

Abstract
RAGE, the Receptor for Advanced Glycation End Products, is a signaling receptor protein of the immunoglobulin superfamily implicated in multiple pathologies. It binds a diverse repertoire of ligands, but the structural basis for the interaction of different ligands is not well understood. We earlier showed that carboxylated glycans on the V-domain of RAGE promote the binding of HMGB1 and S100A8/A9. Here we study the role of these glycans on the binding and intracellular signaling mediated by another RAGE ligand, S100A12. S100A12 binds carboxylated glycans, and a subpopulation of RAGE enriched for carboxylated glycans shows more than ten fold higher binding potential for S100A12 than total RAGE. When expressed in mammalian cells, RAGE is modified by complex glycans predominantly at the first glycosylation site (N25IT) that retains S100A12 binding. Glycosylation of RAGE and maximum binding sites for S100A12 on RAGE are also cell type dependent. Carboxylated glycan-enriched population of RAGE forms higher order multimeric complexes with S100A12, and this ability to multimerize is reduced upon deglycosylation or by using non-glycosylated sRAGE expressed in E.coli. mAbGB3.1, an antibody against carboxylated glycans, blocks S100A12 mediated NF-κB signaling in HeLa cells expressing full length RAGE. These results demonstrate that carboxylated N-glycans on RAGE enhance binding potential and promote receptor clustering and subsequent signaling events following oligomeric S100A12 binding.
Keywords: S100A12, RAGE, receptor clustering, intracellular signaling, glycans
INTRODUCTION
RAGE is a cell surface signaling receptor implicated in multiple pathologies including inflammation, cancer, diabetes and amyloidoses [Bierhaus and Nawroth, 2009; Sparvero et al., 2009; Yan et al., 2009b]. Although first described as a receptor for advanced glycation end products (AGE), it is now clear that RAGE is a multi-ligand receptor for an expanding array of ligands including several members of the S100 family, HMGB1 and amyloid β peptides among others [Donato, 2007; Sparvero et al., 2009]. Much of its pathological function is linked to its ability to activate multiple signaling pathways and downstream effectors such as NF-κB in response to ligand binding. In addition, since RAGE gene has NF-κB binding sites in its promoter, ligand-mediated activation of signaling pathways results in upregulation of the receptor and sustained cell activation at sites where ligands accumulate, leading to exaggerated host response and uncontrolled pathology [Bierhaus et al., 2005; Bierhaus et al., 2001]. RAGE is thus considered a target for therapy in a diverse group of disorders [Yan et al., 2009a]. However, design of therapeutics targeted against RAGE in different diseases would require deeper understanding of both the structural organization of the receptor molecule and the basis for its ligand-promiscuity.
RAGE contains a V-type domain, two C-type Ig-like domains, (C1 and C2), a single trans-membrane spanning helix, and a cytosolic domain essential for signaling. NMR studies combined with CD, light scattering and limited proteolysis suggest that V and C1 domains are an integrated structural unit (VC1), whereas the C2 domain is an independent domain attached to VC1 trough a flexible linker [Dattilo et al., 2007]. Many splice variants of RAGE exist, and have recently been classified as RAGE, RAGE_v1 to RAGE_v19 [Hudson et al., 2008]. The prevalent ones are full length RAGE (RAGE), secreted RAGE that lacks the cytoplasmic and transmembrane domains (sRAGE, RAGE-v1) and N-terminal truncated RAGE (RAGE_v2). The relative expression of the isoforms is tissue specific. Through its ability to scavenge RAGE ligands, sRAGE is believed to act a decoy receptor by regulating signaling mediated by activation of full length RAGE.
By virtue of its multi-domain structure and ability to recognize different classes of ligands, RAGE behaves as a pattern recognition receptor (PRR) akin to innate immune receptors such as Toll-like receptors (TLRs) in orchestrating immune responses. However, unlike other PRRs that predominantly bind exogenous ligands, RAGE binds endogenous ligands, especially those considered to be damage associated molecular pattern molecules (DAMPs) [Srikrishna and Freeze, 2009]. Recent studies have focused on the molecular mechanisms of RAGE binding to specific ligands. These studies show that AGEs, HMGB1, Aβ peptides, S100B, S100A1, S100A2 and S100A5 bind to the V domain, S100A12 binds to V-C1 domains, and S100A6 interacts with V-C2 domain [Dattilo et al., 2007; Kiryushko et al., 2006; Leclerc et al., 2009; Leclerc et al., 2007; Ostendorp et al., 2007; Xie et al., 2008]. Studies on S100 protein-RAGE interactions also suggest that multimerization of ligand and receptor occurs and that formation of these higher ordered complexes may be essential for signal transduction [Donato, 2007; Moroz et al., 2002; Ostendorp et al., 2007]. In addition to contribution by protein interaction domains, post-translational modifications such as glycosylation of the receptors, or acetylation or phosphorylation of ligands could also play important roles in defining specificity of interactions, multimerization and downstream signaling. Though the role of glycans in formation of signaling complexes is well documented [Demetriou et al., 2001; Sacchettini et al., 2001], glycans are often considered as impediments in high-resolution protein structural analysis. Many of the current structural studies on RAGE are performed on sRAGE protein or sRAGE domains often expressed in bacteria, yeast or insect cells that lack the complex glycosylation machinery of the mammalian systems. Studies on glycan-deficient RAGE may not therefore accurately reflect the appropriate in vivo ligand binding, multimerization, or signaling pathways.
Our studies suggest that N-glycan modifications of RAGE may serve as unique ligand binding sites and may contribute to some of the pleiotropic binding ability of the receptor. RAGE has two N-glycosylation sites on the V-domain and both sites are occupied by complex and hybrid N-glycans [Turovskaya et al., 2008]. Several years ago, we identified a novel group of anionic N-glycans that contain an immunogenic carboxylate group unrelated to sialic or uronic acids [Norgard-Sumnicht et al., 1995]. These carboxylated glycans contain glutamic or aspartic acids possibly in amide linkage to the core N-glycans [Srikrishna et al., 2005a]. The glycans show restricted expression on mouse and human cells of myeloid lineage including monocytes, macrophages and dendritic cells and on endothelial cells [Srikrishna et al., 2001b; Srikrishna et al., 2005b]. They are absent or undetectable on normal epithelial cells, but expressed on several tumor cells [Turovskaya et al., 2008]. They bind DAMP molecules HMGB1, S100A8/A9 and annexin I [Srikrishna et al., 2002; Srikrishna et al., 2001a]. We found that a subpopulation of RAGE molecules from bovine lung is modified by carboxylated glycans [Srikrishna et al., 2002; Turovskaya et al., 2008] and that binding of HMGB1 to RAGE partially depends on carboxylated glycans [Srikrishna et al., 2002]. In recent studies, we reported that the subpopulation of RAGE enriched for carboxylated glycans showed 100-fold increase in binding potential (Bmax/Kd) for S100A8/A9, suggesting that carboxylated glycans form critical binding sites for these ligands on RAGE [Turovskaya et al., 2008]. We also found that inhibiting carboxylated glycan-dependent interactions of DAMP molecules using mAbGB3.1, an anti-glycan antibody blocked onset of T cell mediated colitis [Srikrishna et al., 2005b], colitis-dependent colon cancer [Turovskaya et al., 2008] and recruitment of myeloid-derived suppressor cells (MDSC) to secondary lymphoid organs in tumor-bearing mice [Sinha et al., 2008].
In the present study, we show that the binding of the inflammatory mediator S100A12 to RAGE is enhanced by carboxylated glycans expressed on RAGE. S100A12 is expressed in neutrophils and monocytes [Foell et al., 2007; Guignard et al., 1995]. It is highly upregulated in several human inflammatory diseases, including Crohn’s disease, rheumatoid arthritis, cystic fibrosis, Kawasaki disease and other inflammatory states [Foell et al., 2007]. It was the first member of the S100 family shown to interact with RAGE [Hofmann et al., 1999]. RAGE ligation by S100A12 on leukocytes and endothelial cells activates NF-κB and expression of proinflammatory genes [Hofmann et al., 1999]. Crystal structure of calcium bound S100A12 shows dimeric and hexameric states [Moroz et al., 2002; Moroz et al., 2003]. Recent studies show that both calcium and zinc are essential for S100A12 oligomerization and its interaction with RAGE [Moroz et al., 2009a; Moroz et al., 2009b]. S100A12 was first shown to bind to the V-domain of mouse sRAGE with a KD of 90 nM [Hofmann et al., 1999]. This sRAGE was expressed in insect cells, which typically lack complex N-linked glycans. S100A12 shows a binding affinity of 80nM to human sRAGE-Fc fusion protein expressed in NS0 myeloma cells, which synthesize complex N-glycans [Kiryushko et al., 2006]. Using extensive NMR, fluorescence, and other biophysical studies Xie et al showed that S100A12 did not bind to the isolated V-domain but showed strong binding to the C1 domain in the presence of Ca with a binding affinity of 90 nM [Xie et al., 2007]. However, these RAGE domains were expressed in E.coli, which lacks N-glycosylation machinery. Recently, using surface plasmon resonance, a binding affinity of 170 nM was determined for oligomeric S100A12 and V-domain of sRAGE expressed in E.coli [Leclerc et al., 2009]. We reasoned that the reported discrepancies in domain recognition and binding affinities for S100A12 were related to the use of non-glycosylated or partially glycosylated RAGE. Our present findings not only show that glycans on RAGE enhance binding of S100A12, but also suggest that they could in fact promote receptor clustering and downstream signaling.
MATERIALS AND METHODS
The following materials were from the sources indicated: Carbograph SPE, Alltech, Deerfield, IL. Concanavalin A Sepharose, Superdex TM200, and Na 125I, Amersham Biosciences, Piscataway, NJ. Iodobeads Iodinating reagent, Pierce, Rockford, IL; cis-reporter plasmid pNF-κB-Luc, Stratagene, La Jolla, CA; and pSV-β-galactosidase control vector, luciferase and β galactosidase assay systems, Promega, Madison, WI. Expression and purification of recombinant human S100A12 is as described [Foell et al., 2003]. Synthetic S100A12 used in preliminary experiments was kindly provided by Dr. Carolyn Geczy, University of New South Wales, Sydney, Australia. S100A11 was a kind gift from Dr. Jean-Christophe Deloulme, INSERM, France. RAGE was purified from bovine lung and enriched for the subpopulation expressing carboxylated glycans using mAbGB3.1 as described earlier [Turovskaya et al., 2008]. Full length human RAGE-pcDNA3.1 constructs, and human sRAGE produced by baculovirus expression sytem in expressSF+ cells were generously provided by Novartis Tsukuba Research Institute, Tsukuba, Japan. Tail deleted human RAGE-pcDNA3.1 constructs were a kind gift from Dr. Heikki Rauvala, University of Helsinki, Finland. Human His-tagged PNGaseF expression plasmid was a gift from Dr. J. Shaun Lott, Massey University, Palmerston, New Zealand. PNGaseF was expressed in E. coli and purified in our laboratory.
Binding of S100A12 to carboxylated glycopeptides immobilized on ELISA plates
GAG-free, desialylated carboxylate-enriched glycopeptides were isolated from bovine lung by proteinase K digestion and fractionated by charge on a DEAE column as described [Srikrishna et al., 2001a]. For binding assays, wells of a 96-well plate were coated with 100 ng of BSA-coupled carboxylated glycans in PBS for 4 h at 37°C. They were washed, blocked with 3% BSA in PBS overnight at 4°C, and incubated with increasing concentrations of recombinant human S100A12 in the presence or absence of soluble carboxylated or carboxylate-neutralized glycopeptides in HBSS containing 1 mM CaCl2 and 1% BSA overnight at 4°C. After washing, wells were incubated with anti S100A12 for 2h at room temperature followed by alkaline phosphatase conjugated secondary antibody, and development with p-nitrophenyl phosphate substrate.
Kinetic analysis of carboxylated glycan-S100A12 complex formation in solution
Complex formation of S100A12 with soluble carboxylate-enriched glycopeptides (moderately charged (eluted by 0.1M NaCl) and highly charged (eluted by 0.3M NaCl) isolated from bovine lung) was carried out in 10mM phosphate buffer, pH 7.2 containing 0.15M NaCl with and without 0.5mM Ca2+ in a volume of 100 μl in a quartz microplate. The glycans (100 μM final concentration) were mixed with S100A12 (20 μM final concentration) and time course of complex formation as light scattering was measured at 420 nm.
Western blot analysis
Proteins were separated on SDS-PAGE gels under reducing conditions and electro-blotted onto nitrocellulose membranes. The membranes were blocked overnight with 3% BSA in PBS, washed with PBS containing 0.05% Tween 20, and incubated with rabbit anti-RAGE in PBS containing 1% BSA and 0.05% Tween 20 for 1–2 h at room temperature. This was followed by incubation with alkaline phosphatase conjugated anti-rabbit IgG. Bound proteins were visualized by incubating with 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (BCIP/NBT).
Ligand binding assays
S100A12 or S100A11 was added at increasing concentrations to the wells of a 96-well plate containing immobilized total RAGE, carboxylated glycan-enriched RAGE, enriched RAGE deglycosylated under non-denaturing conditions or different purified sRAGEs. Incubations were done in HBSS containing 1 mM CaCl2 and 1% BSA overnight at 4°C. Bound S100 proteins was quantified using respective anti-S100 antibodies, followed by alkaline phosphatase conjugated secondary antibody and p-nitrophenyl phosphate (PNP) substrate, against standard S100A12 or S100A11. Bovine RAGE or sRAGE RAGE bound to the plates was quantified independently with anti-RAGE, using respective calibration curves. Non-specific binding was determined by incubation in wells blocked with BSA alone or by using only secondary antibody controls. Non-linear regression analysis was done using the Prism software (GraphPad prism).
Ligand binding studies was also done using labeled S100A12 as follows: S100A12 was radio-iodinated using Na125I and Iodo beads iodinating reagent to a specific activity of 2.5×105 cpm/μg. Total and mAbGB3.1 enriched RAGE were immobilized on nitrocellulose strips and blocked with 3% BSA. Strips were incubated with increasing concentrations of 125I S100A12 in the presence or absence of excess cold S100A12 or indicated amounts of mAbGB3.1, control antibody, or sRAGEs from different sources in HBSS containing 1 mM CaCl2 and 1% BSA overnight at 4°C. Strips were washed and bound label was measured using a gamma counter. Iodinated S100A12 were analyzed on 12% SDS-PAGE gels or on native gels, transferred onto nitrocellulose and detected by autoradiography.
Generation of mammalian cells stably expressing His-Tagged sRAGE
The secretion signal of human sRAGE was replaced with the Igkappa light chain secretion signal for enhanced expression/secretion by cloning sRAGE (sequence for mature protein, omitting the native secretion signal) into pSecTag2b (Invitrogen). The construct containing Igkappa secretion signal and RAGE extracellular domain was subcloned into the EcoR1 site of the moloney murine leukemia virus (MoMulV) derived vector pcz-CFG5.1-IRES2-EGFP to generate pcz-CFG5.1-sRAGE-IRES2-EGFP. Recombinant retroviral particles were generated by the three vector packaging system as described [Temme et al., 2003]. 293T cells were co-transfected with an expression construct for gag-pol (pHIT60), MoMuLV-retroviral constructs and the vesicular stomatitis virus G-protein (kindly provided by Dr. D. Trono, University of Geneva, Switzerland). Viral supernatants were harvested after transfection, pooled, filtered and used to infect HeLa, H4, HL60, HEK293 and U373 cells. Supernatants were tested for sRAGE secretion by western blots. Positive clones were further established by limiting dilution of cells. Clones expressing sRAGE were grown in DMEM medium containing 2% fetal bovine serum and penicillin and streptomycin. sRAGE was purified from the culture supernatants on a Ni-NTA column, and purity confirmed by SDS-PAGE followed by Coomassie brilliant blue staining.
Individual glycosylation sequences of sRAGE were mutated by site directed mutagenesis of the expression vector pcz-CFG5.1-sRAGE using the ‘transformer’ site-directed mutagenesis kit (BD Clontech) and the selection primer SelektNruI 5-GCGCTGCTTCGGGATGTACGGGCCAG-3 together with appropriate mutation primers according to the manufacturer’s instructions. Mutation primer RA_Mut_N25Q (5′-p-CCGGCCGGTGCTCAACAGATCACAGCCCGGAT-3′) abolished the glycosylation signal at position 25 of the mature protein, whereas mutation primer RA_Mut_N81Q (5′-p-GCTCGTGTCCTTCCCCAGGGCTCCCTCTTCCTT-3′) destroyed the glycosylation site at aa 81. The resulting sequences were verified by sequencing. HeLa cells stably expressing His-Tagged sRAGEs carrying the individual mutations were established using retroviral transductions as described above.
Determination of mAbGB3.1 reactivity
mAbGB3.1 reactivity of the different RAGEs was quantitated by ELISA. The protein was immobilized on wells of 96-well plate, and after blocking with 3% BSA in PBS was incubated with mAbGB3.1 at 1μg/ml overnight at 4°C. Bound antibody was detected using anti-mouse Ig alkaline phosphatase conjugate and PNP. mAbGB3.1 reactivity was defined in arbitrary units, 1 unit being the reactivity of one ng bovine RAGE under a defined set of ELISA conditions.
Metabolic labeling of sRAGE and isolation and analysis of glycans
HeLa cells expressing sRAGE were metabolically labeled using [2-3H] mannose at 20 μCi/ml and sRAGE from cell culture supernantant was purified as above. Glycans were released by PNGaseF. Undigested protein served as control. Released glycans were isolated, and desialylation, methyl esterification, and ion-exchange and lectin chromatography analysis using QAE Sephadex and Concanavalin A Sepharose respectively were carried out as described [Srikrishna et al., 2005a].
Electron microscopy and image analysis
sRAGE samples (purified sRAGE, and mAbGB3.1-enriched sRAGE) were diluted into their respective dilution buffers (~ 0.08mg/ml)), applied to a carbon coated electron microscopy grid and stained with 2% uranyl acetate. Images were obtained under low-dose conditions using a Tecnai 12 microscope (FEI electron optics) equipped with a Lab6 filament at 120 kV at a nominal magnification of 67,000 × and at ~1 um defocus. The micrographs were digitized on a SCAI scanner (Z/I Imaging) at 7-mm raster and compressed to a final pixel size of 0.6 nm. 516 negatively stained particles were picked from 38 micrographs. These particles were subjected to correlation-based, reference-free K means clustering in two dimensions and sorted into 5 self-consistent classes using the imaging processing software EMAN. The sorted images were iteratively refined and averaged for 50 rounds in order to remove any possible bias of the image chosen as the initial alignment reference.
Analytical size exclusion chromatography
A 20 ml column of Superdex™ 200 gel filtration column was pre-equilibrated with HBSS containing 0.05%NP-40, and calibrated using molecular weight marker protein standards bovine thyroglobulin (670 kDa), ferritin (440 kDa), IgG (150 kDa), bovine albumin (66 kDa), and ribonuclease (13.7 Da). 100-μl of solution containing 125I labeled S100A12 (50 nM) and RAGE from different sources (5 nM) in HBSS/NP-40 was loaded onto the column and eluted at a flow rate of 0.3 ml/min. 0.15 ml fractions were collected and counted for S100A12 radioactivity. RAGE was measured in the fractions by ELISA.
Generation of stable transfectants expressing full length RAGE and RAGE lacking the C-terminal signaling domain
HeLa cells were cultured in RPMI 1640 medium containing 10% bovine calf serum, 100 units/ml penicillin, and 50 μg/ml streptomycin at 37°C in a humidified incubator (5% CO2, 95% air). Cells were transfected with constructs expressing full length RAGE or cytoplasmic tail deleted (signaling deficient) RAGE using transfectin (BioRad) according to the manufacturer’s instructions. 72 h after transfection the cells were selected in medium containing G418 (2mg/ml). After a 2–3-week growth in G418-containing medium, G418-resistant clones were further cloned by limiting dilution and expanded. The expression of RAGE was verified using western blots and flow cytometry.
Flow cytometry
Stable HeLa transfectants expressing full length or tail deleted RAGE in culture were detached using PBS containing 10mM EDTA and incubated with rat monoclonal anti-RAGE in HBSS containing 1% BSA followed by PE-conjugated anti-rat IgG. They were analyzed by flow cytometry with a FACSort (Becton Dickinson, Mountain View, CA) equipped with CellQuest software, and gated by the side scatter and forward scatter filters.
NF-κB reporter assay
Stable RAGE transfectants were transiently transfected with pNF-κB-Luc, containing a Luciferase cDNA under a regular TATA Box and an enhancer element with five NF-κB binding sites (Stratagene, La Jolla, CA). To correct for variations in transfection efficiency, cells were simultaneously transfected with pSV-β-Galactosidase control vector. 12h after transfection, cells were serum-starved for 12h, and stimulated with S100A12 in the presence or absence of inhibitors. 24h after activation, cells were harvested and enzyme activities were measured in lysates. The ratio of Luc/Gal served to normalize for Luc activity. Transfected unactivated cells accounted for endogenous activity, and activity in tail deleted RAGE accounted for RAGE-unrelated background.
RESULTS
Carboxylated glycan-enriched RAGE shows increased binding potential for S100A12
We recently reported that a subpopulation of bovine lung RAGE modified by carboxylated glycans showed a >30-fold increase in the molar binding and >100 fold increase in binding potential (Bmax/Kd) to S100A8/A9, suggesting that carboxylated glycans form critical binding sites for S100A8/A9 on RAGE ([Turovskaya et al., 2008] and Table 1). Structure-function studies suggest that murine S100A8 is a functional homolog of human S100A12 [Ravasi et al., 2004]. We therefore reasoned that carboxylated glycans expressed on RAGE or associated receptors may promote similar binding and downstream signaling in S100A12 mediated interactions. In fact, we found that immunopurified bovine lung RAGE bound purified human S100A12 with a Kd of approximately 51.8± 20.6 nM, and a Bmax of 1.2± 0.2 moles/mole RAGE (with a binding potential Bmax/Kd of 0.03± 0.01). The subpopulation of RAGE enriched for carboxylated glycans using mAbGB3.1 as described earlier [Turovskaya et al., 2008] also bound S100A12 with a Kd of 133± 46.4 nM, showing that the binding affinities are minimally affected. However, this subpopulation (referred to as mAbGB3.1-enriched RAGE) showed Bmax of 44.5± 8.5 moles of S100A12/mole RAGE, >30 fold higher than seen for total RAGE, with a 10–15 fold higher binding potential (Bmax/Kd of 0.36 ± 0.11) (Figure 1A and Table 1). Moreover, this fraction contained 70% of the total S100A12 binding activity originally determined for the starting preparation of total RAGE. PNGaseF digestion under non-denaturing conditions removed 90% of the S100A12 binding to mAbGB3.1-enriched RAGE fraction (Figure 1A). Mock digestions of RAGE retain full binding activity. We confirmed the above findings by an independent approach using radiolabeled S100A12 (not shown).
Table 1.
S100 proteins and RAGE: binding equilibrium
| Total bovine RAGE | Glycan Enriched bovine RAGE | |||||
|---|---|---|---|---|---|---|
| Purified human ligands | Bmax (mole/mole) | Kd (nM) | Bmax/Kd (×10−4) | Bmax (mole/mole) | Kd (nM) | Bmax/Kd (×10−4) |
| Av.Binding potential | Av.Binding potential | |||||
| S100A8/A9* | 0.0114±0.002 | 34.4±13 | 4 | 0.402±0.03 | 7.62±1.83 | 560 |
| >30 fold ↑ | >100 fold ↑ | |||||
| S100A12 | 1.2±0.2 | 51.8±20.6 | 300 | 44.5±8.5 | 133±46.4 | 3600 |
| >30 fold ↑ | >10 fold ↑ | |||||
| S100A11 | 0.04±0.003 | 53.1±7.8 | 0.7 | 0.03±0.002 | 30.9±3.6 | 1.1 |
The parameters for S100A8/A9 were published earlier [Turovskaya et al., 2008]
Figure 1. Carboxylated glycans on RAGE mediate binding to S100A12, but not to S100A11.

To determine binding equilibrium of purified human S100A12 (A) or S100A11 (B) to purified bovine RAGE, increasing amounts of the ligands were added to total RAGE, mAbGB3.1 enriched RAGE, or RAGE deglycosylated using PNGaseF under non-denaturing conditions that removed both N-glycans. RAGE on plate was quantified using anti-RAGE. Bound ligands were quantified using specific antibodies. Data were fitted to non-linear regression analysis using GraphPad Prism. Each point is the mean ± SD of two determinations.
RAGE extracellular region has three disulfide linkages, one bond in each domain (V, C1 and C2 respectively). Dattilo et al showed that 15N-1H HSQC spectra of V-domain acquired in the presence of 1mM DTT show a spectrum characteristic of an unfolded protein [Dattilo et al., 2007]. The homogenization buffer used for our bovine lung RAGE isolation contained 10mM DTT [Turovskaya et al., 2008]. Homogenization of whole bovine lung tissue was conducted at low temperatures, which does not cause reduction of internal disulfides in the C1C2 domains. In addition, we avoided any reduction of the exposed disulfide bond in the V domain by initiating refolding immediately after homogenization through elimination of DTT (20-fold buffer dilution followed by dialysis, ammonium sulfate precipitation and subsequent purification steps including an affinity chromatography step. However, to eliminate the possibility of any reduced disulfide reduction and protein unfolding, we measured free SH- groups in bovine RAGE using Ellman’s reagent (5, 5′-dithiobis (2 nitro benzoate or DTNB) before and after complete denaturation (denaturation and reduction was carried out using 0.2% SDS and 1% 2-mercaptoethanol). Free cysteine was determined using a standard curve with DTT. While the denatured protein had 6 moles of reduced thiols per mole of purified protein corresponding to three disulfide bonds in the native protein, the native purified protein that we used in our binding assays had none (not shown).
To verify if glycan-dependent binding to RAGE is unique for S100A8/A9 and S100A12, we examined the binding of another S100 protein, S100A11, up-regulated in osteoarthritic cartilage [Cecil et al., 2005]. S100A11 induces chondrocyte hypertrophy via RAGE-mediated signaling, and the interactions can be blocked by anti-RAGE. However, it is not clear whether S100A11 mediated its effects by direct interaction with RAGE or through associated proteins. We found that purified human S100A11 directly binds bovine RAGE with a Kd of approximately 53.1± 7.8 nM, and a Bmax of 0.04± 0.003 moles/mole RAGE (Figure 1B and Table 1). However, mAbGB3.1-enriched RAGE did not show enhanced S100A11 binding such as seen with S100A8/A9 and S100A12 (Kd of approximately 30.9± 3.6 nM, and a Bmax of 0.03± 0.002 moles/mole RAGE), showing that carboxylated glycans may not mediate the binding of S100A11 to RAGE. The above results suggest that though many S100 family members bind RAGE, they may bind to different structural domains or regions on the receptor, some involving carboxylated N-glycans on the V domain and others not, thus providing differential binding specificity and likely dictating different intracellular signaling events. As mentioned above, S100A11 exerts RAGE dependent signaling in keratinocytes and chondrocytes. However, based on our low stoichiometric binding it is likely that in vivo other proteins in addition to RAGE could also contribute to S100A11 dependent signaling.
S100A12 forms complexes with carboxylated glycans
A lectin type interaction of S100A12 to glycans might modulate its binding to RAGE. To test whether S100A12 directly bound to glycans, we first incubated S100A12 with BSA-coupled desialylated carboxylate-enriched glycopeptides purified from bovine lung [Srikrishna et al., 2001a] immobilized on microtiter plate wells. We found that human S100A12 bound to the glycans with a Kd of approximately 29± 7 nM (Figure 2A). The specificity of S100A12 binding to these glycans was demonstrated by a dose-dependent inhibition in presence of soluble carboxylated glycopeptides from bovine lung, with an IC50 of approximately 10μM, but much less effectively by carboxylate-neutralized glycopeptides with less than 40% inhibition at 100μM (Figure 2B).
Figure 2. S100A12 shows specific binding to carboxylated glycans.

Purified recombinant human S100A12 (A) was incubated with BSA-conjugated carboxylate-enriched bovine lung glycopeptides coated onto microtiter plates and binding was detected using anti-S100A12. Non-linear regression transforms of binding data using GraphPad Prism are presented. (B) Inhibitions of binding were carried out in presence of increasing concentrations of free carboxylated or carboxylate-neutralized glycans. Each point is the mean two determinations. C. Time course of complexation between S100A12 and carboxylated glycans (0.1M, moderately charged, 0.3M highly charged) in the presence or absence of calcium was measured by light scattering at 420 nm.
Since mAbGB3.1-enriched RAGE carries much higher binding sites for S100A12 per unit protein, it is possible that carboxylated glycans provide critical epitope density and/or multivalency to promote multimeric ligand binding or glycan-lectin complex formation [Brewer et al., 2002; Dam and Brewer, 2003; Sacchettini et al., 2001]. To test this hypothesis in vitro, we mixed S100A12 with free moderately charged and highly charged carboxylated glycans in solution and looked for evidence of complex formation and rate of clustering by measuring light scattering. We found that there was a rapid onset of light scattering after mixing the glycans with S100A12 (Fig 2C). Glycans containing higher charges (eluted by 0.3M NaCl) were more effective than less-charged glycans (eluted by 0.1M NaCl). For highly charged glycans, complex formation was enhanced in presence of calcium. The kinetics of interactions with the glycans suggests that a rapid interaction with the glycans is enhanced by oligomerization of S100A12 in presence of calcium. Carboxylate-neutralized glycans were much less effective in mediating complex formation (not shown). In control experiments we found that Ca2+ does not cause large turbidity or complex formation of bovine lung glycopeptides alone in the absence of S100A12. These findings demonstrate that S100A12 could function as a multivalent lectin in binding carboxylated glycans.
Expression of human sRAGE in mammalian cells and oligosaccharide analysis
We used bovine RAGE as representative of RAGE purified from a natural source. Though human RAGE ectodomain is 80% identical to bovine RAGE ectodomain, the ligand binding V-domains where both N-glycosylation sites are located on human and bovine RAGE are 90% identical. Even so, binding studies using human S100A12 against bovine RAGE may not be completely representative. Therefore we performed binding studies using human ectodomain of RAGE (sRAGE) expressed in mammalian and non-mammalian cells against human S100A12. sRAGE, the secreted or soluble form of RAGE, contains the entire extracellular module, but lacks the transmembrane domain and is produced by alternative splicing of RAGE mRNA [Hudson et al., 2008]. It is believed that sRAGE functions as a decoy receptor that binds RAGE ligands and prevents them from binding to the cell surface receptor, thereby functioning as a regulator of RAGE-mediated signaling and cell perturbation.
We first studied the glycosylation patterns of human sRAGEs expressed in different systems and compared their ability to bind to mAbGB3.1 and S100A12. We expressed His tagged human sRAGE in HEK293, U373, H4, HL60 and HeLa cells using retroviral vectors and purified the expressed protein on Ni-NTA columns. In addition, we also tested RAGE generated and purified from a baculovirus expression system in insect cells when potential N-linked glycosylation sites may be either fully or partially glycosylated or not glycosylated at all. Most insect cells can process N-linked glycans up to the initial addition of GlcNAc residues to the antenna to form incomplete complex-type chains. Western blots of the purified proteins from different sources showed multiple bands perhaps representing different glycoforms (Fig 3A). We found that both N-glycosylation sites on all the different sRAGEs were modified: by Endo H sensitive chains, and Endo H resistant, PNGase F sensitive chains (Fig 3A). However, mAbGB3.1 reactivity of the different preparations varied considerably, with highest mAbGB3.1 reactivity seen in RAGE expressed in HeLa and HL60 cells (Fig 3B). This suggests that glycosylation of RAGE is cell type specific.
Figure 3. Glycosylation of sRAGEs expressed in different cells.
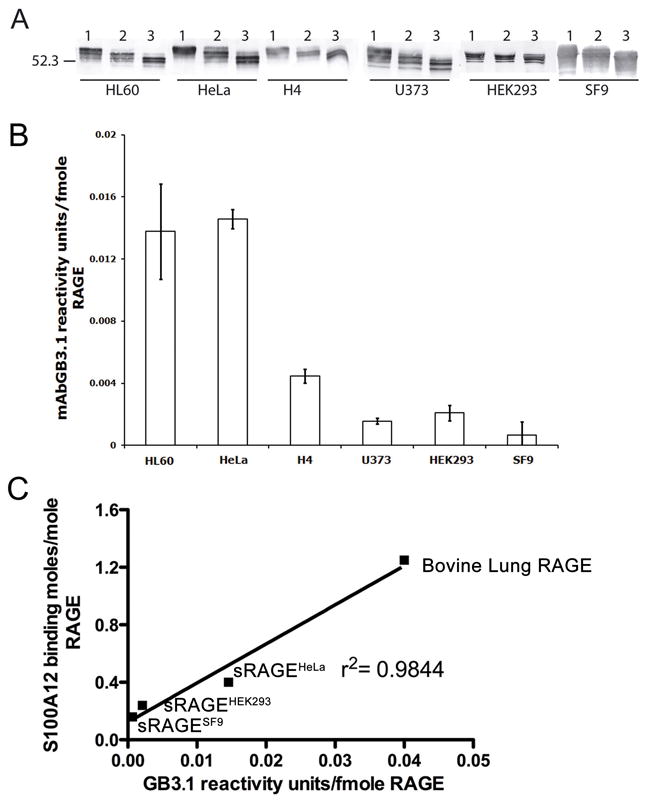
A. sRAGEs were expressed in different mammalian cells using retroviral vectors and in SF9 cells and purified as described under Methods. Purity was confirmed by SDS-PAGE and Coomassie brilliant blue staining. Western blots show that sRAGEs from different sources are all glycosylated. Lane 1: undigested. Lane 2: Endo H digested Lane 3: PNGase F digested. B. mAbGB3.1 reactivity of the different preparations as determined by ELISA. C. There is direct correlation between mAbGB3.1 reactivity of the human sRAGEs and bovine RAGE, and their maximum binding sites for human S100A12.
To analyze the nature of the glycans on total and mAbGB3.1-enriched sRAGEs, we used sRAGE expressed in HeLa cells. Expression and secretion of soluble RAGE by cells and relative ease of purification from culture medium enabled us to metabolically label glycans on RAGE by [2-3H] mannose and study their characteristics. We released 3H mannose labeled glycans from total purified sRAGE or mAbGB3.1 enriched sRAGE by PNGase F, purified on carbograph and analyzed by QAE Sephadex ion-exchange and Con A lectin chromatography.
The radiolabel from total RAGE glycans was distributed among complex chains (multi-antennary, bi-antennary), and hybrid, and high mannose chains (Table 2). Complex chains contain 3 Man residues, hybrid chains contain an average of 5 Man and high mannose, an average of 6–7 Man. Based on the 2–3 fold higher mannose content in high mannose-type glycans, each RAGE molecule is therefore likely to have at least one complex-type chain, with another being complex, hybrid or high-mannose chain. This is consistent with one of the glycan chains being sensitive to Endo H. In contrast, the majority of glycans (95%) from mAbGB3.1-enriched RAGE were complex glycans (with 76% being multiantennary chains) compared to 57% from unfractionated sRAGE (12% being multiantennary).
Table 2.
Analysis of glycans from sRAGE and its glycosylation mutants
| Con A profile % of total glycans | QAE profile % of total glycans | ||||
|---|---|---|---|---|---|
| Complex glycans | Hybrid or high mannose glycans | Neutral glycans | Glycans with 1–2 charges | Glycans with 1–2 charges after desialylation | |
| WT sRAGEHeLa | 57 ± 8 (12% MA) | 43 ± 8 | 40 ± 2 | 60 ± 2 | 5 ± 2 |
| WT carboxylated- glycan-enriched sRAGEHeLa | 95 ± 2 (76% MA) | 5 ± 2 | 35 ± 6 | 65 ± 6 | 18 ± 4 |
| Site 1 glycans (sRAGEHeLa mutant 2) | 82.5 ± 7.8 | 17.5 ± 7.8 | 19 ± 4 | 81 ± 4 | 9 ± 2 |
| Site 2 glycans (sRAGEHeLa mutant 1) | 71 ± 3 | 29 ± 3 | 23 ± 2 | 77 ± 2 | 6 ± 2 |
Each value represents the mean ± SD of two determinations. MA: multi-antennary complex glycans (tri, tetra, penta)
QAE Sephadex anion-exchange chromatography analysis showed that about 60% of the label on total RAGE was anionic (Table 2). 5% of label remained charged after mild acid treatment to remove sialic acids. In contrast, about 18% of label on glycans from mAbGB3.1-enriched RAGE still remained charged after desialylation. ~35% of the remaining charges became neutral by treatment with methyl iodide, showing that they were associated with non-sialyl carboxylic acids. Charges remaining after methyl esterification could be due other charged species such as sulfates, or to incomplete esterification of carboxylates, since under the conditions used, the maximum efficiency of esterification of 3H Neu5Ac is about 72%.
These results show that mAbGB3.1-enriched sRAGE carries predominantly multiantennary, charged glycans, and is enriched for the species carrying carboxylated glycans.
When we compared the kinetics of purified human S100A12 binding to different sRAGEs, the Bmax for ligand binding varied considerably depending on the cell type. In fact, there was a direct correlation between mAbGB3.1 reactivity of the individual sRAGEs and maximum binding sites on each receptor for S100A12, strongly suggesting that carboxylated glycans on RAGE form a large part of S100A12 recognition (Fig 3C). It is also evident from the regression curve that when mAbGB3.1 reactivity is extrapolated to zero, S100A12 binding is not totally lost, suggesting that either the RAGE peptide itself or other N-glycans on RAGE could share part of S100A12 recognition site. Thus S100A12 could be a multivalent molecule with different binding sites or domains on RAGE. This corroborates recent reports that show S100A12 interacts with both V and C1 domains of RAGE [Leclerc et al., 2009; Xie et al., 2007]. Binding of 125I S100A12 to total RAGE was more effectively inhibited by sRAGE from HeLa cells as compared to sRAGE from other sources (not shown), showing that expression of carboxylated glycans is important for the inhibitory potency of sRAGEs in interactions involving S100A12. sRAGEHeLa showed a Bmax for S100A12 of 0.4± 0.04 moles/mole RAGE, while mAbGB3.1-enriched subpopulation (1–2% of total sRAGE) showed a Bmax of 1.93± 0.38 moles/mole RAGE, ~5 fold higher, again suggesting that carboxylated glycans on RAGE provide additional binding sites for S100A12.
Studies on N-glycosylation mutants of sRAGE
To further investigate the role of individual N-glycan chains of RAGE on S100A12 binding and to determine whether one or both are modified by carboxylated glycans, we employed site-directed mutagenesis to generate mutant proteins (N25Q and N81Q) lacking N-glycosylation at either of the potential sites. Mutation resulted in reduction of molecular mass from the wild type protein consistent with loss of N-linked glycosylation. Both mutants showed mAbGB3.1 reactivity equivalent to that seen in wild type sRAGE, suggesting that both sites can be potentially modified by carboxylated glycans. We were unsuccessful in our attempts to express the double-mutant N25Q/N81Q with complete absence of N-linked glycans.
Mutant 1 with site 2 modified by glycans showed slightly reduced binding (Bmax 0.17± 0.05 moles/mole RAGE) compared to wild type sRAGE, while mutant 2 with site 1 modified by glycans showed slightly higher Bmax (0.5± 0.05 moles/mole RAGE) compared to wild type sRAGE. This suggests that though site 2 can be potentially modified by carboxylated glycans, it may be less accessible for binding to S100A12. It is also likely that conformational changes induced in the mutants by removal of one or the other glycan chain modulates binding. Site 2 glycans were also less accessible for PNGaseF digestion under non-denaturing conditions, which readily removed glycans in site 1. Again deglycosylation of wild type sRAGE and mutant 2 decreased S100A12 binding by >90% (not shown).
We released 3H mannose labeled glycans from total purified mutant sRAGEs and analyzed them by QAE Sephadex ion-exchange and Con A lectin chromatography. The radiolabel from mutant sRAGEs was distributed predominantly among complex chains (Table 2). 29% of label from site 2 and 18% label from site 1 was distributed among hybrid and high mannose, compared to 43% in wild type RAGE. QAE analysis showed that about ~20% of the radiolabel on the mutant RAGEs were neutral, compared to 40% seen in wild type sRAGE. About 6–9% of label on glycans from both mutant RAGEs still remained charged after desialylation.
These results show that both glycosylation sites on RAGE can be potentially modified by carboxylated glycans and in wild type RAGE site 1 glycan is almost always fully processed, while glycosylation on site 2 is variable (complex, hybrid, or high mannose). These findings also suggest that glycosylation on site 1 may influence processing at the second site. This could be due to an altered conformation that limits accessibility to processing enzymes in the N-glycosylation pathway. This is also in agreement with N-glycosylation pattern of sRAGE purified from mouse lung [Hanford et al., 2004].
Analysis of RAGE-S100A12 complexes
Multivalent ligands can cluster cell surface receptors. Results from our turbidimetric experiments mentioned above (Fig 2C) are consistent with the formation of complexes of oligomeric S100A12 with glycans, suggesting that carboxylated glycans can promote receptor ligand assemblies on cell surfaces. To explore this, we first employed high-resolution electron microscopy (EM) to elucidate the oligomerization states of total and mAbGB3.1-enriched RAGE. Electron micrographs from negatively stained sRAGE specimens revealed a mixture between small and large particles (Fig 4A). The small particles were roughly globular with dimensions that might approximate the mass of sRAGE dimers (white rectangular), assuming a compact spherical shape. The large particles fall into few visual classes, yellow boxes showing various size structures most likely corresponding to different oligomeric complexes, in keeping with recent studies showing that RAGE exists as a homo-oligomer in the native inactive state [Xie et al., 2008], and white circles corresponding to the more homogeneous class of particles enriched in the mAbGB3.1-fraction (Fig 4A inset),
Figure 4. RAGE forms higher-order oligomers.

A. Field of view of negatively stained sRAGE and in inset mAbGB3.1-enriched fraction as captured by EM imaging. Protein densities are white on a dark background. For display purposes, some of the oligomers in this image have been marked using white boxes for small oligomers, possibly dimers, and white circles for slightly larger oligomers which are enriched in the mAbGB31. fraction. Yellow circles mark larger, multi size oligomers. Scale bar is 100 nm. The mAbGB3.1-enriched fraction (inset) seems to be more homogeneous with molecular distribution corresponding to the white circles B. Results from reference-free, K-means clustering. Class averages of five slef consistent classes with the percentage of classified images. The image marked with sym represents the symmetrized average (2-fold) of the most self-consistent, 70% of the data. All images display similar morphology with a central mass and two equivalent lobes attached. C. Comparison of the symmetrized average with the surface representation of four homology models of sRAGE molecules suggests that this enriched-fraction consists of four or more sRAGE oligomerized receptor.
To further explore the effects of mAbGB3.1 purification/glycosylation on the oligomerization state of sRAGE, we selected 516 particles for in depth image analysis, Reference-free clustering procedures [Ludtke et al., 1999; Penczek et al., 1992], further established the structural homogeneity of the mAbGB3.1-enriched fraction. Unlike in the total fraction, we observed here an enriched population of oligomers with average dimensions of about 35 × 35 nm (Fig 4B). Alignment and averaging using these EM images revealed that most of the oligomers fall into two very similar classes (~70%), while remaining particles do express same size and overall morphology. These observations suggest the formation of a stable organized multimer rather than random aggregates, supporting the concept of receptor multimerization. In average the oligomers consist of a central density flanked by two equivalent additional lobe-like masses. All classes show evidence for approximate two-fold symmetry. Comparison of a symmetrized average (Fig 4B, marked sym) with a homology model of sRAGE [Dattilo et al., 2007] shows that a minimum of four sRAGE molecules is required to account for the density (Fig 4C). Owing to the fact that we have two-dimensional projections rather than three-dimensional density, it is possible that more than four monomers (through stacking) contribute to the density. However, the two-fold symmetry of the projection suggests that the number of monomers contributing should be divisible by two (i.e. tetramer, hexamer, octamer, decamer, etc.). Our studies confirm those of others [Xie et al., 2007] showing that sRAGE domains exist as tetramers even in a fully glycosylated native form, and that a reversible equilibrium could exist between large and small homo-oligomers of the protein. However, more definitive structural studies are required to determine the stoichiometry of oligomerized sRAGE and to determine internal symmetry of the oligomers.
Next, we analyzed the formation and stoichiometry of RAGE-S100A12 complexes using gel filtration. When mixed at receptor ligand ratios of 1:10, unfractionated RAGE forms complexes with human S100A12 corresponding to ratios of at least (RAGE)3 (S100A12)6, while the mAbGB3.1-enriched population forms higher order complexes of hexamer:dodecamer and above, when tested with bovine RAGE and sRAGEHeLa (Fig 5). The complexes form rapidly (within 5 minutes) even at higher mixing ratios of 1:100 receptor to ligand (not shown). This ability to form multimeric complexes is reduced, but not completely eliminated, by PNGaseF deglycosylation. sRAGE expressed in E.coli without N-glycans also shows reduced formation of higher order complexes. The perceived differences in size of observed assemblies in EM and by gel filtration could be due to qualitative estimates of molecular weights of proteins and of multivalent complexes which are dependent on the hydrodynamic properties, shape and glycosylation states of the proteins. Our estimate of the molecular weight is based on the behavior of RAGE on PAGE gels (~50kDa for the fully glycosylated denatured protein). Earlier NMR studies suggest that sRAGE eluting as 150kDa protein on a similar gel filtration column is in fact a tetramer, and that a reversible equilibrium could exist between multimers in the absence of ligand [Xie et al., 2007]. Despite the differences, it is evident from our studies that higher order RAGE-S100A12 assemblies are promoted by carboxylated glycans.
Figure 5. Analysis of RAGE-S100A12 complex formation.

Purified bovine RAGE, mAbGB3.1 enriched bovine RAGE, deglycosylated mAbGB3.1-enriched bovine RAGE, human sRAGE expressed in HeLa cells and and mAbGB3.1-enriched sRAGEHeLa, and human sRAGE expressed in E.coli were analyzed as such or mixed with radioiodinated human S100A12 and the complexes formed were analyzed on precalibrated Superdex 200 gel filtration column (fractionation range 10,000–600,000). Fractions were collected and RAGE quantitated by ELISA. S100A12 was measured by quantitating radioactivity (not shown). Molecular weights of the complexes were determined from a calibration curve using marker protein standards (shown by arrows on the first panel for bovine RAGE) 1: bovine thyroglobulin (670 kDa) 2: ferritin (440 kDa) 3: IgG (150 kDa) 4: bovine albumin (66 kDa), and 5: ribonuclease (13.7 Da). RAGE multimerization is evident in solution. Unfractionated RAGE forms complexes with S100A12 corresponding to ratios of at least RAGE3: S100A126 (MW 220 kDa), while mAbGB3.1 enriched populations form higher order complexes of hexamer:dodecamer and above. Deglycosylation significantly reduces the ability to form complexes.
mAbGB3.1 blocks NF-κB activation induced by S100A12
Receptor clustering by multivalent ligands can activate intracellular signaling pathways. S100A12 binding to RAGE mediates downstream signaling events in cells leading to NF-κB activation. To determine the functional significance of glycan dependent multimerization in mediating intracellular signaling following S100A12 ligation, we expressed the full-length RAGE or the non-signaling, cytoplasmic domain deleted mutant of RAGE in HeLa cells to generate stable transfectants. Expression of RAGE on cells was confirmed by western blot and FACS analysis (Fig 6A and 6B). Using these stable transfectants we studied NF-κB activation using transient transfection with a luciferase expression plasmid containing four tandem copies of the NF-κB consensus sequence fused to a TATA-like promoter region. After transfection, cells were serum-starved and stimulated for 24 hrs with S100A12 (5 μg/ml). We determined luciferase activity in the cell lysates and normalized for transfection efficiency by the amount of β-gal activity expressed by the co-transfected control plasmid pSV-β-Gal. Non-stimulated transfected cells were used as control of basal expression. As shown in Fig 6C, S100A12 stimulated NF-κB-dependent transcription in the cells transfected with full-length RAGE. Moderate NF-κB-dependent transcription seen in cells transfected with signaling incompetent, cytoplasmic tail deleted RAGE represented RAGE-independent endogenous activity. Preincubation with mAbGB3.1 prior to stimulation decreased NF-κB expression almost to levels seen in cells transfected with cytoplasmic tail deleted RAGE, while an isotype control antibody had no or minimal effect. At the same molar concentrations, sRAGEHeLa more effectively inhibited NF-κB activation than sRAGE expressed in insect cells which does not have the mAbGB3.1 epitope, again showing that glycosylation of the individual sRAGEs determine their inhibition potency. To account for any activation mediated by LPS in our preparations, we used S100A12 rendered endotoxin-free by purification over endotrap columns. The protein retained minor amounts of LPS. Endotoxin-free preparations usually represent endotoxin levels lower than 0.125 EU/ml. Our preparation had 0.24 EU/ml. To rule out any possible activation by residual LPS, in separate studies, we performed S100A12 mediated signaling experiments in which boiling the preparations or clearing them from S100A12 by immunoprecipitation completely abrogated the effects of S100A12, while polymyxin B treatment, which neutralizes the effect of LPS, did not produce any changes in S100A12 mediated signaling, indicating that it is S100A12 and not any residual LPS which is responsible for the activation (Foell D et al, unpublished).
Figure 6. Activation of NF-κB by S100A12 ligation of RAGE is inhibited by mAbGB3.1 and sRAGE.

Stable transfectants of HeLa cells expressing full length and cytoplasmic tail deleted (signaling deficient) RAGE were generated as described. Expression of RAGE in cells was confirmed by A. Western blotting using anti-RAGE, Lane 1, untransfected HeLa cells; Lane 2, Full length RAGE transfectants and Lane 3, Tail deleted RAGE transfectants and by B. FACS analysis. Unstained cells are represented in the background. Low-level endogenous expression is seen in untransfected cells. C. Activation of NF-κB by S100A12 ligation of RAGE is inhibited by mAbGB3.1 and sRAGE: HeLa cells expressing full length or tail deleted RAGE were transiently transfected with an NF-κB-responsive cis-reporter gene and a pSV-β-Galactosidase control vector. S100A12 activation in the presence and absence of inhibitors and analysis of enzymes in lysates was done as described. The ratio of Luc/Gal served to normalize for Luc activity. Luc/Gal ratio in transfected unactivated control, which accounted for endogenous activity, was considered 100%. Activity in tail deleted RAGE accounted for RAGE-unrelated background. All values represent the mean ± S.D. (n = 2).
DISCUSSION
The basis for the ability of RAGE to bind structurally different ligands leading to activation of distinct signaling pathways is not well understood. Studies show that different ligands interact either with the V domain, VC1 domain, or VC2 domain [Dattilo et al., 2007; Kiryushko et al., 2006; Leclerc et al., 2009; Leclerc et al., 2007; Ostendorp et al., 2007; Xie et al., 2008]. We had earlier shown that carboxylated N-glycans in the V-domain on a subpopulation of RAGE provide important structural determinants for binding of two of its ligands, HMGB1 and S100A8/A9 [Srikrishna et al., 2002]. Deglycosylation reduces binding potential of HMGB1 to RAGE two-fold [Srikrishna et al., 2002]. Carboxylated glycans increase the binding potential of S100A8/A9 to RAGE more than 100 fold ([Turovskaya et al., 2008] and Table 1). Here we show that carboxylated-glycan enriched RAGE shows a ten-fold higher binding potential for S100A12 than total RAGE. All three aforementioned RAGE ligands show lectin-like properties since all of them directly bind to carboxylated glycans. However, not all RAGE ligands require the presence of glycans for binding. For example, the binding of S100B, S100A1, S100A2 and S100A11 to RAGE is not glycosylation-dependent ([Leclerc et al., 2009] and Fig 1B). Deglycosylation, in fact, promotes the binding of AGE modified proteins [Osawa et al., 2007], suggesting that structural differences within the domains determine binding to RAGE of different ligands.
As we mentioned earlier, there are several discrepancies in existing reports on the domains involved in the interaction of RAGE and S100A12 and of binding affinities [Hofmann et al., 1999; Kiryushko et al., 2006; Leclerc et al., 2009; Xie et al., 2007]. Most report requirement of V and C1 domains of RAGE for S100A12 binding. Dattilo et al [Dattilo et al., 2007] point out that studies using C1C2 construct expressed in E.coli without the V-domain [Xie et al., 2007] have to be reexamined since C1 requires V domain for full stability and structural integrity and that C1C2 may be structurally unstable and may exhibit aberrant function. Aside from the reported differences in domain recognition, the aforementioned studies using RAGE expressed in E.coli or insect systems, as well as ours using fully glycosylated RAGE expressed in mammalian cells, demonstrate that binding affinities (KD) for S100A12 are in the nanomolar ranges for both deglycosylated, partially glycosylated and fully glycosylated RAGE. A comparison of binding affinities alone would therefore lead to the conclusion that glycosylation plays a minimal role in ligand binding. Our earlier studies also show that RAGE can be readily deglycosylated with PNGase F, indicating that glycans are solvent accessible. This again implies that the glycans do not interact with or modify the structure of folded protein domains, and seems to reinforce the argument that non-glycosylated or partially glycosylated constructs are valid for the study of RAGE structure and ligand binding. While this may be true for some RAGE ligands as mentioned above, our findings using S100A12 strongly indicate that binding affinities do not reflect their binding potentials, nor the in vivo effects that glycans can have on the cell surface in promoting receptor multimerization and formation of signaling assemblies when certain RAGE ligands bind. In addition, a common polymorphism of RAGE, G82S, which occurs at a glycosylation site, has been shown to destabilize the tertiary structure of the protein [Xie et al., 2008], which could potentially affect binding of one or more ligands.
We show that mAbGB3.1-enriched RAGE exhibits several fold increase in binding potentials for S100A8/A9 and S100A12. Both these ligands are able to form higher order homo and hetero oligomers, providing multiple interaction sites with the receptor. By its ability to bind both V and C1 domains, S100A12 also displays multiple binding modes. Multivalent ligands are potent effectors in promoting specific signaling responses by their ability to cluster receptors. The densities of the binding epitope of a multivalent ligand, and/or the density of its binding sites on its receptor, influence factors in receptor signaling beyond binding affinity. Receptor clustering depends on stoichiometry of complex formation and number of receptors in a cluster, rate of cluster formation and proximity of receptor [Cairo et al., 2002; Gestwicki et al., 2002]. Many studies show that oligomerization of receptors is a common mechanism for signaling [Lemmon and Schlessinger, 1998]. Receptor multimerization has been demonstrated for cytokine receptors, which bind soluble oligomeric ligands. Several cytokines have carbohydrate-binding properties including, IL-1, IL-2, IL-3, IL-4, IL-6 and IL-7 [Cebo et al., 2002]. While carbohydrate interactions by themselves generally tend to be of low affinity, a multivalent and multimeric soluble ligand interacting with a multivalent glycan could promote formation of lattices or clusters that increases the avidity of binding [Dam and Brewer, 2003]. This clustering is thermodynamically favorable and has been elegantly demonstrated with galectins [Brewer et al., 2002; Sacchettini et al., 2001]. One of the factors that could govern lattice formation is multivalency provided by multiple branching of the glycans [Demetriou et al., 2001; Lau et al., 2007]. As mentioned under results, we find that 76% of the glycans on mAbGB3.1-enriched RAGE is multi-antennary (tri, tetra and penta-antennary). S100A12 is hexameric, and multiple binding sites could be provided on a single RAGE domain by the display of multi-antennary glycan chains modified by one or more carboxylated-glycan epitopes. Our size exclusion chromatography studies support the formation of these large complexes. This is not unusual or sterically unlikely. Multiantennary complex carbohydrates have been shown by several studies to bind multiple lectin molecules [Dam and Brewer, 2009]. For example four soybean agglutinin molecules, which have a diameter exceeding 64 angstroms each, can bind four glycans on a tetrantennary chain. This is because oligosaccharide conformation is not always rigid, and oligosaccharides have a flexible conformation depending on the stereochemistry of linkages. This is certainly true of proteins such as RAGE whose glycan modifications are exposed. In fact, a new definition of lectin receptor in biological system is now emerging which considers “relative Kds” based on density and number of glycan epitopes on cell surface proteins, rather than single dissociation constant for lectin binding to monovalent glycans or proteins [Dam and Brewer, 2009].
Based on 3H-mannose labeling studies and immunoprecipitation with mAbGB3.1, we find that only a small population of total RAGE expressed in cells is modified by carboxylated glycans. This suggests that either the biosynthetic machinery that adds the modification is limited, or that the glycan expression on RAGE is tightly regulated or both. This enriched fraction contained 70% of the total S100A12 activity of total RAGE. Therefore, it is likely that this small percentage of molecules could promote initial high affinity binding to multivalent ligands such as S100A12 and S100A8/A9, and increase initial rate of complexation, leading to subsequent domain interactions and formation of a signaling assembly (Figure 7). In support of this, in turbidimetric experiments, which allow for quantitation of the relative rates of clustering, we found that the rate of clustering induced by S100A12 was directly related to increased density of carboxylated glycans (Fig 2C). This glycan-induced clustering appears important for signaling, since mAbGB3.1 blocks S100A12 mediated activation of NF-κB in RAGE expressing cells (Fig 6). Pre-assembly of the receptor (Fig 4) may facilitate rapid cellular responses to ligand-induced stimulation. Whether this ligand-receptor assembly occurs on organized membrane microdomains remains to be determined. This also brings up the question whether other proteins that are potentially modified by carboxylated glycans could also bind S100A12 and cause formation and aggregation of heterogeneous protein complexes on the cell surface. However, studies with galectins show that multivalent carbohydrate binding proteins could selectively cross link a single species of glycoprotein [Brewer et al., 2002]. RAGE glycan microstructure thus seems to play an important role in controlling the outcome of ligand binding, and provides one important aspect of regulation of this multi-functional protein.
Figure 7. Schematic representation of RAGE receptor clustering and formation of signaling assembly following carboxylated glycan-mediated S100A12 binding.
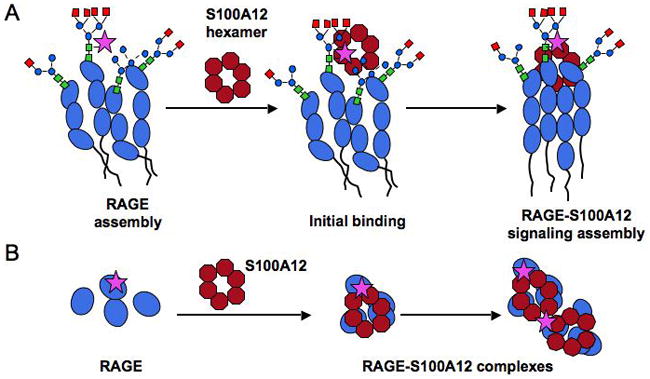
A Lateral view: Our EM studies as well as studies from other labs suggest the presence of a pre-assembled RAGE multimer on cell surfaces. The V-domains are modified by hybrid, high mannose and complex glycan chains, with a subpopulation modified by carboxylated glycans (marked by a star). Ca (and Zn) promotes hexamerization of extracellular S100A12 released from cells in response to inflammatory and other stimuli. Carboxylated glycans could provide initial high affinity binding sites on RAGE for S100A12, and promote increased initial rate of complexation, leading to subsequent domain interactions and formation of a signaling assembly. Multiple binding sites could be provided on a single RAGE domain by the display of multi-antennary glycan chains modified by one or more carboxylated-glycan epitopes (only one epitope is shown for sake of space and clarity). Whether this ligand-receptor assembly occurs on organized membrane microdomains remains to be determined B. A view from the top of the cell membrane showing preassembled receptor and ligand-induced formation of signaling complexes. By providing multi-valency or multiple binding sites, caboxylated-glycans could promote higher order complexes (as seen in Fig 5) with mAbGB3.1-enriched RAGE.
We found that both N-glycosylation sites on RAGE retain the ability to express carboxylated glycans and bind S100A12. The first glycosylation site (N25IT) is modified by a fully processed N-glycan chain that retains S100A12 binding and mAbGB3.1 reactivity. Glycosylation at the second site (N81GS) in the native (wild type) protein is variable (complex, hybrid or high mannose). In the absence of glycosylation on site 1, site 2 is modified by a more fully-processed chain suggesting greater accessibility to the processing enzymes. RAGE lacking site 1 glycans shows slightly reduced binding to S100A12. Though site 2 can be potentially modified by carboxylated glycans, it may be less accessible for binding to S100A12. It is also likely that conformational changes induced in the mutants by removal of one or the other glycan chain modulates binding. The differing patterns at site 2 suggest that glycosylation of site 1 affects the glycosylation of site 2. Our recent analysis of N-glycans on site 1 of sRAGE expressed in HeLa cells suggests considerable heterogeneity of glycan structures (Houliston et al, unpublished). The glycosylation of individual sequons in glycoproteins is affected by various factors, including the rate of trafficking through the secretory pathway, and the accessibility of the N-glycans to glycosidases and glycosyl transferases.
Our studies imply that sRAGEs produced by bacterial or insect expression systems and commonly used in in vitro studies and in mouse models may be differentially glycosylated or partially glycosylated and may not provide strong inhibitors of S100A8/A9, S100A12 or HMGB1 mediated RAGE signaling. In support of this, Rouhiainen et al showed that bovine lung derived RAGE is a stronger inhibitor of transendothelial migration of monocytes than recombinant sRAGE expressed in insect cells [Rouhiainen et al., 2004], and attributed the difference to differential glycosylation of the two sRAGEs. A large number of S100 proteins interact with RAGE and the molecular mechanisms of this interaction are only beginning to be understood. Though our studies suggest that carboxylated glycans on RAGE could modulate the interaction of S100A12 and S100A8/A9, we have not extensively looked at other S100 proteins to be able to pinpoint unique structural motifs for interaction with carboxylated-glycan modified subpopulation of RAGE. By multiple alignment of C-terminal HMGB1 domain with various S100 proteins, Huttunen et al showed that RAGE-binding region of HMGB1 has remarkable resemblance with the amino terminal of many S100 proteins [Huttunen et al., 2002]. Many of the amino acids in this alignment for S100A8/A9, S100A12, S100A11 and other S100 proteins are identical or similar by nature. It would therefore be preliminary to speculate on specific binding motifs on S100 proteins until more extensive studies are done.
In conclusion, our studies provide strong evidence that carboxylated glycans expressed on RAGE mediate S100A12-dependent signaling. The mechanism by which RAGE clustering is mediated by ligand-glycan interaction on cell surfaces will be the focus of future studies.
Acknowledgments
Funded by: National Institutes of Health Grants R01-CA92608 (HF and GS) and R21-CA127780 (GS), Dr. Howard and Barbara Milstein Endowment fund, a gift from Howard and Carole Goldfeder (HF) and Department of Defense Office of the Congressionally Directed Medical Research Programs Proposal 09082003 (DH and NV).
We thank Dr. Robert Houliston and Dr. Yehia Mechref for unpublished data on RAGE glycans, Dr. Lars Bode, Dr. Gowtham Subbarao and Dr. Lena Brive for preliminary experiments, Violet Abraham and Sabine Heinicke for help with generating bovine lung glycopeptides and recombinant viruses respectively, and Mien Nguyen, Jessica Clima, Jacob Haling and Biji Varughese for help with sRAGE purifications. We also thank Dr. Robert Houliston, Dr. Yoshi Ichikawa and Mie Ichikawa for critical reading of this manuscript.
Abbreviations
- DAMP
Damage Associated Molecular Pattern
- EM
Electron Microscopy
- HMGB1
High Mobility Box Group1 protein
- MDSC
Myeloid Derived Suppressor Cells
- NF-kB
Nuclear factor kappa-B
- NMR
nuclear magnetic resonance
- PAGE
polyacrylamide gel electrophoresis
- PRR
Pattern Recognition Receptor
- RAGE
Receptor for Advanced Glycation End Products
- TLR
Toll-like receptor
References
- Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–86. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Nawroth PP. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia. 2009;52:2251–63. doi: 10.1007/s00125-009-1458-9. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- Brewer CF, Miceli MC, Baum LG. Clusters, bundles, arrays and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr Opin Struct Biol. 2002;12:616–23. doi: 10.1016/s0959-440x(02)00364-0. [DOI] [PubMed] [Google Scholar]
- Cairo CW, Gestwicki JE, Kanai M, Kiessling LL. Control of multivalent interactions by binding epitope density. J Am Chem Soc. 2002;124:1615–9. doi: 10.1021/ja016727k. [DOI] [PubMed] [Google Scholar]
- Cebo C, Vergoten G, Zanetta JP. Lectin activities of cytokines: functions and putative carbohydrate-recognition domains. Biochim Biophys Acta. 2002;1572:422–34. doi: 10.1016/s0304-4165(02)00323-9. [DOI] [PubMed] [Google Scholar]
- Cecil DL, Johnson K, Rediske J, Lotz M, Schmidt AM, Terkeltaub R. Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J Immunol. 2005;175:8296–302. doi: 10.4049/jimmunol.175.12.8296. [DOI] [PubMed] [Google Scholar]
- Dam TK, Brewer CF. Carbohydrate-lectin cross-linking interactions: structural, thermodynamic, and biological studies. Methods Enzymol. 2003;362:455–86. doi: 10.1016/S0076-6879(03)01031-0. [DOI] [PubMed] [Google Scholar]
- Dam TK, Brewer CF. Lectins as Pattern Recognition Molecules. The effects of epitope density in innate immunity. Glycobiology. 2009 doi: 10.1093/glycob/cwp186. [DOI] [PubMed] [Google Scholar]
- Dattilo BM, Fritz G, Leclerc E, Kooi CW, Heizmann CW, Chazin WJ. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry. 2007;46:6957–70. doi: 10.1021/bi7003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- Donato R. RAGE: a single receptor for several ligands and different cellular responses: the case of certain S100 proteins. Curr Mol Med. 2007;7:711–24. doi: 10.2174/156652407783220688. [DOI] [PubMed] [Google Scholar]
- Foell D, Kucharzik T, Kraft M, Vogl T, Sorg C, Domschke W, Roth J. Neutrophil derived human S100A12 (EN-RAGE) is strongly expressed during chronic active inflammatory bowel disease. Gut. 2003;52:847–53. doi: 10.1136/gut.52.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J Am Chem Soc. 2002;124:14922–33. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- Guignard F, Mauel J, Markert M. Identification and characterization of a novel human neutrophil protein related to the S100 family. Biochem J. 1995;309 (Pt 2):395–401. doi: 10.1042/bj3090395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanford LE, Enghild JJ, Valnickova Z, Petersen SV, Schaefer LM, Schaefer TM, Reinhart TA, Oury TD. Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE) J Biol Chem. 2004;279:50019–24. doi: 10.1074/jbc.M409782200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, Grant PJ, Schmidt AM. Identification, classification, and expression of RAGE gene splice variants. Faseb J. 2008;22:1572–80. doi: 10.1096/fj.07-9909com. [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Fages C, Kuja-Panjula J, Ridley AJ, Rauvala H. Receptor for Advanced Glycation End Products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002;62:4805–11. [PubMed] [Google Scholar]
- Kiryushko D, Novitskaya V, Soroka V, Klingelhofer J, Lukanidin E, Berezin V, Bock E. Molecular mechanisms of Ca(2+) signaling in neurons induced by the S100A4 protein. Mol Cell Biol. 2006;26:3625–38. doi: 10.1128/MCB.26.9.3625-3638.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, Dennis JW. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–34. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Leclerc E, Fritz G, Vetter SW, Heizmann CW. Binding of S100 proteins to RAGE: An update. Biochim Biophys Acta. 2009;1793:993–1007. doi: 10.1016/j.bbamcr.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem. 2007;282:31317–31. doi: 10.1074/jbc.M703951200. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Transmembrane signaling by receptor oligomerization. Methods Mol Biol. 1998;84:49–71. doi: 10.1385/0-89603-488-7:49. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Moroz OV, Antson AA, Dodson EJ, Burrell HJ, Grist SJ, Lloyd RM, Maitland NJ, Dodson GG, Wilson KS, Lukanidin E, Bronstein IB. The structure of S100A12 in a hexameric form and its proposed role in receptor signalling. Acta Crystallogr D Biol Crystallogr. 2002;58:407–13. doi: 10.1107/s0907444901021278. [DOI] [PubMed] [Google Scholar]
- Moroz OV, Blagova EV, Wilkinson AJ, Wilson KS, Bronstein IB. The Crystal Structures of Human S100A12 in Apo Form and in Complex with Zinc: New Insights into S100A12 Oligomerisation. J Mol Biol. 2009a;391:536–51. doi: 10.1016/j.jmb.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Moroz OV, Burkitt W, Wittkowski H, He W, Ianoul A, Novitskaya V, Xie J, Polyakova O, Lednev IK, Shekhtman A, Derrick PJ, Bjoerk P, Foell D, Bronstein IB. Both Ca2+ and Zn2+ are essential for S100A12 protein oligomerization and function. BMC Biochem. 2009b;10:11–28. doi: 10.1186/1471-2091-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroz OV, Dodson GG, Wilson KS, Lukanidin E, Bronstein IB. Multiple structural states of S100A12: A key to its functional diversity. Microsc Res Tech. 2003;60:581–92. doi: 10.1002/jemt.10300. [DOI] [PubMed] [Google Scholar]
- Norgard-Sumnicht KE, Roux L, Toomre DK, Manzi A, Freeze HH, Varki A. Unusual anionic N-linked oligosaccharides from bovine lung. Journal of Biological Chemistry. 1995;270:27634–45. doi: 10.1074/jbc.270.46.27634. [DOI] [PubMed] [Google Scholar]
- Osawa M, Yamamoto Y, Munesue S, Murakami N, Sakurai S, Watanabe T, Yonekura H, Uchigata Y, Iwamoto Y, Yamamoto H. De-N-glycosylation or G82S mutation of RAGE sensitizes its interaction with advanced glycation endproducts. Biochim Biophys Acta. 2007;1770:1468–74. doi: 10.1016/j.bbagen.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Ostendorp T, Leclerc E, Galichet A, Koch M, Demling N, Weigle B, Heizmann CW, Kroneck PM, Fritz G. Structural and functional insights into RAGE activation by multimeric S100B. Embo J. 2007;26:3868–78. doi: 10.1038/sj.emboj.7601805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penczek P, Radermacher M, Frank J. Three-dimensional reconstruction of single particles embedded in ice. Ultramicroscopy. 1992;40:33–53. [PubMed] [Google Scholar]
- Ravasi T, Hsu K, Goyette J, Schroder K, Yang Z, Rahimi F, Miranda LP, Alewood PF, Hume DA, Geczy C. Probing the S100 protein family through genomic and functional analysis. Genomics. 2004;84:10–22. doi: 10.1016/j.ygeno.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–82. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- Sacchettini JC, Baum LG, Brewer CF. Multivalent protein-carbohydrate interactions. A new paradigm for supermolecular assembly and signal transduction. Biochemistry. 2001;40:3009–15. doi: 10.1021/bi002544j. [DOI] [PubMed] [Google Scholar]
- Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–75. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, Rutledge R, Lin B, Amoscato AA, Zeh HJ, Lotze MT. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. 2009;7:17. doi: 10.1186/1479-5876-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikrishna G, Brive L, Freeze H. Novel carboxylated N-glycans contain oligosaccharide-linked glutamic acid. Biochemical & Biophysical Research Communications. 2005a;332:1020–7. doi: 10.1016/j.bbrc.2005.05.056. [DOI] [PubMed] [Google Scholar]
- Srikrishna G, Freeze HH. Endogenous Damage Associated Molecular Pattern (DAMP) molecules at the crossroads of inflammation and cancer. Neoplasia. 2009;11:615–28. doi: 10.1593/neo.09284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikrishna G, Huttunen HJ, Johansson L, Weigle B, Yamaguchi Y, Rauvala H, Freeze HH. N-glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. Journal of Neurochemistry. 2002;80:998–1008. doi: 10.1046/j.0022-3042.2002.00796.x. [DOI] [PubMed] [Google Scholar]
- Srikrishna G, Panneerselvam K, Westphal V, Abraham V, Varki A, Freeze HH. Two proteins modulating transendothelial migration of leukocytes recognize novel carboxylated glycans on endothelial cells. Journal of Immunology. 2001a;166:4678–88. doi: 10.4049/jimmunol.166.7.4678. [DOI] [PubMed] [Google Scholar]
- Srikrishna G, Toomre D, Manzi A, Panneerselvam K, Freeze H, Varki A, Varki N. A novel anionic modification of N-glycans on mammalian endothelial cells is recognized by activated neutrophils and modulates acute inflammatory responses. Journal of Immunology. 2001b;166:624–632. doi: 10.4049/jimmunol.166.1.624. [DOI] [PubMed] [Google Scholar]
- Srikrishna G, Turovskaya O, Shaikh R, Newlin R, Foell D, Murch S, Kronenberg M, Freeze HH. Carboxylated Glycans Mediate Colitis through Activation of NF-{kappa}B. J Immunol. 2005b;175:5412–22. doi: 10.4049/jimmunol.175.8.5412. [DOI] [PubMed] [Google Scholar]
- Temme A, Rieger M, Reber F, Lindemann D, Weigle B, Diestelkoetter-Bachert P, Ehninger G, Tatsuka M, Terada Y, Rieber EP. Localization, dynamics, and function of survivin revealed by expression of functional survivinDsRed fusion proteins in the living cell. Mol Biol Cell. 2003;14:78–92. doi: 10.1091/mbc.E02-04-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, Varki N, Kronenberg M, Freeze HH, Srikrishna G. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008;29:2035–43. doi: 10.1093/carcin/bgn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A. Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J Biol Chem. 2007;282:4218–31. doi: 10.1074/jbc.M608888200. [DOI] [PubMed] [Google Scholar]
- Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE) J Biol Chem. 2008;283:27255–69. doi: 10.1074/jbc.M801622200. [DOI] [PubMed] [Google Scholar]
- Yan SF, Du Yan S, Ramasamy R, Schmidt AM. Tempering the wrath of RAGE: An emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann Med. 2009a:1–15. doi: 10.1080/07853890902806576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med. 2009b;87:235–47. doi: 10.1007/s00109-009-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]