

Abstract
We previously identified the marked upregulation of integrin β4 in human lung endothelial cells (EC) treated with simvastatin, an HMG coA-reductase inhibitor with vascular-protective and anti-inflammatory properties in murine models of acute lung injury (ALI). We now investigate the role of integrin β4 as a novel mediator of vascular inflammatory responses with a focus on MAPK signaling and the downstream expression of the inflammatory cytokines (IL-6, IL-8) essential for the full elaboration of inflammatory lung injury. Silencing of integrin β4 (siITGB4) in human lung EC resulted in significant increases in both basal and LPS-induced phosphorylation of ERK 1/2, JNK, and p38 MAPK, consistent with robust integrin β4 regulation of MAPK activation. In addition, siITB4 increased both basal and LPS-induced expression of IL-6 and IL-8 mRNA and protein secretion into the media. We next observed that integrin β4 silencing increased basal and LPS-induced phosphorylation of SHP-2, a protein tyrosine phosphatase known to modulate MAPK signaling. In contrast, inhibition of SHP-2 enzymatic activity (sodium stibogluconate) abrogated the increased ERK phosphorylation associated with integrin β4-silencing in LPS-treated EC and attenuated the increases in levels of IL-6 and IL-8 in integrin β4-silenced EC. These findings highlight a novel negative regulatory role for integrin β4 in EC inflammatory responses involving SHP-2-mediated MAPK signaling. Upregulation of integrin β4 may represent an important element of the anti-inflammatory and vascular-protective properties of statins and provides a novel strategy to limit inflammatory vascular syndromes.
Keywords: Integrins, Endothelial Cells, Inflammation
Introduction
We previously reported direct vascular protective effects of simvastatin, an HMG CoA-reductase inhibitor, both in vitro [Chen et al., 2008; Jacobson et al., 2004] and in vivo in a murine model of inflammatory lung injury [Jacobson et al., 2005]. In an effort to characterize the mechanisms underlying these effects, we surveyed differential gene expression in endothelial cells (EC) treated with simvastatin and identified the dramatic upregulation of integrin β4, a finding subsequently corroborated by others [Feng et al., 2004]. Accordingly, we hypothesized that integrin β4 may represent an important mediator of statin effects on EC inflammatory responses.
Integrins exist as transmembrane heterodimers consisting of α and β subunits mediating both inside-out and outside-in signaling and are well recognized as modulators of EC cytoskeletal rearrangement, barrier regulation [Eliceiri et al., 2002; Su et al., 2007] and angiogenesis [Hood et al., 2003; Nikolopoulos et al., 2004], and participate in pro-inflammatory pathways. For example, inhibition of integrin β5 attenuates inflammatory lung injury in separate models of rat ischemia-reperfusion lung injury and murine acute lung injury (ALI) [Su et al., 2007]. Additionally, integrin β2 [Xu et al., 2008] and integrin β6 [Ganter et al., 2008] have also been implicated as mediators of increased lung vascular permeability associated with ALI, although the precise mechanisms of these integrin-mediated effects remain to be fully characterized. While 8 β subunits have been identified, integrin β4 is uniquely characterized by its long cytoplasmic tail of over 1000 amino acids [Hogervorst et al., 1990]. The role of integrin β4 in epithelial hemidesmosome formation [Dans et al., 2001; Spinardi et al., 1993] and tumor invasiveness [Kitajiri et al., 2002; Van Waes et al., 1991] is well substantiated and its expression is associated with a poor prognosis in a variety of cancers [Grossman et al., 2000; Tagliabue et al., 1998]. Integrin β4 complexes only with integrin α6 and serves as a laminin receptor that links intracellularly to the cytoskeleton via plectin, an actin-binding protein [Rabinovitz and Mercurio, 1997; Wiche, 1998]. Notably, integrin β4 has been implicated in activation of the Rho GTPases [O'Connor et al., 2000] as well as phosphatidylinositol 3-kinase (PI3-K) [Shaw et al., 1997], mitogen-activated protein kinases (MAPK) [Abdel-Ghany et al., 2002; Mainiero et al., 1997] and NF-KB signaling [Nikolopoulos et al., 2004], pathways actually highly relevant to the propagation of inflammation and injury. However, few reports have examined EC integrin β4 signaling and no study has yet to explore signaling specific to lung vascular EC.
We investigated the role of integrin β4 as a novel mediator of vascular inflammatory responses with a focus on MAPK signaling and the downstream expression of the inflammatory cytokines, IL-6 and IL-8, known to be essential for the full elaboration of inflammatory lung injury [Dolinay et al., 2008; Lee et al., 2006; Loppnow and Libby, 1989; Schuh and Pahl, 2009; Strieter et al., 1989]. EC silencing of integrin β4 (siITGB4) resulted in significant increases in both basal and LPS-induced phosphorylation of ERK 1/2, JNK, and p38 MAPK, consistent with robust MAPK activation. In addition, integrin β4 silencing increased both basal and LPS-induced expression of IL-6 and IL-8 mRNA as well as protein levels of IL-6 and IL-8. We further identified a role for SHP-2, a protein tyrosine phosphatase, in mediating integrin β4 effects on MAPK signaling and IL-6 and IL-8 expression. Together, these findings highlight a novel regulatory role for integrin β4 in EC inflammatory responses involving SHP-2-mediated MAPK signaling.
Materials and Methods
Materials and reagents
Non-specific siRNA (nsRNA) and siRNA specific for integrin β4 (siITGB4) were purchased from Dharmacon (Layfayette, CO) and Ambion (Austin, TX), respectively. All antibodies were purchased from Cell Signaling (Danvers, MA). An RT-PCR kit was purchased from Qiagen (Valencia, CA) and the primers for IL-6 and IL-8 were purchased from IDT (Coralville, IA). ELISA kits for IL-6 and IL-8 were purchased from Biolegend (San Diego, CA) and Abcam (Cambridge, MA), respectively. All other reagents were purchased from Sigma (St. Louis, MO) unless otherwise specified.
Cell culture
Human pulmonary artery EC were purchased from Clonetics (San Diego, CA) and were cultured in EGM-2 supplemented with 2% FBS, hydrocortisone, hFGF, VEGF, ascorbic acid, hEGF, GA-1000, Heparin, R3-IGF-1 (Clonetics). The cells were incubated in 75 cm2 flask and cultured at 37°C in 5% CO2 and 95% air. All cells were used at passages 4-8.
Silencing RNA
The target sequence for siITGB4 was 5′-GAGCUGCACGGAGUGUGUCtt-3′. The target sequence for nsRNA used as a control (Dharmacon) was 5′-UAGCGACUAAACACAUCAA-3′. EC were plated on 6-well plates (60-80% confluent) or 12-well plates and were transfected with nsRNA or siITGB4 at the indicated concentrations using siPORT™ Amine (Ambion). After incubating for 48-72 h, the cells or medium were harvested for protein or ELISA assay.
RT-PCR
EC were grown in 6-well plates and silenced with siRNA as described above. After 48 h cells were treated with LPS for 4 h. The cells were then harvested and total RNA was extracted using a commercially available kit according to the manufacture's instruction. Equal amounts of RNA were used for reverse transcription and 2 μl RT product was then used for PCR. After the strands were allowed to denature for 10 min at 94°C, PCR was performed at 94°C (30 sec), 56 °C (60 sec), and 75 °C (60 sec). The PCR primers used were: IL-6 forward: 5-TCAATGAGGAGACTTGCCTGGT-3, backward: 5-ACAGCTCTGGCTTGTTCCTCAC-3; IL-8 forward: 5-CGATGTCAGTGCATAAAGACA, backward: 5-TGAATTCTCAGCCCTCTTCAAAAA-3; GAPDH forward: 5-GTCTTCACCACCATGGAGAA-3, backward: 5-ATCCACAGTCTTCTGGGTGG-3.
Measurement of IL-6 and IL-8 in the media
EC were grown to 80% confluence and silenced with siRNA as described above. After 72 h the cells were treated with LPS for 4 h in 2 ml complete EGM medium. After briefly centrifuging the medium to remove dead cells, the medium was collected to measure IL-6 or IL-8 concentration using commercially available ELISA kits according to the manufacturer's instruction.
Western Blotting
Samples were harvested with RIPA buffer containing proteinase inhibitors and phosphatase inhibitors as per standard protocols. After sonication and centrifugation, the supernant was collected, Laemmli sample buffer added, and was then boiled and subsequently analyzed by SDS-PAGE. After transfer to a nitrocellulose membrane (Bio-Rad Inc., Hercules, CA), Western blotting was performed using appropriate primary antibodies and horseradish peroxidase-conjugated secondary antibodies prior to visualization via chemiluminescence (Amersham Biosciences, Piscataway, NJ). Blot density was determined by Alpha Imager software (Alpha Innotech, San Leandro, CA).
Statistical analysis
Pairwise comparisons were made using student's t test. One-way ANOVA with post hoc Tukey HSD tests were used for groupwise comparisons. P < 0.05 was considered statistically significant. Results are expressed as means +/- SD.
Results
Integrin β4 negatively regulates LPS-induced EC MAPK signaling
Initial studies determined the optimal timing of LPS-mediated MAPK activation with kinase-specific time points of LPS-induced (5 μg/ml) activation of ERK, JNK, and p-38 (Figure 1). These results provided the kinetics required to assess the effect of integrin β4 silencing on maximal and submaximal LPS-mediated MAPK activation.
Figure 1. Differential LPS-induced EC MAPK signaling.
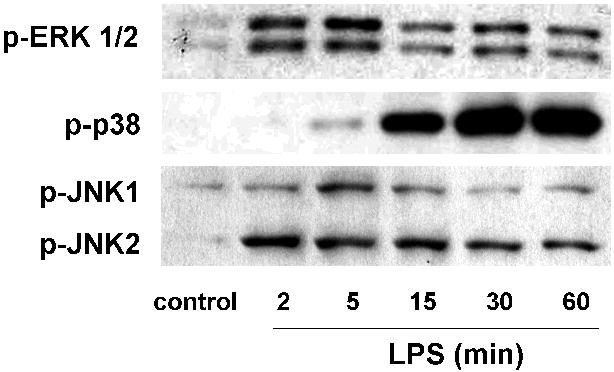
Human pulmonary artery EC were treated with LPS (5 μg/ml) at variable times to identify the optimal timing to evaluate ERK, p38, and JNK phosphorylation. Maximal ERK phosphorylation was noted at 5 min while p38 phosphorylation was marked at 15 min and peaked at 30 min. Total JNK phosphorylation was most prominent at 5-15 min (representative blots shown). The dosing of 5 μg/ml LPS was chosen as it was associated with a consistent response across each MAPK (data not shown).
We next investigated the role of integrin β4 on ERK 1/2 activation and its upstream mediators in EC both under basal conditions and upon stimulation with LPS. Compared to controls, LPS alone (5 μg/ml, 5 min) effected a significant increase in ERK 1/2 phosphorylation (Figure 2). Silencing of EC with siRNA specific for integrin β4 (siITGB4, 100 nM, 3 d) attenuated protein expression by ∼80% and resulted in a significant increase in ERK 1/2 phosphorylation both under basal conditions and in response to LPS compared to controls transfected with non-specific siRNA (nsRNA, 100 nM, 3 d). In addition, silencing of integrin β4 resulted in increased basal and LPS-induced phosphorylation of c-Raf and MEK 1/2, mediators upstream of ERK 1/2 signaling. However, there was no evidence of increased Ras activation (Ras-GTP) in integrin β4-silenced EC suggesting that integrin β4 mediates ERK signaling downstream of Ras (data not shown).
Figure 2. Role of Integrin β4 in EC ERK 1/2 Activation.
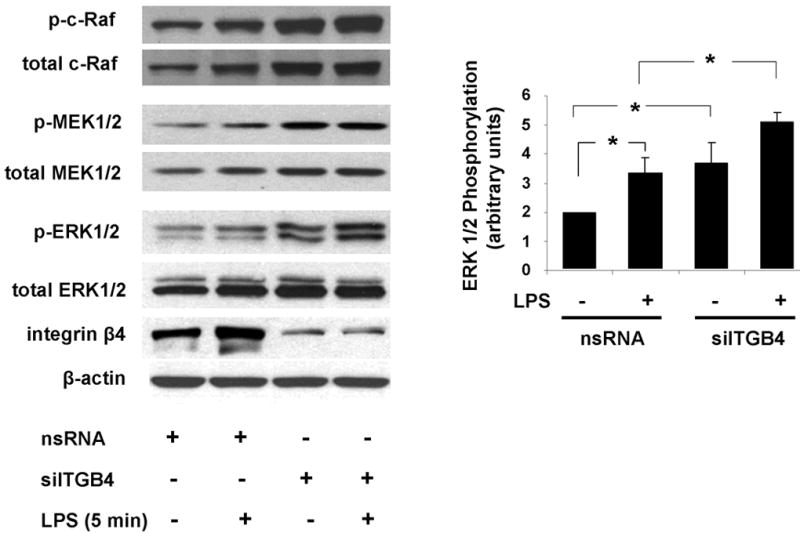
ERK 1/2 phosphorylation was measured in EC transfected with nsRNA (100 nM, 3 d) or siITGB4 (100 nM, 3 d) both under basal conditions and in response to LPS treatment (5 μg/ml, 5 min). Relative to controls, siITGB4 resulted in a significant increase in both basal and LPS-induced ERK 1/2 phosphorylation (* p < 0.05, n = 3 / experimental condition). Similar results were observed with respect to phosphorylation of c-Raf and MEK 1/2.
Similar experiments were conducted to examine the contribution of integrin β4 to basal and LPS-induced EC JNK and p38 MAPK activity. Silencing of integrin β4 resulted in significant increases in both basal and LPS-induced (5 μg/ml, 10 min) JNK phosphorylation (Figure 3). Comparable changes were also detected with respect to p38 phosphorylation with significantly increased levels evident in response to LPS alone (5 μg/ml, 20 min) as well as after silencing of integrin β4, both under basal conditions and after LPS compared to the respective controls (Figure 4). These data support a negative regulatory role for integrin β4 on EC MAPK signaling.
Figure 3. Role of Integrin β4 in EC JNK Activation.
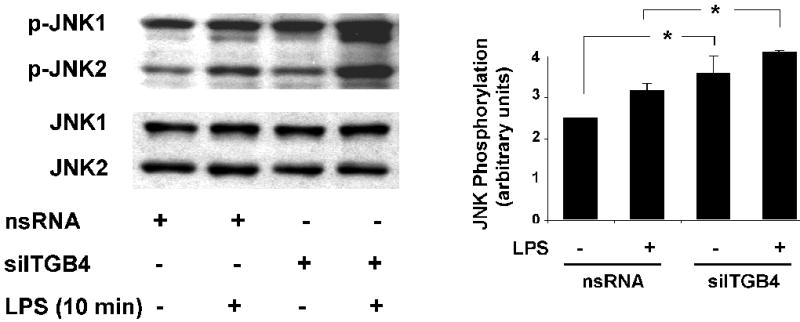
JNK phosphorylation was measured in EC transfected with nsRNA (100 nM, 3 d) or siITGB4 (100 nM, 3 d) both under basal conditions and in response to LPS treatment (5 μg/ml, 10 min). Relative to controls, siITGB4 resulted in a significant increase in both basal and LPS-induced total JNK phosphorylation (* p < 0.05, n = 3 / experimental condition).
Figure 4. Role of Integrin β4 in EC p38 Activation.
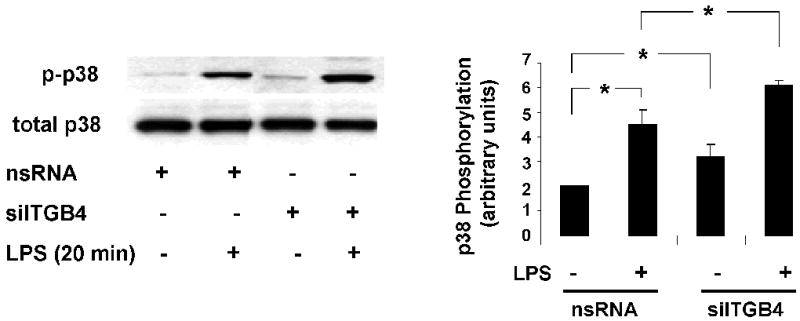
Phosphorylation of p38 MAPK was measured in EC transfected with nsRNA (100 nM, 3 d) or siITGB4 (100 nM, 3 d) both under basal conditions and in response to LPS treatment (5 μg/ml, 10 min). Both basal and LPS-induced (5 μg/ml, 20 min) p38 phosphorylation was significantly increased in integrin β4-silenced cells compared to controls (* p < 0.05, n = 3 / experimental condition).
Reductions in integrin β4 expression are associated with augmented EC IL-6 and IL-8 expression
Upregulation of IL-6 and IL-8 cytokines are relevant to the pathophysiology of ALI and EC inflammation and a downstream product of MAPK activation. We next assessed the role of integrin β4 in EC IL-6 and IL-8 expression and found that IL-6 and IL-8 mRNA were significantly increased in integrin β4-silenced cells (Figure 5A). In addition, the significant increase in IL-6 and IL-8 mRNAs induced by LPS (100 ng/ml, 4 h) was further augmented in integrin β4-silenced cells and reflected by comparable increased levels of IL-6 and IL-8 protein secreted into the media (Figure 5B).
Figure 5. Role of Integrin β4 in EC IL-6 and IL-8 Expression.
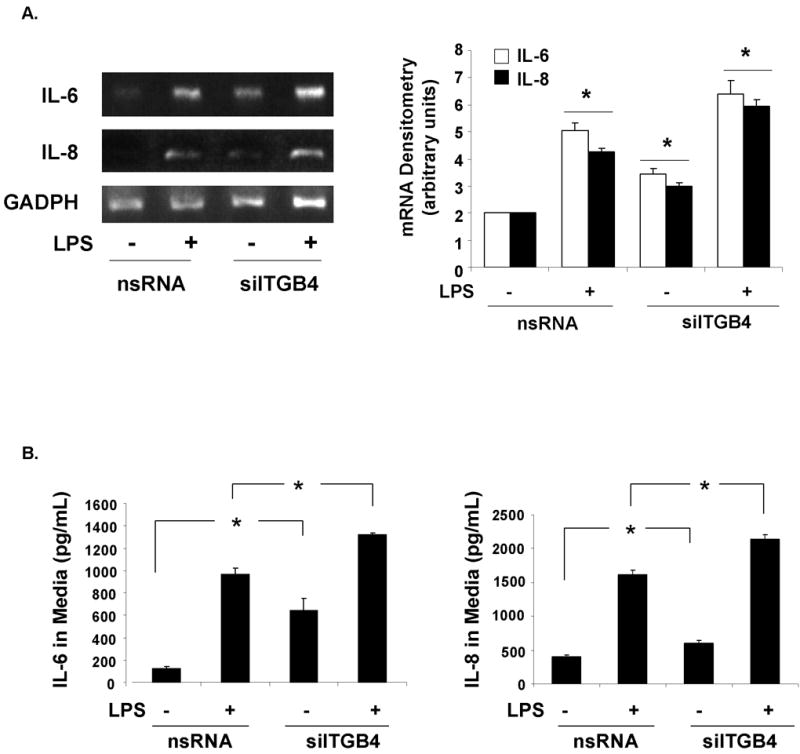
(A) RT-PCR of IL-6 and IL-8 mRNA demonstrate a significant upregulation of both in EC transfected with siITGB4 (100 nM, 3d) compared to controls transfected with nsRNA (100 nM, 3 d) both under basal conditions and upon LPS (100 ng/ml, 4 h) stimulation (* p < 0.05 compared to the respective controls, n = 3 / experimental group). (B) Measurements of IL-6 and IL-8 levels in the media of EC transfected with siITGB4 (100 nM, 3d) were also significantly increased compared to controls (nsRNA) both under basal conditions and upon LPS (100 ng/ml, 4 h) stimulation.
Reduced integrin β4 is associated with augmented SHP2-mediated MAPK signaling
The protein tyrosine phosphatase Src homology 2 phosphatase 2 (SHP-2) is a known mediator of integrin β4 MAPK signaling via activation of Src [Bertotti et al., 2006; Oh et al., 1999]. We examined the role of SHP-2 in integrin β4-regulated EC MAPK signaling. While LPS alone (5 μg/ml, 5 min) was associated with increased SHP-2 phosphorylation, silencing of integrin β4 resulted in a significant increase in both basal and LPS-induced SHP-2 phosphorylation (Figure 6A), effects that were abrogated by sodium stibogluconate (SS, 500 μM, 30 min), a pharmacologic SHP inhibitor. We then assessed the effect of SHP-2 inhibition on EC basal and LPS-induced (5 μg/ml, 5 min) ERK 1/2 phosphorylation in integrin β4-silenced cells (Figure 6B). Stibogluconate treatment (500 μM, 30 min) fully inhibited the augmented LPS-induced ERK 1/2 phosphorylation associated with integrin β4 silencing.
Figure 6. Regulation of Integrin β4-Mediated MAPK Signaling by SHP-2.
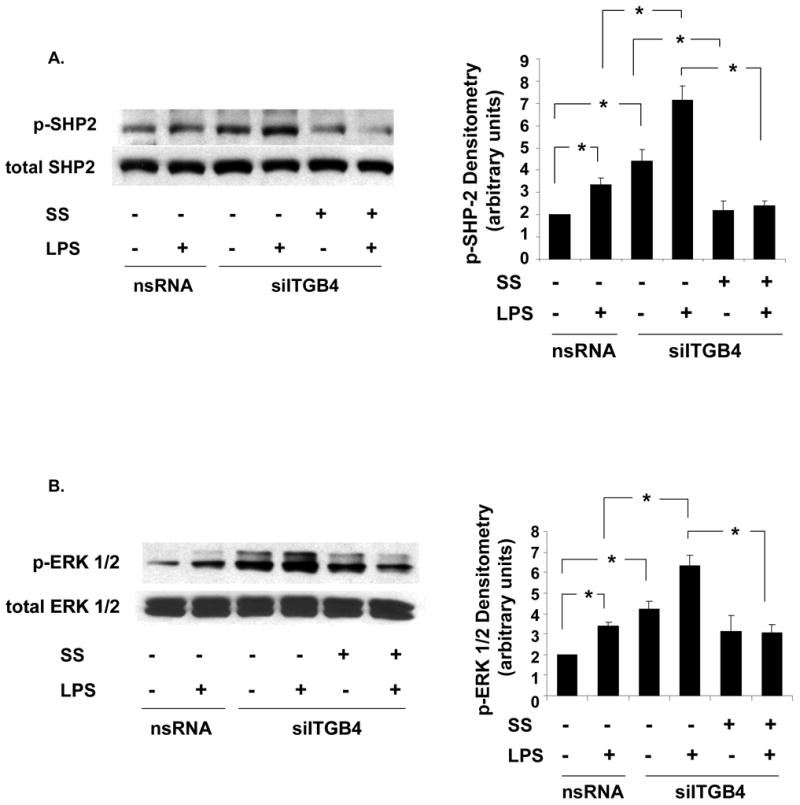
(A) SHP-2 phosphorylation is significantly increased in integrin β4-silenced EC (siITGB4, 100 nm, 3 d) both under basal conditions and in response to LPS (5 μg/ml, 5 min) stimulation while treatment with sodium stibogluconate (SS, 500 μM, 30 min) abrogates these effects (* p < 0.05, n = 3 / experimental condition). (B) Similarly, ERK 1/2 phosphorylation is significantly increased in integrin β4-silenced EC (siITGB4, 100 nm, 3 d) both under basal conditions and in response to LPS (5 μg/ml, 5 min) stimulation while treatment with SS (500 μM, 30 min) fully inhibits LPS-induced ERK phospohrylation in these cells (* p < 0.05, n = 3 / experimental condition).
Integrin β4 negatively regulates IL-6 and IL-8 expression via SHP-2
Finally, we measured the effects of SHP-2 inhibition on increased IL-6 and IL-8 expression in integrin β4-silenced EC. Comparable to the effects on ERK 1/2, treatment with SS (500 μM, 30 min) fully inhibited the augmented LPS-induced (200 ng/ml, 4 h) IL-6 and IL-8 expression in integrin β4-silenced EC (Figure 7). Indeed, the delayed time course of these effects relative to that observed with MAPK signaling is consistent with the additional time required for protein synthesis.
Figure 7. Regulation of Integrin β4-Mediated IL-6 and IL-8 Expression by SHP-2.
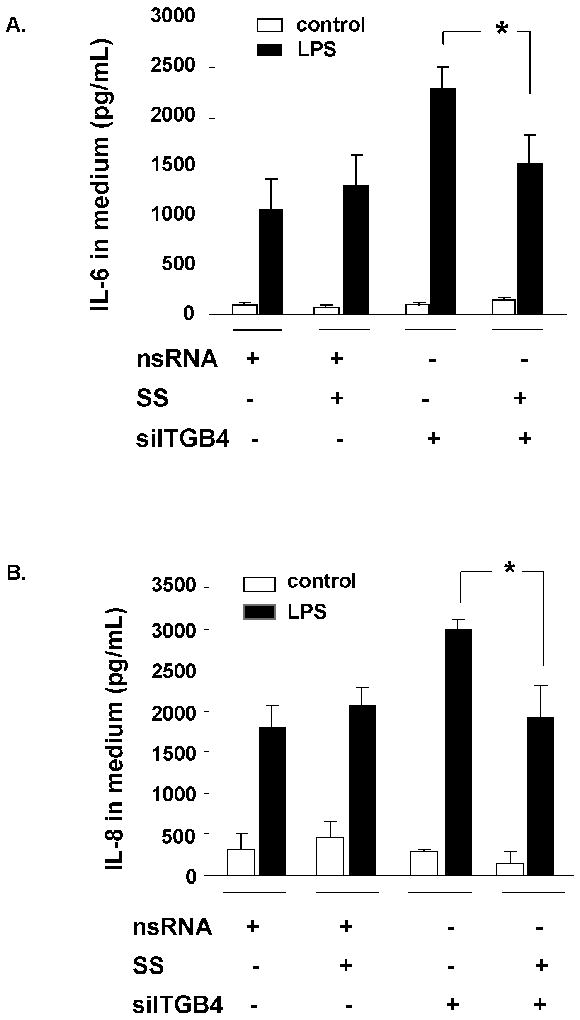
Increased LPS-induced (100 ng/ml, 4 h) IL-6 (A) and IL-8 (B) levels measured in the media of EC transfected with siITGB4 (100 nM, 3d) is significantly inhibited by SS (500 μM, 30 min) treatment (* p < 0.05, n = 3 / experimental condition). SS had no effect in control EC transfected with nsRNA (100 nM, 3d).
Discussion
This study addresses the role of integrin β4, a gene and protein strongly induced by simvastatin treatment in endothelium, as a mechanism for the observed vascular protective and anti-inflammatory properties of statins. Although the role of integrin β4 in EC inflammatory responses has not been previously addressed, we now report the negative regulation of EC MAPK signaling by integrin β4 in the context of LPS stimulation. Integrin β4 functions as a 1brake” on LPS-induced inflammatory pathways. With integrin β4 silencing, the loss of this inhibition results in MAPK activation and increased expression of inflammatory cytokines such as IL-6 and IL-8. The mechanism by which integrin β4 produces MAPK activities and inflammatory cytokine production appears to be associated with the protein tyrosine phosphatase SHP-2.
We recognize potential discrepancies between our findings and other reports of integrin β4 signaling utilizing different cell types and in vitro models. For example, tyrosine phosphorylation of the integrin β4 cytoplasic domain (not evaluated in our studies) has been linked to subsequent binding of SHP-2 and MAPK activation [Bertotti et al., 2006; Dans et al., 2001; Mainiero et al., 2003; Merdek et al., 2007]. Nonetheless, examples of phenotypic differences involving the regulation of specific signaling pathways in different cell types are abundant in the literature and it is certainly no surprise that the attributes of integrin β4 in the context of EC inflammatory responses is distinct (or even opposite) to that characterized in models relevant to cancer biology. Moreover, the idea that endothelial integrin β4 may have unique properties is supported by evidence of their failure to form type I hemidesmosomes as observed in stratified epithelial cells [Hieda et al., 1992; Uematsu et al., 1994].
Our findings with integrin β4 also represent a notable contrast relative to reports involving other integrin β subunits, namely integrin β2 [Xu et al., 2008], β5 [Su et al., 2007], and β6 [Ganter et al., 2008], each of which have been shown to propagate EC inflammatory responses. This may be related to the unique structure of integrin β4, as the extracellular portion of the β4 subunit sequence exhibits only 35% identity with other integrin beta subunits and is the most different among this class of molecules. [Hogervorst et al., 1990]. The transmembrane region is poorly conserved, whereas the cytoplasmic domain shows no substantial identity in any region to the cytoplasmic tails of the known β sequences or to other protein sequences. The exceptionally long cytoplasmic domain of integrin β4 suggests unique interactions of the β4 subunit with cytoplasmic proteins.
Our results firmly link EC integrin β4 to the negative regulation of SHP-2-mediated MAPK signaling and the downstream expression of the inflammatory cytokines IL-6 and IL8 and are consistent with what would be predicted on the basis of the vascular-protective effects of statins in association with the upregulation of integrin β4. Indeed, this is supported by reports of the inhibition of agonist-induced EC IL-8 expression by statins associated with reduced levels of ERK, p38, and JNK phosphorylation [Kibayashi et al., 2005]. In light of our previously published findings of the protective effects of statins in murine ALI [Jacobson et al., 2005] and the mounting evidence suggesting a potential beneficial effect of statins in patient populations at risk for ALI [Falagas et al., 2008], our findings may have direct clinical relevance and ultimately lead to novel therapeutic strategies for ALI directed specifically at integrin β4 signaling.
In summary, we explored potential mechanisms for the influence of integrin β4 on MAPK activities and observed that integrin β4 silencing results in decreased basal and LPS-induced phosphorylation of SHP-2, a protein tyrosine phosphatase known to mediate integrin β4-MAPK signaling. These findings highlight a novel regulatory role for integrin β4 in EC inflammatory responses involving SHP-2-mediated MAPK signaling. Upregulation of integrin β4 by statins may represent an important element of the anti-inflammatory and vascular-protective properties of these drugs and may provide novel strategies designed to limit vascular inflammatory syndromes.
Acknowledgments
Contract Grant Sponsor: NHLBI
Contract Grant Numbers: HL 77134, HL 58064, HL 96887
This work also acknowledges support from the the Dr. Lowell T Coggeshall Endowment (JGNG)
References
- Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. Focal adhesion kinase activated by beta(4) integrin ligation to mCLCA1 mediates early metastatic growth. J Biol Chem. 2002;277:34391–400. doi: 10.1074/jbc.M205307200. [DOI] [PubMed] [Google Scholar]
- Bertotti A, Comoglio PM, Trusolino L. Beta4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J Cell Biol. 2006;175:993–1003. doi: 10.1083/jcb.200605114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Pendyala S, Natarajan V, Garcia JG, Jacobson JR. Endothelial cell barrier protection by simvastatin: GTPase regulation and NADPH oxidase inhibition. Am J Physiol Lung Cell Mol Physiol. 2008;295:L575–83. doi: 10.1152/ajplung.00428.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dans M, Gagnoux-Palacios L, Blaikie P, Klein S, Mariotti A, Giancotti FG. Tyrosine phosphorylation of the beta 4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J Biol Chem. 2001;276:1494–502. doi: 10.1074/jbc.M008663200. [DOI] [PubMed] [Google Scholar]
- Dolinay T, Wu W, Kaminski N, Ifedigbo E, Kaynar AM, Szilasi M, Watkins SC, Ryter SW, Hoetzel A, Choi AM. Mitogen-activated protein kinases regulate susceptibility to ventilator-induced lung injury. PLoS One. 2008;3:e1601. doi: 10.1371/journal.pone.0001601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, Cheresh DA. Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J Cell Biol. 2002;157:149–60. doi: 10.1083/jcb.200109079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas ME, Makris GC, Matthaiou DK, Rafailidis PI. Statins for infection and sepsis: a systematic review of the clinical evidence. J Antimicrob Chemother. 2008;61:774–85. doi: 10.1093/jac/dkn019. [DOI] [PubMed] [Google Scholar]
- Feng C, Ye C, Liu X, Ma H, Li M. Beta4 integrin is involved in statin-induced endothelial cell death. Biochem Biophys Res Commun. 2004;323:858–64. doi: 10.1016/j.bbrc.2004.08.171. [DOI] [PubMed] [Google Scholar]
- Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, Sheppard D, Violette SM, Weinreb PH, Horan GS, Matthay MA, Pittet JF. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res. 2008;102:804–12. doi: 10.1161/CIRCRESAHA.107.161067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman HB, Lee C, Bromberg J, Liebert M. Expression of the alpha6beta4 integrin provides prognostic information in bladder cancer. Oncol Rep. 2000;7:13–6. [PubMed] [Google Scholar]
- Hieda Y, Nishizawa Y, Uematsu J, Owaribe K. Identification of a new hemidesmosomal protein, HD1: a major, high molecular mass component of isolated hemidesmosomes. J Cell Biol. 1992;116:1497–506. doi: 10.1083/jcb.116.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogervorst F, Kuikman I, von dem Borne AE, Sonnenberg A. Cloning and sequence analysis of beta-4 cDNA: an integrin subunit that contains a unique 118 kd cytoplasmic domain. Embo J. 1990;9:765–70. doi: 10.1002/j.1460-2075.1990.tb08171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JD, Frausto R, Kiosses WB, Schwartz MA, Cheresh DA. Differential alphav integrin-mediated Ras-ERK signaling during two pathways of angiogenesis. J Cell Biol. 2003;162:933–43. doi: 10.1083/jcb.200304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson JR, Barnard JW, Grigoryev DN, Ma SF, Tuder RM, Garcia JG. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1026–32. doi: 10.1152/ajplung.00354.2004. [DOI] [PubMed] [Google Scholar]
- Jacobson JR, Dudek SM, Birukov KG, Ye SQ, Grigoryev DN, Girgis RE, Garcia JG. Cytoskeletal activation and altered gene expression in endothelial barrier regulation by simvastatin. Am J Respir Cell Mol Biol. 2004;30:662–70. doi: 10.1165/rcmb.2003-0267OC. [DOI] [PubMed] [Google Scholar]
- Kibayashi E, Urakaze M, Kobashi C, Kishida M, Takata M, Sato A, Yamazaki K, Kobayashi M. Inhibitory effect of pitavastatin (NK-104) on the C-reactive-protein-induced interleukin-8 production in human aortic endothelial cells. Clin Sci (Lond) 2005;108:515–21. doi: 10.1042/CS20040315. [DOI] [PubMed] [Google Scholar]
- Kitajiri S, Hosaka N, Hiraumi H, Hirose T, Ikehara S. Increased expression of integrin beta-4 in papillary thyroid carcinoma with gross lymph node metastasis. Pathol Int. 2002;52:438–41. doi: 10.1046/j.1440-1827.2002.01379.x. [DOI] [PubMed] [Google Scholar]
- Lee PJ, Zhang X, Shan P, Ma B, Lee CG, Homer RJ, Zhu Z, Rincon M, Mossman BT, Elias JA. ERK1/2 mitogen-activated protein kinase selectively mediates IL-13-induced lung inflammation and remodeling in vivo. J Clin Invest. 2006;116:163–73. doi: 10.1172/JCI25711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loppnow H, Libby P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell Immunol. 1989;122:493–503. doi: 10.1016/0008-8749(89)90095-6. [DOI] [PubMed] [Google Scholar]
- Mainiero F, Colombara M, Antonini V, Strippoli R, Merola M, Poffe O, Tridente G, Ramarli D. p38 MAPK is a critical regulator of the constitutive and the beta4 integrin-regulated expression of IL-6 in human normal thymic epithelial cells. Eur J Immunol. 2003;33:3038–48. doi: 10.1002/eji.200323931. [DOI] [PubMed] [Google Scholar]
- Mainiero F, Murgia C, Wary KK, Curatola AM, Pepe A, Blumemberg M, Westwick JK, Der CJ, Giancotti FG. The coupling of alpha6beta4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. Embo J. 1997;16:2365–75. doi: 10.1093/emboj/16.9.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdek KD, Yang X, Taglienti CA, Shaw LM, Mercurio AM. Intrinsic signaling functions of the beta4 integrin intracellular domain. J Biol Chem. 2007;282:30322–30. doi: 10.1074/jbc.M703156200. [DOI] [PubMed] [Google Scholar]
- Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Giancotti FG. Integrin beta4 signaling promotes tumor angiogenesis. Cancer Cell. 2004;6:471–83. doi: 10.1016/j.ccr.2004.09.029. [DOI] [PubMed] [Google Scholar]
- O'Connor KL, Nguyen BK, Mercurio AM. RhoA function in lamellae formation and migration is regulated by the alpha6beta4 integrin and cAMP metabolism. J Cell Biol. 2000;148:253–8. doi: 10.1083/jcb.148.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh ES, Gu H, Saxton TM, Timms JF, Hausdorff S, Frevert EU, Kahn BB, Pawson T, Neel BG, Thomas SM. Regulation of early events in integrin signaling by protein tyrosine phosphatase SHP-2. Mol Cell Biol. 1999;19:3205–15. doi: 10.1128/mcb.19.4.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitz I, Mercurio AM. The integrin alpha6beta4 functions in carcinoma cell migration on laminin-1 by mediating the formation and stabilization of actin-containing motility structures. J Cell Biol. 1997;139:1873–84. doi: 10.1083/jcb.139.7.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh K, Pahl A. Inhibition of the MAP kinase ERK protects from lipopolysaccharide-induced lung injury. Biochem Pharmacol. 2009;77:1827–34. doi: 10.1016/j.bcp.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 1997;91:949–60. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Spinardi L, Ren YL, Sanders R, Giancotti FG. The beta 4 subunit cytoplasmic domain mediates the interaction of alpha 6 beta 4 integrin with the cytoskeleton of hemidesmosomes. Mol Biol Cell. 1993;4:871–84. doi: 10.1091/mbc.4.9.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strieter RM, Kunkel SL, Showell HJ, Remick DG, Phan SH, Ward PA, Marks RM. Endothelial cell gene expression of a neutrophil chemotactic factor by TNF-alpha, LPS, and IL-1 beta. Science. 1989;243:1467–9. doi: 10.1126/science.2648570. [DOI] [PubMed] [Google Scholar]
- Su G, Hodnett M, Wu N, Atakilit A, Kosinski C, Godzich M, Huang XZ, Kim JK, Frank JA, Matthay MA, Sheppard D, Pittet JF. Integrin alphavbeta5 regulates lung vascular permeability and pulmonary endothelial barrier function. Am J Respir Cell Mol Biol. 2007;36:377–86. doi: 10.1165/rcmb.2006-0238OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabue E, Ghirelli C, Squicciarini P, Aiello P, Colnaghi MI, Menard S. Prognostic value of alpha 6 beta 4 integrin expression in breast carcinomas is affected by laminin production from tumor cells. Clin Cancer Res. 1998;4:407–10. [PubMed] [Google Scholar]
- Uematsu J, Nishizawa Y, Sonnenberg A, Owaribe K. Demonstration of type II hemidesmosomes in a mammary gland epithelial cell line, BMGE-H. J Biochem. 1994;115:469–76. doi: 10.1093/oxfordjournals.jbchem.a124361. [DOI] [PubMed] [Google Scholar]
- Van Waes C, Kozarsky KF, Warren AB, Kidd L, Paugh D, Liebert M, Carey TE. The A9 antigen associated with aggressive human squamous carcinoma is structurally and functionally similar to the newly defined integrin alpha 6 beta 4. Cancer Res. 1991;51:2395–402. [PubMed] [Google Scholar]
- Wiche G. Role of plectin in cytoskeleton organization and dynamics. J Cell Sci. 1998;111(Pt 17):2477–86. doi: 10.1242/jcs.111.17.2477. [DOI] [PubMed] [Google Scholar]
- Xu J, Gao XP, Ramchandran R, Zhao YY, Vogel SM, Malik AB. Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating beta2 integrins. Nat Immunol. 2008;9:880–6. doi: 10.1038/ni.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]