument equipped with either an ESI or APCI interface. Azides that failed to ionize under ESI or APCI conditions were treated with Ph3P in THF at 55 °C for 2 h to form the corresponding iminophosphoranes (R–N=PPh3), which were subsequently characterized by HRMS. Melting points were measured on an electrothermal Mel-Temp manual melting point apparatus and are uncorrected. CAUTION: Organic azides are potentially explosive substances and the proposed synthesis employing elevated temperature should only be run on scales less than 1 mmol.
Azide Synthesis; General Procedure
To the boronic acid (1.0 mmol, 1.0 equiv), pinacol boronate (1.0 mmol, 1.0 equiv), or potassium organotrifluoroborate salt (1.0 mmol, 1.0 equiv) in MeOH (5 mL) were added NaN3 (1.5 mmol, 1.5 equiv) and Cu(OAc)2 (0.1 mmol, 0.1 equiv). The soln was vigorously stirred in an 8-mL round-bottom vial at 55 °C under air. The mixture was concentrated onto Celite and purification by flash chromatography (prepacked silica cartridges) afforded the title compounds.
1-Azido-4-methoxybenzene (2a)
Following the general procedure using 4-methoxyphenylboronic acid (1a) or potassium trifluoro(4-methoxyphenyl)borate (7a) for 3 h and chromatography (10% EtOAc–hexane) afforded 2a (110 mg, 74% from 1a; 105 mg, 70% from 7a) as a yellow oil; Rf = 0.6 (hexane–EtOAc, 9:1).
IR: 2109 cm−1.
1H NMR (600 MHz, CD3OD): δ = 3.76 (s, 3 H), 6.92 (d, J = 9.0 Hz, 2 H), 6.96 (d, J = 9.0 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 56.1, 116.4, 121.1, 133.7, 158.8.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C25H22NOP: 384.1512; found: 384.1535.
1-Azido-4-phenoxybenzene (2b)
Following the general procedure using 4-phenoxyphenylboronic acid (1b) for 3 h and chromatography (hexane) afforded 2b (182 mg, 86%) as a brown oil: Rf = 0.2 (hexane).
IR: 2123 cm−1.
1H NMR (600 MHz, CD3OD): δ = 6.92–6.97 (m, 6 H), 7.06 (t, J = 7.2 Hz, 1 H), 7.27–7.30 (m, 2 H).
13C NMR (150 MHz, CD3OD): δ = 119.7, 121.4, 124.3, 124.5, 130.9, 136.4, 155.9, 158.7.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C30H24NOP: 446.1668; found: 446.1707.
4-Azidobiphenyl (2c)
Following the general procedure using biphenyl-4-ylboronic acid (1c) for 3 h and chromatography (hexane) afforded 2c (100 mg, 51%) as a yellow solid; mp 62–64 °C; Rf = 0.3 (hexane).
IR: 2115 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.12 (d, J = 7.8 Hz, 2 H), 7.32 (t, J = 7.2 Hz, 1 H), 7.41 (t, J = 7.2 Hz, 2 H), 7.58 (d, J = 7.8 Hz, 2 H), 7.63 (d, J = 7.8 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 120.6, 127.9, 128.5, 129.6, 130.1, 139.5, 140.7, 141.5.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C30H24NP: 430.1719; found: 430.1756.
1-Azido-4-(hydroxymethyl)benzene (2d)
Following the general procedure using 4-(hydroxymethyl)phenylboronic acid (1d) or potassium trifluoro[4-(hydroxymethyl)phenyl]borate (7d) for 3 h and chromatography (Et2O) afforded 2d (148 mg, 99% from 1d; 60 mg, 40% from 7d) as a yellow oil; Rf = 0.3 (hexane–EtOAc, 7:3).
IR: 2107 cm−1.
1H NMR (600 MHz, CD3OD): δ = 4.57 (s, 2 H), 7.03 (d, J = 8.4 Hz, 2 H), 7.37 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 64.7, 120.0, 129.6, 139.9, 140.5.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C25H22NOP: 384.1512; found: 384.1512.
Methyl 1-Phenyl-1H-1,2,3-triazole-4-carboxylate (3e)
Following the general procedure using phenylboronic acid (1e) for 1.5 h afforded crude 2e. Methyl propiolate (3.0 equiv) and sodium ascorbate (0.1 equiv) were added and the mixture was stirred at r.t. overnight. Chromatography (30% EtOAc–hexane) afforded 3e (130 mg, 64%) as a white solid; mp 178–182 °C; Rf = 0.3 (hexane–EtOAc, 7:3).
1H NMR (600 MHz, CD3OD): δ = 3.96 (s, 3 H), 7.53 (t, J = 7.8 Hz, 1 H), 7.60 (t, J = 7.8 Hz, 2 H), 7.89 (d, J = 7.8 Hz, 2 H), 9.11 (s, 1 H).
13C NMR (150 MHz, CD3OD): δ = 52.8, 122.0, 127.9, 129.9, 131.2, 138.0, 141.4, 161.1.
HRMS (ESI+): m/z [M + H]+ calcd for C10H10N3O2: 204.0768; found: 204.0790.
1-Azido-4-(methylthio)benzene (2f)
Following the general procedure using 4-(methylthio)boronic acid (1f) or potassium trifluoro[4-(methylthio)phenyl]borate (7f) for 3 h and chromatography (Et2O) afforded 2f (162 mg, 98% from 1f; 110 mg, 67% from 7f) as a yellow oil; Rf = 0.6 (hexane–EtOAc, 9:1).
IR: 2115 cm−1.
1H NMR (600 MHz, CD3OD): δ = 2.45 (s, 3 H), 6.98 (d, J = 8.4 Hz, 2 H), 7.28 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 16.4, 120.6, 129.6, 136.6, 138.6.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C25H22NPS: 400.1283; found: 400.1296.
Methyl 1-(4-Fluorophenyl)-1H-1,2,3-triazole-4-carboxylate (3g)
Following the general procedure using 4-fluorophenylboronic acid (1g) for 1.5 h afforded crude 2g. Methyl propiolate (3.0 equiv) and sodium ascorbate (0.1 equiv) were added and the mixture was stirred at r.t. overnight. Chromatography (20% EtOAc–hexane) afforded 3g (148 mg, 67%) as a yellow solid; mp 185–188 °C; Rf = 0.4 (hexane–EtOAc, 7:3).
1H NMR (600 MHz, CD3OD): δ = 3.96 (s, 3 H), 7.35 (d, J = 8.4 Hz, 2 H), 7.93 (d, J = 8.4 Hz, 2 H), 9.10 (s, 1 H).
13C NMR (150 MHz, CD3OD): δ = 52.8, 117.9 (d, 2JC-F = 24 Hz), 124.4 (d, 3JC-F = 8.9 Hz), 128.2, 136.8, 141.5, 162.4, 164.5 (d, 1JC-F = 247 Hz).
HRMS (ESI+): m/z [M + H]+ calcd for C10H9FN3O2: 222.0673; found: 222.0675.
1-Azido-4-iodobenzene (2h)
Following the general procedure using 4-iodophenylboronic acid (1h) for 3 h and chromatography (hexane) afforded 2h (157 mg, 64%) as a yellow oil; Rf = 0.8 (hexane–EtOAc, 8:2).
IR: 2125 cm−1.
1H NMR (600 MHz, CD3OD): δ = 6.87 (d, J = 7.2 Hz, 2 H), 7.70 (d, J = 7.2 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 89.0, 122.3, 140.2, 141.7.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C24H19INP: 480.0373; found: 480.0417.
1-Azido-4-chlorobenzene (2i)
Following the general procedure using 4-chlorophenylboronic acid (1i) or potassium 4-chlorophenyltrifluoroborate (7i) for 3 h and chromatography (Et2O) afforded 2i (117 mg, 76% from 1i; 70 mg, 46% from 7i) as a brown oil; Rf = 0.7 (hexane–EtOAc, 9:1).
IR: 2106 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.04 (d, J = 8.4 Hz, 2 H), 7.37 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 89.0, 122.3, 140.2, 141.7.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C24H19ClNP: 388.1016; found: 388.1024.
1-Azido-4-bromobenzene (2j)
Following the general procedure using 4-bromophenylboronic acid (1j) for 3 h and chromatography (Et2O) afforded 2j (143 mg, 72%) as a yellow oil; Rf = 0.7 (hexane–EtOAc, 9:1).
IR: 2108 cm−1.
1H NMR (600 MHz, CD3OD): δ = 6.99 (d, J = 8.4 Hz, 2 H), 7.51 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 118.8, 122.0, 134.0, 141.0.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C24H19BrNP: 432.0511; found: 432.0533.
4-Azidobenzamide (2k)
Following the general procedure using 4-carbamoylphenylboronic acid (1k) for 3 h and chromatography (70% EtOAc–hexane) afforded 2k (131 mg, 81%) as a white amorphous solid; mp 167–169 °C; Rf = 0.2 (hexanes–EtOAc, 6:4).
IR: 2097 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.14 (d, J = 8.4 Hz, 2 H), 7.90 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 120.0, 130.7, 131.6, 145.3, 171.8.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C25H21N2OP: 397.1464; found: 397.1479.
4-Azidobenzaldehyde (2l)
Following the general procedure using 4-formylphenylboronic acid (1l) for 3 h and chromatography (5% EtOAc–hexane) afforded 2l (124 mg, 84%) as a brown oil; Rf = 0.3 (hexane–EtOAc, 9:1).
IR: 2115 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.22 (d, J = 8.4 Hz, 2 H), 7.91 (d, J = 8.4 Hz, 2 H), 9.90 (s, 1 H).
13C NMR (150 MHz, CD3OD): δ = 120.7, 132.7, 134.9, 147.9, 192.7.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C25H20NOP: 382.1355; found: 382.1393.
4-Azidobenzoic Acid (2m)
Following the general procedure using 4-carboxyphenylboronic acid (1m) or potassium 4-carboxyphenyltrifluoroborate (7m) for 3 h. The mixture was treated with 1 M HCl (10 mL) and extracted with EtOAc (3 × 20 mL). The combined extracts were washed with sat. aq NaCl, dried (Na2SO4), and concentrated in vacuo. Recrystallization of the crude mixture (EtOAc, 10 mL) afforded 2m (150 mg, 92% from 1m; 121 mg, 75% from 7m) as a yellow solid; mp 109–115 °C; Rf = 0.3 (hexanes–EtOAc, 7:3).
IR: 2110 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.15 (d, J = 9.0 Hz, 2 H), 8.04 (d, J = 9.0 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 120.1, 128.9, 132.8, 146.4, 169.2.
HRMS (ESI–): m/z [M – H]− calcd for C7H4N3O2: 162.0309; found: 162.0323.
Methyl 4-Azidobenzoate (2n)
Following the general procedure using 4-(methoxycarbonyl)phenylboronic acid (1n) for 3 h and chromatography (10% EtOAc–hexane) afforded 2n (175 mg, 99%) as a yellow oil; Rf = 0.7 (hexane–EtOAc, 7:3).
IR: 2117 cm−1.
1H NMR (600 MHz, CD3OD): δ = 3.89 (s, 3 H), 7.16 (d, J = 8.4 Hz, 2 H), 8.03 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 52.8, 120.2, 128.2, 132.6, 146.6, 167.9.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C26H22NO2P: 412.1461; found: 412.1477.
4′-Azidoacetophenone (2o)
Following the general procedure using 4-acetylphenylboronic acid (1o) or potassium 4-acetylphenyltrifluoroborate (7o) for 3 h and chromatography (10% EtOAc–hexane) afforded 2o (134 mg, 83% from 1o; 117 mg, 73% from 7o) as a brown solid; mp 40–45 °C; Rf = 0.3 (hexane–EtOAc, 9:1).
IR: 2128 cm−1.
1H NMR (600 MHz, CD3OD): δ = 2.55 (s, 3 H), 7.12 (d, J = 8.4 Hz, 2 H), 7.98 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 26.6, 120.1, 131.6, 135.1, 146.6, 198.9.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C26H22NOP: 396.1512; found: 396.1552.
Methyl 1-[4-(Trifluoromethyl)phenyl]-1H-1,2,3-triazole-4-carboxylate (3p)
Following the general procedure using 4-(trifluoromethyl)phenylboronic acid (1p) for 3 h afforded crude 2p. Methyl propiolate (3.0 equiv) and sodium ascorbate (0.1 equiv) were added and the mixture was stirred at r.t. overnight. Chromatography (20% EtOAc–hexane) afforded 3p (184 mg, 68%) as a yellow solid; mp 193–196 °C; Rf = 0.3 (hexane–EtOAc, 7:3).
1H NMR (600 MHz, CD3OD): δ = 3.97 (s, 3 H), 7.94 (d, J = 8.4 Hz, 2 H), 8.15 (d, J = 8.4 Hz, 2 H), 9.27 (s, 1 H).
13C NMR (150 MHz, CD3OD): δ = 52.8, 122.4, 128.2, 128.3 (q, 3JC-F = 4.8 Hz), 132.4 (d, 2JC-F = 33.0 Hz), 140.7, 141.7, 162.2; CF3 signal not observed.
HRMS (ESI+): m/z [M + H]+ calcd for C11H9F3N3O2: 272.0641; found: 272.0647.
4-Azidobenzonitrile (2q)
Following the general procedure using 4-cyanophenylboronic acid (1q) or 4-cyanophenylboronic acid pinacol ester (4q) for 3 h and chromatography (Et2O) afforded 2q (141 mg, 98% from 1q; 118 mg, 82% from 4q) as a light brown solid; mp 56–59 °C; Rf = 0.4 (hexane–EtOAc, 9:1).
IR: 2112 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.23 (d, J = 8.4 Hz, 2 H), 7.74 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 109.3, 119.5, 121.2, 135.2, 146.8.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C25H19N2P: 379.1359; found: 379.1370.
1-Azido-4-(methylsulfonyl)benzene (2r)
Following the general procedure using 4-(methylsulfonyl)phenylboronic acid (1r) for 3 h and chromatography (10% EtOAc–hexane) afford 2r (166 mg, 84%) as a brown oil; Rf = 0.2 (hexane–EtOAc, 7:3).
IR: 2132 cm−1.
1H NMR (600 MHz, CD3OD): δ = 3.11 (s, 3 H), 7.29 (d, J = 8.4 Hz, 2 H), 7.95 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 44.5, 120.9, 130.6, 138.2, 147.4.
HRMS (ESI+) (iminophosphorane adduct) : m/z [M − N2 + PPh3 + H]+ calcd for C25H22NO2PS: 432.1182; found: 432.1184.
1-Azido-4-nitrobenzene (2s)
Following the general procedure using 4-nitrophenylboronic acid (1s) for 3 h and chromatography (Et2O) afforded 2s (140 mg, 85%) as a yellow solid; mp 63–66 °C; Rf = 0.4 (hexane–EtOAc, 9:1).
IR: 2128 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.27 (d, J = 8.4 Hz, 2 H), 8.28 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 120.9, 126.7, 146.2, 148.6.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C24H19N2O2P: 399.1257; found: 399.1251.
3-Azidobiphenyl (2t)
Following the general procedure using biphenyl-3-ylboronic acid (1t) for 3 h and chromatography (hexane) afforded 2t (90 mg, 46%) as a colorless oil; Rf = 0.4 (hexane).
IR: 2101 cm−1.
1H NMR (600 MHz, CD3OD): δ = 6.98 (d, J = 7.2 Hz, 1 H), 7.16 (s, 1 H), 7.30–7.34 (m, 2 H), 7.37 (m, 3 H), 7.52 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 118.5, 118.8, 124.8, 128.1, 128.9, 130.0, 131.4, 141.4, 141.9, 144.4.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C30H24NP: 430.1719; found: 430.1757.
2-Azidobiphenyl (2u)
Following the general procedure using biphenyl-2-ylboronic acid (1u) for 3 h and chromatography (hexane) afforded 2u (80 mg, 41%) as a colorless oil; Rf = 0.3 (hexane).
IR: 2107 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.19 (t, J = 7.8 Hz, 1 H), 7.26 (d, J = 7.8 Hz, 1 H), 7.29–7.32 (m, 2 H), 7.36–7.39 (m, 5 H).
13C NMR (150 MHz, CD3OD): δ = 120.1, 126.2, 128.5, 129.1, 130.0, 130.6, 132.3, 135.3, 138.4, 139.7.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C30H24NP: 430.1719; found: 430.1713.
1-Azido-N-pivaloylaniline (2v)
Following the general procedure using 2-(pivaloylamino)phenylboronic acid (1v) for 3 h and chromatography (5% EtOAc–hexane) afforded 2v (207 mg, 95%) as a yellow oil; Rf = 0.5 (hexane–EtOAc, 9:1).
IR: 2129 cm−1.
1H NMR (600 MHz, CD3OD): δ = 1.31 (s, 9 H), 7.11 (t, J = 7.8 Hz, 1 H), 7.17 (overlapping d, J = 7.8 Hz, 1 H), 7.19 (overlapping t, J = 7.8 Hz, 1 H), 7.79 (d, J = 7.8 Hz, 1 H).
13C NMR (150 MHz, CD3OD): δ = 27.9, 40.7, 119.6, 126.2, 126.3, 127.2, 130.5, 133.2, 179.5.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C29H29N2OP: 453.2090; found: 453.2116.
4-Azidoaniline (2w)
Following the general procedure using 4-aminophenylboronic acid pinacol ester (4w) for 24 h and chromatography (10% EtOAc–hexane) afforded 2w (127 mg, 95%) as a brown oil; Rf = 0.3 (hexanes–EtOAc, 9:1).
IR: 2108 cm−1.
1H NMR (600 MHz, CD3OD): δ = 6.73 (d, J = 8.4 Hz, 2 H), 6.80 (d, J = 8.4 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 117.8, 120.8, 130.9, 146.6.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C24H21N2P: 369.1515; found: 369.1528.
4-Azido-N,N-dimethylaniline (2x)
Following the general procedure using 4-(dimethylamino)phenylboronic acid pinacol ester (4x) for 24 h and chromatography (15% EtOAc–hexane) afforded 2x (130 mg, 80%) as a yellow solid; mp 35–39 °C; Rf = 0.3 (hexanes–EtOAc, 7:3).
IR: 2087 cm−1.
1H NMR (600 MHz, CD3OD): δ = 2.88 (s, 6 H), 6.77 (d, J = 9.0 Hz, 2 H), 6.89 (d, J = 9.0 Hz, 2 H).
13C NMR (150 MHz, CD3OD): δ = 41.3, 115.4, 120.7, 129.9, 150.1.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C26H25N2P: 397.1828; found: 397.1796.
4-Azido-1-(tert-butoxycarbonyl)-1H-pyrazole (2y)
Following the general procedure using 1-(tert-butoxycarbonyl)-1H-pyrazole-4-boronic acid pinacol ester (2y) for 1 h and chromatography (15% EtOAc–hexane) afforded 2y (50 mg, 24%) as a yellow oil; Rf = 0.2 (hexane–EtOAc, 9:1).
IR: 2119 cm−1.
1H NMR (600 MHz, CD3OD): δ = 1.64 (s, 9 H), 7.73 (s, 1 H), 8.10 (s, 1 H).
13C NMR (150 MHz, CD3OD): δ = 26.8, 86.3, 91.3, 120.6, 136.8, 147.1.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C26H26N3O2P: 444.1835; found: 444.1880.
5-Azido-1-(tert-butoxycarbonyl)-1H-indole (2z)
Following the general procedure using 1-(tert-butoxycarbonyl)-1H-indole-5-boronic acid pinacol ester (1z) for 24 h and chromatography (10% EtOAc–hexane) afforded 2z (135 mg, 52%) as a brown oil; Rf = 0.7 (hexane–EtOAc, 8:2).
IR: 2111 cm−1.
1H NMR (600 MHz, CD3OD): δ = 1.67 (s, 9 H), 6.59 (d, J = 4.2 Hz, 1 H), 6.99 (dd, J = 8.4, 2.4 Hz, 1 H), 7.25 (d, J = 2.4 Hz, 1 H), 7.64 (d, J = 4.2 Hz, 1 H), 8.11 (d, J = 8.4 Hz, 1 H).
13C NMR (150 MHz, CD3OD): δ = 28.5, 85.2, 108.0, 111.7, 116.6, 117.2, 128.3, 133.2, 134.1, 136.2, 150.8.
HRMS (ESI+) (iminophosphorane adduct): m/z [M − N2 + PPh3 + H]+ calcd for C31H29N2O2P: 493.2039; found: 493.2007.
4-Azidopyridine (6a)
Following the general procedure using pyridin-4-ylboronic acid (5a) for 24 h and chromatography (30% EtOAc–hexane) afforded 6a (41 mg, 34%); as a yellow oil Rf = 0.2 (hexanes–EtOAc, 7:3).
IR: 2112 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.14 (d, J = 5.4 Hz, 2 H), 8.47 (br s, 2 H).
13C NMR (150 MHz, CD3OD): δ = 115.9, 151.5, 151.6.
HRMS (ESI+): m/z [M + H]+ calcd for C5H5N4: 121.0509; found: 121.0509.
5-Azidopyrimidine (6b)
Following the general procedure using pyrimidin-5-ylboronic acid (5b) for 24 h and chromatography (30% EtOAc–hexane) afforded 6b (44 mg, 36%) as a white solid; mp 52–57 °C; Rf = 0.3 (hexane–EtOAc, 7:3).
IR: 2110 cm−1.
1H NMR (600 MHz, CD3OD): δ = 8.60 (s, 2 H), 8.92 (s, 1 H).
13C NMR (150 MHz, CD3OD): δ = 138.4, 149.1, 155.3.
HRMS (ESI+): m/z [M + H]+ calcd for C4H4N5: 122.0461; found: 122.0475.
8-Azidoquinoline (6c)
Following the general procedure using quinolin-8-ylboronic acid (5c) for 24 h and chromatography (15% EtOAc–hexane) afforded 6c (60 mg, 35%) as a white solid; mp 54–57 °C; Rf = 0.1 (hexane–EtOAc, 9:1).
IR: 2120 cm−1.
1H NMR (600 MHz, CD3OD): δ = 6.98 (d, J = 7.8 Hz, 1 H), 7.15 (d, J = 8.4 Hz, 1 H), 7.32 (t, J = 7.8 Hz, 1 H), 7.40 (dd, J = 8.4, 4.2 Hz, 1 H), 8.14 (d, J = 8.4 Hz, 1 H), 8.70 (d, J = 4.2 Hz, 1 H).
13C NMR (150 MHz, CD3OD): δ = 111.8, 117.0, 122.5, 128.7, 130.5, 137.6, 139.6, 145.8, 148.5.
HRMS (ESI+): m/z [M + H]+ calcd for C9H7N4: 171.0665; found: 171.0677.
4-Azidoisoquinoline (6d)
Following the general procedure using isoquinolin-4-ylboronic acid (5d) for 24 h and chromatography (10% EtOAc–hexane) afforded 6d (73 mg, 43%) as a white solid; mp 54–56 °C; Rf = 0.2 (hexane–EtOAc, 9:1).
IR: 2123 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.75 (t, J = 7.8 Hz, 1 H), 7.83 (t, J = 7.8 Hz, 1 H), 8.09 (overlapping d, J = 9.0 Hz, 1 H), 8.10 (overlapping d, J = 8.4 Hz, 1 H), 8.39 (s, 1 H), 9.04 (s, 1 H).
13C NMR (150 MHz, CD3OD): δ = 122.6, 128.8, 130.0, 130.3, 130.5, 132.5 (2 C), 134.5, 149.8.
HRMS (ESI+): m/z [M + H]+ calcd for C9H7N4: 171.0665; found: 171.0682.
Acknowledgments
This research was supported by a grant from the NIH (R01AI070219) and funding from the Center for Drug Design, Academic Health Center, University of Minnesota to C.C.A.
References
- 1.Tron GC, Pirali T, Billington RA, Canonico PL, Sorba G, Genazzani AA. Med Res Rev. 2008;28:278. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- 2.For some representative recent examples, see: Gupte A, Boshoff HI, Wilson DJ, Neres J, Labello NP, Somu RV, Xing C, Barry CE, Aldrich CC. J Med Chem. 2008;51:7495. doi: 10.1021/jm8008037.Giffin MJ, Heaslet H, Brik A, Lin YC, Cauvi G, Wong CH, McRee DE, Elder JH, Stout CD, Torbett BE. J Med Chem. 2008;51:6263. doi: 10.1021/jm800149m.Gill C, Jadhav G, Shaikh M, Kale R, Ghawalkar A, Nagargoje D, Shiradkar M. Bioorg Med Chem Lett. 2008;18:6244. doi: 10.1016/j.bmcl.2008.09.096.Klein M, Dinér P, Dorin-Semblat D, Doering C, Grotli M. Org Biomol Chem. 2009;7:3421. doi: 10.1039/b906482f.Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. J Med Chem. 2009;52:4623. doi: 10.1021/jm900410u.Upadhayaya RS, Kulkarni GM, Vasireddy NR, Vandavasi JK, Dixit SS, Sharma V, Chattopadhyaya J. Bioorg Med Chem. 2009;17:4681. doi: 10.1016/j.bmc.2009.04.069.
- 3.Avermaria F, Zimmermann V, Bräse S. Synlett. 2004:1163. [Google Scholar]
- 4.Barral K, Moorhouse AD, Moses JE. Org Lett. 2007;9:1809. doi: 10.1021/ol070527h. [DOI] [PubMed] [Google Scholar]
- 5.Zhu W, Ma D. Chem Commun. 2004:888. doi: 10.1039/b400878b. [DOI] [PubMed] [Google Scholar]
- 6.Morgan J, Pinhey JT. J Chem Soc, Perkin Trans 1. 1990:715. [Google Scholar]
- 7.Huber ML, Pinhey JT. J Chem Soc, Perkin Trans 1. 1990:721. [Google Scholar]
- 8.Tao CZ, Cui X, Li J, Liu AX, Liu L, Guo QX. Tetrahedron Lett. 2007;48:3525. [Google Scholar]
- 9.Chan DMT, Monaco KL, Wang RP, Winters MP. Tetrahedron Lett. 1998;39:2933. [Google Scholar]
- 10.Lam PYS, Clark CG, Saubern S, Adams J, Winters MP, Chan DMT, Combs A. Tetrahedron Lett. 1998;39:2941. [Google Scholar]
- 11.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165. [Google Scholar]
- 12.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- 13.Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 14.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Chan DMT, Lam PYS. Recent Advances in Copper-Promoted C-Heteroatom Bond Cross-Coupling Reactions with Boronic Acids and Derivatives. In: Hall DG, editor. Boronic Acids. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 16.Bertz SH, Cope S, Murphy M, Ogle CA, Taylor BJ. J Am Chem Soc. 2007;129:7208. doi: 10.1021/ja067533d. [DOI] [PubMed] [Google Scholar]
- 17.Pudlo M, Csányi D, Moreau F, Hajós G, Riedl Z, Sapi J. Tetrahedron. 2007;63:10320. [Google Scholar]
- 18.Cho YA, Kim DS, Ahn HR, Canturk B, Molander GA, Ham J. Org Lett. 2009;11:4330. doi: 10.1021/ol901669k. [DOI] [PMC free article] [PubMed] [Google Scholar]