

Abstract
A randomized, controlled clinical trial established the efficacy and safety of short-term use of hydroxyurea in adult sickle cell anemia. To examine the risks and benefits of long-term hydroxyurea usage, patients in this trial were followed for 17.5 years during which they could start or stop hydroxyurea. The purpose of this follow-up was to search for adverse outcomes and estimate mortality. For each outcome and for mortality, exact 95% confidence intervals were calculated, or tests were conducted at α = 0.05 level (p-value <0.05 for statistical significance). Although the death rate in the overall study cohort was high (43.1%; 4.4 per 100 person-years), mortality was reduced in individuals with long-term exposure to hydroxyurea. Survival curves demonstrated a significant reduction in deaths with long-term exposure. Twenty-four percent of deaths were due to pulmonary complications; 87.1% occurred in patients who never took hydroxyurea or took it for <5 years. Stroke, organ dysfunction, infection and malignancy were similar in all groups. Our results, while no longer the product of a randomized study because of the ethical concerns of withholding an efficacious treatment, suggest that long-term use of hydroxyurea is safe and might decrease mortality.
Introduction
Hydroxyurea was introduced as a treatment for sickle cell anemia (homozygosity for HBB glu6val) more than 25 years ago based on its ability to increase fetal hemoglobin (HbF) level.[1] In a randomized, controlled trial in adults (the Multicenter Study of Hydroxyurea in Sickle Cell Anemia or MSH), hydroxyurea was associated with a reduction in acute painful episodes and acute chest syndrome events, and HbF levels increased.[2-4] Observational studies in children suggested that hydroxyurea was safe and beneficial.[5] Because of the concern that hydroxyurea, a cell cycle-specific agent, might cause longer-term adverse effects [6], MSH patients were followed after the controlled trial ended. Although patients were followed prospectively using a predetermined protocol, ethical tenets required that treatment no longer be randomized. Therefore, hydroxyurea was used according to patient and physician discretion and all subsequent results must be interpreted with this caveat. After 9 years of follow-up, we found that mortality was related to HbF concentration and the rate of acute painful episodes and acute chest syndrome, and neither untoward reproductive outcomes nor an increased incidence of neoplasia were observed. Analysis of this first follow-up interval suggested that patients who took hydroxyurea for at least 1 year had a 40% reduction in mortality.[7]
To further examine the risks and benefits of long-term hydroxyurea we extended follow-up of MSH patients to 17.5 years. This second follow-up protocol was different and its focus was to; identify serious complications of treatment, detect organ dysfunction, assess all cause mortality, and classify causes of death. As in the initial 9 year follow-up, patients were no longer randomized and most, but not all patients elected to either continue hydroxyurea at doses recommended by their treating physicians, or to start hydroxyurea anew. Laboratory tests were done for clinical purposes and were not required for the extended follow-up. Because of this design, our observational study cannot provide the same strength of evidence for harm or benefit as would a randomized, controlled trial with a single well-defined outcome measure. Nevertheless, it can screen for findings that might warrant further investigation. Our results are consistent with the initial post-randomization follow-up interval and continue to suggest that long-term use of hydroxyurea is safe and might reduce mortality.
Methods
The objective of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia (MSH) Patients' Follow-Up was to determine whether there were long-term adverse outcomes of treatment of sickle cell anemia with hydroxyurea occurring between the date patients were enrolled in the MSH clinical trial and end of the patient follow-up (August 27, 2009). Patients were seen annually when data on hydroxyurea use and reportable events were collected.[7] Although this observational study was prospective in nature, hydroxyurea use and clinical events were determined retrospectively on an annual basis. The retrospective nature of this data makes the determination of actual hydroxyurea dose less reliable than assessment of monthly drug use.
Exploratory statistical methods were used to screen a large number of potential adverse outcomes. For each outcome, exact 95% confidence intervals were calculated, or tests were conducted at α = 0.05 level (p-value <0.05 for statistical significance). Significant findings should only be regarded as indicators of association. Therefore, this study will not provide the same strength of evidence for harm or benefit as would be obtained from a randomized trial with a single well-defined outcome measure, but can screen for findings that might warrant further investigation.
Patients were enrolled in the MSH clinical trial at an approximately uniform rate over the course of 15 months, and were followed in the MSH Patients' Follow-Up to a common closing date. Thus, even if there were no patients lost to observation (e.g., through death) for some outcomes, follow-up times would be unequal. For this reason, event rates are reported as “number per 100 person-years.” Two-sided exact 95% confidence limits were calculated when one or more occurrences of a given type of event were observed. If no occurrences were observed, a one-sided exact 95% confidence limit was calculated. Exact 95% confidence intervals for the difference between event rates were used to perform pair-wise comparisons between the “a priori” cumulative hydroxyurea exposure groups (two-sided).
The Product-Limit (Kaplan-Meier) method [8] was used to examine the relationship between cumulative hydroxyurea usage throughout the course of the patient's involvement with the clinical trial and the patient follow-up study with mortality. The following tests were used to detect differences in mortality between the various cumulative hydroxyurea usage categories: The log-rank, Gehan-Breslow Generalized Wilcoxon, and Tarone-Ware tests. The log-rank test is sensitive to differences in the right tails of survival distributions (i.e., at later times), the Gehan-Breslow Generalized Wilcoxon test gives more weight to earlier failures (deaths), and the Tarone-Ware test falls in between.[9-14]
The Kruskal-Wallis test [15] was used to compare the baseline categorical distributions of the patients who were in each of the cumulative hydroxyurea exposure categories considered in our analysis. Analysis of variance was used to compare continuous measurements. The cumulative hydroxyurea use categories are based on each patient's known cumulative exposure to hydroxyurea from enrollment in the MSH clinical trial and throughout the entire MSH Patients' Follow-Up. The following “a priori” defined cumulative hydroxyurea exposure categories were used: never (zero years of cumulative exposure); years<5 (denoted as <5 years); 5≤years<10 (denoted as 5-10 years); 10≤years<15 (denoted as 10-15 years; years ≥15 (denoted as ≥15 years).
These studies were approved by the Institutional Review Boards of all participating institutions.
Results
The MSH patients were followed for 17 years and 7 months (January 28, 1992 through August 27, 2009). Two hundred ninety-nine patients made up the original MSH study population. Table I presents the characteristics of the patients at baseline according to cumulative hydroxyurea exposure at the end of follow-up. No significant differences were present in baseline data when patients were grouped by cumulative hydroxyurea exposure. No significant difference in baseline age was found among cumulative hydroxyurea exposure groups (p=0.48) suggesting that the reduction in mortality found in the long-term cumulative hydroxyurea exposure categories is not an effect of age.
Table I.
Characteristics of the Patients at Base Line, According to Cumulative Hydroxyurea Exposure. Baseline rows show values obtained prior to randomization to receive hydroxyurea or a placebo. Columns shw the cumulative hydroxyurea use.
| Characteristic | Never (1) | Yrs<5 (2) | 5<=yrs<10 (3) | 10<=yrs<15 (4) | Yrs>=15 (5) | p-value * |
|---|---|---|---|---|---|---|
| N=44 | N=140 | N=55 | N=40 | N=20 | ||
| Age | ||||||
| <20 | 4 (9.1) | 10 (7.1) | 5 (9.1) | 2 (5.0) | 0 | 0.4792 |
| 20<=age<30 | 24 (54.5) | 60 (42.9) | 25 (45.5) | 13 (32.5) | 10 (50.0) | |
| 30<=age<40 | 12 (27.3) | 56 (40.0) | 17 (30.9) | 21 (52.5) | 9 (45.0) | |
| 40<=age<50 | 2 (4.5) | 13 (9.3) | 6 (10.9) | 4 (10.0) | 1 (5.0) | |
| 50<=age<60 | 2 (4.5) | 1 (0.7) | 2 (3.6) | 0 | 0 | |
| Gender | ||||||
| Male | 21 (47.7) | 61 (43.6) | 33 (60.0) | 20 (50.0) | 11 (55.0) | 0.1871 |
| Female | 23 (52.3) | 79 (56.4) | 22 (40.0) | 20 (50.0) | 9 (45.0) | |
| No. of crises in the year before study entry (n, % of patients) | ||||||
| 3-5 | 22 (50.0) | 53 (37.8) | 22 (40.0) | 20 (50.0) | 11 (55.0) | 0.6557 |
| 6-9 | 13 (29.5) | 35 (25.0) | 13 (23.6) | 9 (22.5) | 4 (20.0) | |
| >=10 | 9 (20.4) | 52 (37.1) | 20 (36.4) | 11 (27.5) | 5 (25.0) | |
| Complications of sickle cell anemia (n,% of patients) | ||||||
| Chest syndrome | 29 (65.9) | 97 (69.3) | 34 (61.8) | 20 (50.0) | 15 (75.0) | 0.2626 |
| Ankle ulcer | 12 (27.3) | 43 (30.7) | 17 (30.9) | 13 (32.5) | 2 (10.0) | 0.6387 |
| Aseptic necrosis of bone | 9 (20.4) | 34 (24.3) | 13 (23.6) | 5 (12.5) | 2 (10.0) | 0.2082 |
| Mean reported for lab parameters | Pair-wise comparison significant at the 0.05 level # | |||||
| Hemoglobin (g/dl) | 8.5 | 8.4 | 8.6 | 8.3 | 9.3 | 1-5 2-5 3-5 4-5 |
| MCV (μm3) | 92.2 | 93.2 | 93.7 | 93.0 | 96.4 | None |
| WBC (10-3/mm3) | 12.0 | 12.2 | 13.5 | 12.0 | 12.6 | 1-3 2-3 3-4 |
| Platelets (10-3/mm3) | 492.4 | 463.1 | 476.1 | 501.3 | 487.4 | None |
| Reticulocytes (10-3/mm3) | 319.3 | 309.4 | 351.0 | 341.2 | 354.2 | 2-3 2-5 |
| Fetal Hemoglobin (%) | 5.9 | 4.5 | 5.3 | 5.3 | 6.0 | 1-2 |
| F cells (%) | 35.1 | 29.6 | 35.2 | 34.1 | 35.9 | 2-3 |
| Dense cells (%) | 15.3 | 14.2 | 13.1 | 12.7 | 13.5 | None |
p-value (2-sided) determined by the Kruskal-Wallis test
p-value (2-sided) determined by least significant difference; ANOVA.
Table IIA shows that overall, 43.1% of the original MSH cohort has died (129/299). This corresponds to a rate of 4.4 per 100 person-years. Also shown in Table IIA is the cumulative event rates for the 17.5 years MSH experience. There were 14 patients who had at least one reported incident of a stroke (a cumulative total of 18 episodes; 6.0%; a rate of 0.6 per 100 person-years); 16 patients with renal dysfunction (17.4%; a rate of 1.8 per 100 person-years); 7 patients with hepatic dysfunction (6.0%; a rate of 0.6 per 100 person-years); 3 patients who had a reported malignancy (1.0%; a rate of 0.1 per 100 person-years); and 23 patients who had at least one reported incident of sepsis/infection (18.4%; a rate of 1.9 per 100 person-years). Of the 3 patients with malignancies, 1 woman died due to metastatic cancer (uterine cancer: malignant mixed Müllerian tumor). She was in the 5-10 cumulative hydroxyurea exposure category. Another woman was diagnosed with breast cancer (she was in the <5 years cumulative hydroxyurea exposure category) and a third woman was diagnosed with metastatic adenocarcinoma, primary unknown (she was in the 5-10 years cumulative hydroxyurea exposure category).
Table II. Cumulative Event Rates during the MSH Trial and Follow-Up by Cumulative Hydroxyurea Exposure.
(A) The first column shows death and events by total enrollment in the MSH trial, column 2 shows death and events by patients randomized to hydroxyurea (HU) during the MSH trial, and column 3 shows death and events by patients randomized to placebo (PL) during the MSH trial. The remaining columns consider the ‘a priori’ defined cumulative hydroxyurea exposure categories. (B) Event Rates per 100 Person-years (Exact 95% confidence limits for events; n per 100 person-years)
| Years on HU in MSH Trial, Follow Up, and Extension I | ||||||||
|---|---|---|---|---|---|---|---|---|
| Event | TOTAL | HU | PL | Never | yr<5 | 5<=yr<10 | 10<=yr<15 | yr>=15 |
| 299 | 152 | 147 | 44 | 140 | 55 | 40 | 20 | |
| Total years from MSH enrollment to death or last live Follow-Up | 2909 | 1536 | 1373 | 321 | 1152 | 593 | 506 | 337 |
| Death | 129 | 60 | 69 | 16 | 78 | 26 | 9 | 0 |
| n / N (%) | 43.1 | 39.5 | 46.9 | 36.4 | 55.7 | 47.3 | 22.5 | 0.0 |
| n / 100 person-years | 4.4 | 3.9 | 5.0 | 5.0 | 6.8 | 4.4 | 1.8 | 0.0 |
| Stroke | 18 | 11 | 7 | 0 | 12 | 3 | 0 | 3 |
| n / N (%) | 6.0 | 7.2 | 4.8 | 0.0 | 8.6 | 5.5 | 0.0 | 15.0 |
| n / 100 person-years | 0.6 | 0.7 | 0.5 | 0.0 | 1.0 | 0.5 | 0.0 | 0.9 |
| Renal Disease | 52 | 29 | 23 | 9 | 27 | 10 | 5 | 1 |
| n / N (%) | 17.4 | 19.1 | 15.6 | 20.5 | 19.3 | 18.2 | 12.5 | 5.0 |
| n / 100 person-years | 1.8 | 1.9 | 1.7 | 2.8 | 2.3 | 1.7 | 1.0 | 0.3 |
| Hepatic Disease | 18 | 7 | 11 | 5 | 9 | 3 | 1 | 0 |
| n / N (%) | 6.0 | 4.6 | 7.5 | 11.4 | 6.4 | 5.5 | 2.5 | 0.0 |
| n / 100 person-years | 0.6 | 0.5 | 0.8 | 1.6 | 0.8 | 0.5 | 0.2 | 0.0 |
| Malignancy | 3 | 2 | 1 | 0 | 1 | 2 | 0 | 0 |
| n / N (%) | 1.0 | 1.3 | 0.7 | 0.0 | 0.7 | 3.6 | 0.0 | 0.0 |
| n / 100 person-years | 0.1 | 0.1 | 0.1 | 0.0 | 0.1 | 0.3 | 0.0 | 0.0 |
| Sepsis / Infection | 55 | 28 | 27 | 3 | 27 | 15 | 10 | 0 |
| n / N (%) | 18.4 | 18.4 | 18.4 | 6.8 | 19.3 | 27.3 | 25.0 | 0.0 |
| n / 100 person-years | 1.9 | 1.8 | 2.0 | 0.9 | 2.3 | 2.5 | 2.0 | 0.0 |
Stroke: 14 patients with 18 episodes
Renal Disease: 16 patients with 52 episodes
Hepatic Disease: 7 patients with 18 episodes
Malignancy: 3 patients with 3 episodes
Sepsis/Infection: 23 patients with 55 episodes
The exact 95% confidence interval for mortality and morbidity by years of cumulative hydroxyurea exposure are shown in Table IIB. The exact 95% confidence intervals for the difference between event rates (3 pair-wise comparisons) indicated possible evidence for reduced mortality when long-term exposure to hydroxyurea occurred.
Figure 1 illustrates the cumulative mortality for the MSH patients with respect to the “a priori” defined cumulative hydroxyurea exposure categories. The tests used to detect differences in mortality between the various cumulative hydroxyurea usage categories (the log-rank, Gehan-Breslow Generalized Wilcoxon, and Tarone-Ware tests) all had a p-value<.0001 providing additional evidence for a possible reduction in mortality with long-term exposure to hydroxyurea.
Figure 1.
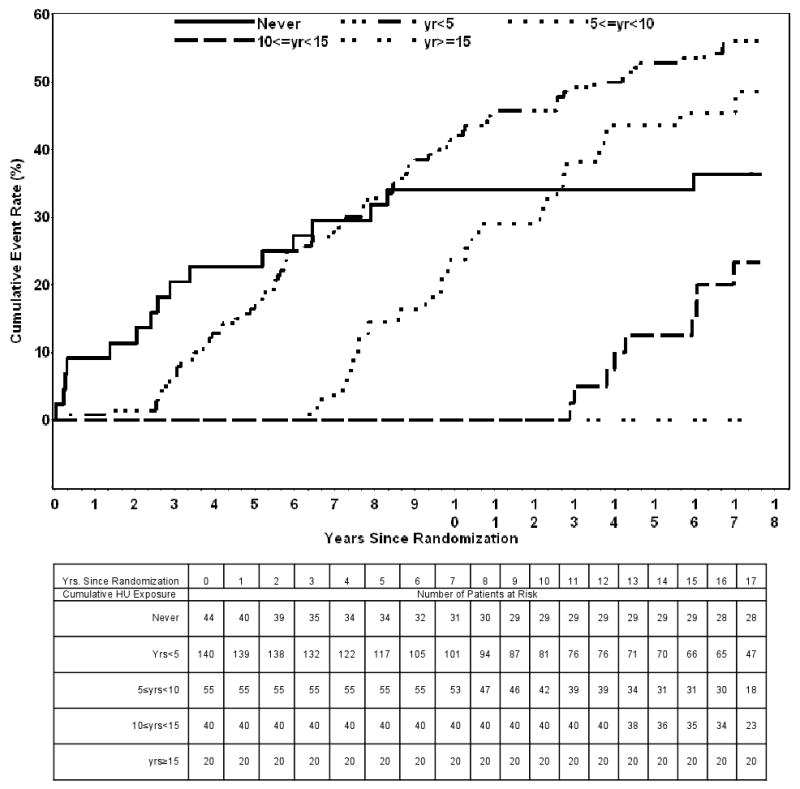
Cumulative Mortality During the MSH Follow-Up by Cumulative Hydroxyurea Exposure.
Cause of death was also assessed with respect to cumulative hydroxyurea exposure (Table III). There were no reported deaths for the group of patients who had a cumulative hydroxyurea exposure of ≥15 years. Twenty-four percent (31/129) of the deaths were due to pulmonary complications. The majority (87.1%) of these 31 deaths occurred in patients never exposed to hydroxyurea and with <5 years exposure. Only 2 deaths due to pulmonary complications occurred in the patient group that had a cumulative hydroxyurea exposure of 5-10 years, 2 deaths occurred in the group 10-15 years, and none occurred in the group years ≥15 years. Twenty-five (19.4%) of the reported deaths were not classified because they were determined with the use of the Social Security Death Index (SSDI) that does not provide a cause of death.
Table 3.
Event Rates per 100 Person-years [Exact 95% Confidence Limits for Events]., Cause of Death According to Cumulative Hydroxyurea Exposure. (129 deaths)
| Event | Never (1) 321 Person-Years | yrs<5 (2) 1152 Person Years | 5≤yrs<10 (3) 593 Person-Years | 10≤yrs<15 (4) 506 Person-Years | yrs≥15 (5) 337 Person-Years |
|---|---|---|---|---|---|
| Death | 4.98 [2.85, 8.09] | 6.77 [5.35, 8.45] | 4.39 [2.87, 6.44] | 1.78 [0.81, 3.38] | 0 [0, 1.09] |
| Stroke | 0 [0, 1.15] | 1.04 [0.54, 1.82] | 0.51 [0.10, 1.48] | 0 [0, 0.73] | 0.89 [0.18, 2.60] |
| Renal Disease | 2.80 [1.28, 5.32] | 2.34 [1.54, 3.41] | 1.69 [0.81, 3.11] | 0.99 [0.32, 2.31] | 0.30 [0.01, 1.65] |
| Hepatic Disease | 1.56 [0.52, 3.64] | 0.78 [0.36, 1.48] | 0.51 [0.10, 1.48] | 0.20 [0.01, 1.10] | 0 [0, 1.09] |
| Malignancy | 0 [0, 1.15] | 0.09 [0, 0.48] | 0.34 [0.04, 1.22] | 0 [0, 0.73] | 0 [0, 1.09] |
| Sepsis/Infection | 0.94 [0.19, 2.73] | 2.34 [1.54, 3.41] | 2.53 [1.42, 4.18] | 1.98 [0.95, 3.63] | 0 [0, 1.09] |
The exact 95% confidence intervals for a difference between event rates (per 100 person-years) indicated a significant difference between the following cumulative HU exposure groups:
Death: 1 and 4, 1 and 5, 2 and 3, 2 and 4, 2 and 5, 3 and 4, 3 and 5, 4 and 5.
Stroke: 1 and 2, 2 and 4.
Renal Disease: 1 and 5, 2 and 4, 2 and 5, 3 and 5.
Hepatic Disease: 1 and 5, 2 and 5.
Malignancy: None.
Sepsis/Infection: 1 and 2, 2 and 5, 3 and 5, 4 and 5.
Discussion
A consensus exists that hydroxyurea should be more widely used.[16, 17] Our follow-up of more than 17 years provides additional evidence that in adults with sickle cell anemia, where treatment is started in the 4th decade of life, this drug is safe and its use also appears to be associated with reduced mortality. (Figure 1, Table III) Supporting these observations, in 330 Greeks with sickle cell disease, predominantly HbS-β+ and HbS-β0 thalassemia (less than 20% of treated patients had sickle cell anemia), 131 received hydroxyurea, based on the severity of symptoms of disease.[18] Treated patients were compared with less symptomatic cases and were followed for a median time of 8 years, compared with 5 years for untreated patients. Along with a reduction of the incidence of painful episodes, blood transfusion, hospitalizations, and incidence of acute chest syndrome, the probability of 10 year survival was 86% in treated cases compared with 65% in those not treated. As in our study, hydroxyurea treatment was safe.
The long-term effects of hydroxyurea started early in life on morbidity and mortality is a question unlikely to ever be answered by a randomized controlled trial. It is therefore critical to examine our results and the results of other observational trials given the limitations of the study design; some shortcomings of the MSH trial and follow-up have already been discussed.[7, 19] MSH results might not apply to patients with different constellations of symptoms, different ages and individuals with HbSC disease. In the follow-up study, comparisons of patients were no longer randomized so patients who might receive more medical care while taking hydroxyurea might have lived longer for reasons other than their hydroxyurea treatment. MSH patients were adults with frequent painful episodes, which are one predictor of mortality; as the study progressed, patient numbers fell due to death and losses to follow-up.[20] The 69 (23.1%) patients lost to follow-up after 17.5 years were still alive, however their updated hydroxyurea usage data, clinical outcomes and reproductive events were not available. Cumulative hydroxyurea exposure was reported as a range of years because for many study participants some hydroxyurea use was not always reported. Also, hydroxyurea use was often intermittent. Laboratory data were not collected according to protocol so HbF level and mean corpuscular volume cannot be used to gauge compliance with treatment. Notwithstanding these caveats, our observational data provides a “real world” understanding of long-term hydroxyurea exposure, albeit in patients who initially volunteered for a controlled clinical trial. It also speaks to effectiveness and safety since the conditions of a randomized clinical trial seldom reflect clinical practice.
The cumulative hydroxyurea exposure for each patient is an accurate minimum exposure. For example, a patient with “≥15 years” has at least 15 years total drug exposure while one with “10-15” has at least 10 years total exposure (Tables II, III). Thus, reasonable conclusions can be drawn regarding long-term hydroxyurea exposure. Short-term and mid-term “cumulative hydroxyurea exposure” groups could have a maximum exposure greater than that defined. For example, a patient with “5-10 years” has an accurate minimum of 5 years but the maximum could be 10 years or greater. Cumulative hydroxyurea exposure categories “never”, “<5 years”, and “5-10 years have been discussed.[7]
In all MSH studies, teratogenic changes were not present in the pregnancies that resulted in live births regardless of whether the mother or the father was treated.[21] Among newborns there were no developmental delays up to age 36 months.[20] Of 22 offspring of study participants (a subset of the known MSH offspring), aged from 6 months to 17 years, Vineland Adaptive Behavior Scales showed that they were functioning in the average range of development. Since all offspring were not studied the these results are not definitive.[22]
After 17.5 years of follow-up, hydroxyurea was associated with improved survival without accompanying serious adverse events. Importantly, there was no indication that the incidence of serious complications like stroke, infections or neoplasia were increased by exposure to this drug (Table II). The absence of serious complications coupled with the suggestion that mortality was reduced raises the question of whether the indications for treatment of adults with sickle cell anemia should be expanded to include individuals without frequent vasoocclusive events. For example, patients with rare painful episodes but other complications like priapism [23], liver disease [24], proteinuria [25], severe chronic anemia associated with mild azotemia [26], or pulmonary hypertension [27] could be reasonable candidates for long-term hydroxyurea treatment. The intensity of hemolysis might contribute to sickle vasculopathy and is associated with these disease complications and with survival.[28, 29] As one example of the often silent but inexorable progression of sickle vasculopathy, pulmonary hypertension is usually not clinically evident but is a major risk factor for mortality.[30-32] In the Greek study, a fall in serum LDH level—a reasonable marker of intravascular hemolysis—in response to hydroxyurea, along with HbF were the best predictors of survival.
HbF levels are associated with decreased morbidity and mortality in sickle cell anemia and sufficiently high concentrations of HbF evenly distributed among sickle erythrocytes can totally abrogate the disease pathophysiology.[20, 33, 35] Agents that stimulate higher HbF levels, might alter the fundamental course of disease by inhibiting HbS polymerization and its untoward effects. In hydroxyurea-treated patients, as HbF increases, hemolysis is usually reduced, as reflected by an increased hemoglobin concentration with reduced bilirubin level and reticulocyte count.[2, 3, 36] Although some questions remain about the ameliorative effect of HbF on some disease events [30-32, 37] and if increased HbF is the only beneficial effect of hydroxyurea.[38-41], with early treatment, certain features of sickle vasculopathy might be reversible.[42-44] If sufficiently high levels of HbF can be induced in most erythrocytes early in life, the progression of sickle vasculopathy and its associated morbidity and mortality might be forestalled.
A randomized, double-blind placebo controlled trial of hydroxyurea in unselected babies with sickle cell anemia has been completed (Clinical Trials.gov; NCT 00006400).[45] Preliminary analysis showed that the hematological changes and clinical results were very closely aligned with those of hydroxyurea-treated MSH patients.(Wong, W, 4th Annual Sickle Cell Disease Research and Education Symposium, Feb 14-19, 2010, Hollywood FL) Although these and other short term studies have a very favorable safety profile, the intermediate and long-term adverse effects of hydroxyurea given to young patients and continued for decades remains to be determined.
Untreated sickle cell anemia is a lethal disease with a high mortality rate, especially in patients with frequent vasculopathic and vasoocclusive events. Even with so-called “mild disease,” it is the rare patient that escapes these complications.[29, 33, 47] Besides drug therapy, transfusions and stem cell transplantation can be efficacious but each has complications.[48] Severely affected adult patients in the fourth decade of life, like those enrolled in the MSH and in the Greek trial, are arguably the least likely group of individuals to benefit from hydroxyurea or any drug whose mechanism of action is to induce higher levels of HbF. Nevertheless, in the MSH follow-up, over 17.5 years of observation, individuals with the greatest exposure to the drug appeared to have improved survival raising the question of whether the indications for this drug should be expanded. The relentless progression of vasculopathic complications of disease, suggest that most adult patients with sickle cell anemia and HbS-β0 thalassemia could be treated with hydroxyurea with likely benefit while pediatric studies to date strongly argue for beginning this drug even earlier in life before the development of irreversible vasculopathy and organ damage.
Acknowledgments
Supported by NHLBI Contract NO1-HB-6712
Footnotes
Authorship contributions: MHS, WFM and MAW wrote and revised the manuscript. WFM and MAW were responsible for the statistical analyses and for developing the study protocol. All other authors revised the manuscript and were involved in patient follow-up.
Disclosures: There are no conflicts of interest to report.
References
- 1.Steinberg MH. Management of sickle cell disease. N Engl J Med. 1999;340:1021–1030. doi: 10.1056/NEJM199904013401307. [DOI] [PubMed] [Google Scholar]
- 2.Charache S, Terrin ML, Moore RD, et al. Design of the multicenter study of hydroxyurea in sickle cell anemia. Controlled Clin Trials. 1995;16:432–446. doi: 10.1016/s0197-2456(95)00098-4. [DOI] [PubMed] [Google Scholar]
- 3.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332:1317–1322. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 4.Steinberg MH, Lu ZH, Barton FB, et al. Fetal hemoglobin in sickle cell anemia: Determinants of response to hydroxyurea. Blood. 1997;89:1078–1088. [PubMed] [Google Scholar]
- 5.Strouse JJ, Lanzkron S, Beach MC, et al. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics. 2008;122:1332–1342. doi: 10.1542/peds.2008-0441. [DOI] [PubMed] [Google Scholar]
- 6.Timson J. Hydroxyurea. Mutat Res. 1975;32:115–132. doi: 10.1016/0165-1110(75)90002-0. [DOI] [PubMed] [Google Scholar]
- 7.Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–1651. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 8.Klein JP, Moeschberger ML. Survival Analysis: Techniques for Censored and Truncated Data. New York: Springer-Verlag; 1997. [Google Scholar]
- 9.Gail MH, Green SB, Byar DP. Comparison of four tests for equality of survival curves in the presence of stratification and censoring. Biometrika. 1979;66:419–428. [Google Scholar]
- 10.Harrington DP, Fleming TR. A class of rank test procedures for censored survival data. Biometrika. 1982;69:553–566. [Google Scholar]
- 11.Kalbfleisch JD, Prentice RL. The Statistical Analysis of Failure Time Data. New York: John Wiley & Sons, Inc.; 1980. [Google Scholar]
- 12.Peto R, Peto J. Asymptotically efficient rank invariant test procedures. J Royal Stat Soc, A. 1972;135:185–206. [Google Scholar]
- 13.Lee ET, Desu MM, Gehan EA. A Monte Carlo study of the power of some two-sample tests. Biometrika. 1975;62:425–432. [Google Scholar]
- 14.Tarone R, Ware J. On distribution-free tests for equality of survival distributions. Biometrika. 1977;64:156–160. [Google Scholar]
- 15.Siegel S, Castellan NJ. Nonparametric statistics for the behavioral sciences. 2nd. McGraw-Hill; NY: 1988. [Google Scholar]
- 16.Brawley OW, Cornelius LJ, Edwards LR, et al. National Institutes of Health Consensus Development Conference statement: hydroxyurea treatment for sickle cell disease. Ann Intern Med. 2008;148:932–938. doi: 10.7326/0003-4819-148-12-200806170-00220. [DOI] [PubMed] [Google Scholar]
- 17.Lanzkron S, Strouse JJ, Wilson R, et al. Systematic review: Hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med. 2008;148:939–955. doi: 10.7326/0003-4819-148-12-200806170-00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle-cell syndromes: results of a 17-year, single center trial (LaSHS) Blood. 2009 Nov 10; doi: 10.1182/blood-2009-05-221333. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 19.DeBaun MR, Field JJ. Limitations of clinical trials in sickle cell disease: a case study of the Multi-center Study of Hydroxyurea (MSH) trial and the Stroke Prevention (STOP) trial. Hematology Am Soc Hematol Educ Program. 2007:482–488. doi: 10.1182/asheducation-2007.1.482. [DOI] [PubMed] [Google Scholar]
- 20.Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease-rates and risk factors. N Engl J Med. 1991;325:11–16. doi: 10.1056/NEJM199107043250103. [DOI] [PubMed] [Google Scholar]
- 21.Ballas SK, McCarthy WF, Guo N, et al. Exposure to hydroxyurea and pregnancy outcomes in patients with sickle cell anemia. J Natl Med Assoc. 2009;101:1046–1051. doi: 10.1016/s0027-9684(15)31072-5. [DOI] [PubMed] [Google Scholar]
- 22.Armstrong FD, Steinberg M, Ballas S, et al. Developmental outcomes of offspring of adults treated with hydroxyurea on the multicenter study of hydroxyurea. ASH. 2009 abstract 1543. [Google Scholar]
- 23.Saad ST, Lajolo C, Grilli S, et al. Follow-up of sickle cell disease patients with priapism treated by hydroxyurea. Am J Hematol. 2004;77:45–49. doi: 10.1002/ajh.20142. [DOI] [PubMed] [Google Scholar]
- 24.Jeng MR, Rieman MD, Naidu PE, et al. Resolution of chronic hepatic sequestration in patients with homozygous sickle cell disease receiving hydroxyurea. J Pediatr Hematol Oncol. 2003;25:257–260. doi: 10.1097/00043426-200303000-00015. [DOI] [PubMed] [Google Scholar]
- 25.Fitzhugh CD, Wigfall DR, Ware RE. Enalapril and hydroxyurea therapy for children with sickle nephropathy. Pediatr Blood cancer. 2005;45:982–985. doi: 10.1002/pbc.20296. [DOI] [PubMed] [Google Scholar]
- 26.Little JA, McGowan VR, Kato GL, et al. Combination erythropoietin-hydroxyurea therapy in sickle cell disease: experience from the National Institutes of Health and a literature review. Haematologica. 2006;91:1076–1083. [PMC free article] [PubMed] [Google Scholar]
- 27.Olnes M, Chi A, Haney C, et al. Improvement in hemolysis and pulmonary arterial systolic pressure in adult patients with sickle cell disease during treatment with hydroxyurea. Am J Hematol. 2009;84:530–532. doi: 10.1002/ajh.21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor JG, Nolan VG, Mendelsohn L, Kato GJ, Gladwin MT, Steinberg MH. Chronic hyper-hemolysis in sickle cell anemia: association of vascular complications and mortality with infrequent vasoocclusive pain. PLoS One. 2008;3(5):e2095. doi: 10.1371/journal.pone.0002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350:886–895. doi: 10.1056/NEJMoa035477. [DOI] [PubMed] [Google Scholar]
- 31.Ataga KI, Moore CG, Jones S, et al. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br J Haematol. 2006;134:109–115. doi: 10.1111/j.1365-2141.2006.06110.x. [DOI] [PubMed] [Google Scholar]
- 32.Machado RF, Anthi A, Steinberg MH, et al. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA. 2006;296:310–318. doi: 10.1001/jama.296.3.310. [DOI] [PubMed] [Google Scholar]
- 33.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:639–644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 34.Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: Incidence and risk factors. Blood. 1994;84:643–649. [PubMed] [Google Scholar]
- 35.Murray N, Serjeant BE, Serjeant GR. Sickle cell-hereditary persistence of fetal haemoglobin and its differentiation from other sickle cell syndromes. Br J Haematol. 1988;69:89–92. doi: 10.1111/j.1365-2141.1988.tb07607.x. [DOI] [PubMed] [Google Scholar]
- 36.Charache S, Barton FB, Moore RD, et al. Hydroxyurea and sickle cell anemia - Clinical utility of a myelosuppressive “switching” agent. Medicine (Baltimore) 1996;75:300–326. doi: 10.1097/00005792-199611000-00002. [DOI] [PubMed] [Google Scholar]
- 37.Aleem A, Jehangir A, Owais M, et al. Echocardiographic abnormalities in adolescent and adult Saudi patients with sickle cell disease. Saudi Med J. 2007;28:1072–1075. [PubMed] [Google Scholar]
- 38.Huang Z, Louderback JG, King SB, Ballas SK, Kim-Shapiro DB. In vitro exposure to hydroxyurea reduces sickle red blood cell deformability. Am J Hematol. 2001;67:151–156. doi: 10.1002/ajh.1098. [DOI] [PubMed] [Google Scholar]
- 39.Benkerrou M, Delarche C, Brahimi L, et al. Hydroxyurea corrects the dysregulated L-selectin expression and increased H(2)O(2) production of polymorphonuclear neutrophils from patients with sickle cell anemia. Blood. 2002;99:2297–2303. doi: 10.1182/blood.v99.7.2297. [DOI] [PubMed] [Google Scholar]
- 40.Cokic VP, Andric SA, Stojilkovic SS, Noguchi CT, Schechter AN. Hydroxyurea nitrosylates and activates soluble guanylyl cyclase in human erythroid cells. Blood. 2008;111:1117–1123. doi: 10.1182/blood-2007-05-088732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood. 1996;88:4701–4710. [PubMed] [Google Scholar]
- 42.Dham N, Ensing G, Minniti C, et al. Prospective echocardiography assessment of pulmonary hypertension and its potential etiologies in children with sickle cell disease. Am J Cardiol. 2009;104:713–720. doi: 10.1016/j.amjcard.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee MT, Small T, Khan MA, Rosenzweig EB, Barst RJ, Brittenham GM. Doppler-defined pulmonary hypertension and the risk of death in children with sickle cell disease followed for a mean of three years. Br J Haematol. 2009;146:437–441. doi: 10.1111/j.1365-2141.2009.07779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pashankar FD, Carbonella J, Bazzy-Asaad A, Friedman A. Longitudinal follow up of elevated pulmonary artery pressures in children with sickle cell disease. Br J Haematol. 2009;144:736–741. doi: 10.1111/j.1365-2141.2008.07501.x. [DOI] [PubMed] [Google Scholar]
- 45.Thompson BW, Miller ST, Rogers ZR, et al. The pediatric hydroxyurea phase III clinical trial (BABY HUG): challenges of study design. Pediatr Blood Cancer. doi: 10.1002/pbc.22269. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voskaridou E, Kalotychou V, Loukopoulos D. Clinical and laboratory effects of long-term administration of hydroxyurea to patients with sickle-cell/β-thalassaemia. Br J Haematol. 1995;89:479–484. doi: 10.1111/j.1365-2141.1995.tb08352.x. [DOI] [PubMed] [Google Scholar]
- 47.Sebastiani P, Nolan VG, Baldwin CT, et al. A network model to predict the risk of death in sickle cell disease. Blood. 2007;110:2727–2735. doi: 10.1182/blood-2007-04-084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of Hemoglobin: Genetics, Pathophysiology, Clinical Management. 2nd. Cambridge: Cambridge University Press; 2009. [Google Scholar]