

Abstract
Classical splenic marginal zone lymphomas are CD5−, CD10−, CD23−, CD43− and usually IgD+ with biphasic white pulp nodules. However, the 2008 WHO classification accepts splenic marginal zone lymphomas with monophasic marginal zone-like white pulp nodules and recognizes a group of unclassifiable splenic small B-cell lymphomas. To explore the relationship of classical splenic marginal zone lymphomas to these other less well-defined splenic lymphomas, a multiparameter study of 47 splenic marginal zone lymphomas and unclassifiable splenic small B-cell lymphomas was performed. 17/31 splenic marginal zone lymphomas were biphasic and 14 monophasic (90–100% marginal zone-like white pulp nodules). Sixteen cases were unclassifiable splenic small B-cell lymphomas, most lacking a marginal zone-type component. There were many clinical similarities between the three groups, including similar survivals. Monophasic and unclassifiable cases were less likely to have a typical splenic marginal zone lymphoma phenotype (28.6%, 23.1%) compared to biphasic cases (86.7%), usually due to IgD negativity (p<0.003). 34/42 (81%) cases had cytogenetic abnormalities by fluorescence in situ hybridization and 17/20 (85%) by classical cytogenetics. The most frequent fluorescence in situ hybridization abnormalities among the splenic marginal zone lymphomas were del(7)(q31) (26%), +12 (25%) and +3q27 (27%) and among the unclassifiable cases +12 (50%) and +3q27 (36%). 5/6 unclassifiable cases with exclusively small non-marginal zone-like lymphocytes involving both white and red pulp had +12 compared to 9/34 other cases (p<0.02). CDK6 (2 cases) and BCL3 (1 case) rearrangements were only seen in the unclassifiable group. These results support including both biphasic and monophasic cases as splenic marginal zone lymphomas, but suggest the lack of a non-marginal zone-like population in the monophasic group is associated with some biologic differences. They also demonstrate a relatively large proportion of unclassifiable cases, including a group with frequent +12.
Keywords: splenic marginal zone lymphoma, spleen, B-cell lymphoma, marginal zone lymphoma, fluorescence in situ hybridization
INTRODUCTION
The 2001 World Health Organization (WHO) classification defines splenic marginal zone lymphoma (SMZL) as a B-cell neoplasm composed of small lymphocytes that surround and replace the white pulp (WP) follicles and merge with a peripheral zone of larger marginal zone (MZ) like cells.(1) The 2008 WHO monograph however, acknowledges that some SMZL lack a central core of smaller lymphocytes with only monophasic (MP) MZ-like WP nodules.(2, 3) Whether the MP cases have any distinctive features is uncertain. SMZL also has characteristic phenotypic features (usually IgD+, CD5−, CD23−, CD10−, and CD43−), but these findings are neither uniformly found nor specific.(2, 4–6) Although del(7)(q31) is a characteristic feature of SMZL, it is not completely specific for SMZL and is present only in a minority of cases.(4, 7–11) Other cytogenetic abnormalities have also been described that are even less specific.(4, 7–12) Interpretation of the literature, however, is problematic since SMZL is sometimes used as a wastebasket for many splenic small B-cell lymphomas that cannot be otherwise categorized.
The 2008 WHO classification recognizes these difficulties in part by creating a new category of splenic B-cell lymphoma/leukemia, unclassifiable. (13) This group of lymphomas includes splenic small B-cell neoplasms that do not fulfill the criteria for SMZL or any of the other well recognized B-cell neoplasms and includes two provisional entities: splenic diffuse red pulp small B-cell lymphoma (SDRPSBL) and hairy cell leukemia variant (HCL-v).(13, 14) The relationship of these provisional entities to each other and to SMZL remains to be determined. Other small B-cell lymphomas that may have a splenic presentation have also been reported such as those with IGH/BCL3 or CDK6 translocations, including some still classified as SMZL.(11, 15, 16)
For all these reasons, a multiparameter study of SMZL and other unclassifiable splenic small B-cell lymphomas was performed to investigate whether cases that fulfill the current criteria for MP SMZL have distinctive features and also to look at the unclassifiable cases in a search for the existence of other distinct entities or to suggest which might simply be morphologic/phenotypic variants of SMZL.
MATERIALS AND METHODS
Case Selection and Clinical Review
Splenic small B-cell lymphomas with a splenectomy specimen that did not fulfill the criteria for chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), mantle cell lymphoma, follicular lymphoma, or hairy cell leukemia were selected. Available clinical data from these selected patients was reviewed. This investigation was approved by the institutional review boards at the University of Pittsburgh and the Cleveland Clinic Foundation. It is acknowledged that because cases without a splenectomy specimen were excluded from this study, it may not reflect the actual relative distribution of types of splenic small B-cell lymphomas.
Morphologic and Immunophenotypic Review
Hematoxylin and eosin-stained sections and all existing immunohistochemical stains and flow cytometric data were reviewed. The following histopathologic features were evaluated: percent biphasic (BP) white pulp nodules composed of a central zone of small lymphocytes with or without a central germinal center surrounded by a peripheral zone of MZ-like cells, percent MP white pulp nodules composed of MZ-like cells, predominance of red pulp (RP) involvement, and presence of WP atrophy. Based on the above features, the cases were separated into 3 groups as follows: BP - >10% BP white pulp nodules (1 case excluded from this category because of a typical CLL phenotype); MP- >90% MP white pulp nodules; unclassifiable (UC) - all other cases. Plasmacytic differentiation (based on light chain restriction in plasma cells by immunohistochemistry), presence of epithelioid histiocyte clusters, presence of prominent RP nodules, extramedullary hematopoiesis, and presence of prominent rings of T-cells surrounding WP nodules (based on CD3 stain) were also recorded. The median time from original diagnosis to splenectomy (and range), known in 45 cases, was as follows: BP cases - 1 month (0–60); MP cases – 1 month (0–96) and UC cases 0 months (0–19). Prior treatment was documented in 3/13 BP cases (no data in 4), in 1/10 MP cases (no data in 4), and in 1/11 UC cases (no data in 5). Peripheral blood smears were also reviewed when available.
Tissue Microarray (TMA) construction
TMA were contructed with 2–5 1mm cores from each of 43 spleens where paraffin embedded tissue was available, and, when possible, with 2 cores of involved splenic hilar lymph nodes. Six cores from 3 separate control spleens not involved by lymphoma were included in each TMA.
Immunohistochemistry
Immunohistochemical stains were performed on cases with sufficient material. unless already available, using the Ventana BenchMark XT instrument and antibodies to detect the following: CD20 (Ventana, Tucson, AZ), CD3 (Dako, Glostrup, Denmark), CD5 (Cell Marque), CD23 (The Binding Site, Birmingham, U.K.), CD10 (Cell Marque), cyclin D1 (Ventana), BCL2 (Cell Marque), BCL6 (Dako), IRF4/MUM1 (Dako), CD43 (Ventana), IgD (Dako), and kappa and lambda light chain (Lab Vision, Fremont, CA). Cases considered positive showed positivity on the majority of neoplastic cells. Cases considered negative lacked definitive staining of the neoplastic cells. An immunohistochemical stain for p53 (Dako) was performed on the tissue microarray. Only cases with a majority of strongly positive cells were considered positive.
Cytogenetic fluorescence in situ hybridization (FISH) studies
FISH studies were performed on TMAs using the following probes: CEP7, D7S486, LSI BCL6 dual color breakapart, LSI MALT1 dual color breakapart, LSI BCL2 dual color breakapart, LSI EGR1/D5S23, CEP18, CEP12, and CEPX (Abbot Molecular Inc, Des Plaines, IL), LSI BCL3 dual color breakapart (Dako North America, Inc, Carpinteria, CA) and LSI CDK6 breakapart (courtesy of EDM composed of SpectrumOrange- (BACS: CTB-10G5, RP5-1099C19, CTB-85C5, and RP5-850G1) and SpectrumGreen-labeled (BACS: CTB-104I4, GS1-440B14, and GS1-165I4) DNA probes that hybridize to flanking regions of the CDK6 break point). The slides were deparaffinized, co-denatured with probe, allowed to hybridize overnight, unbound probe was washed away, and slides were mounted with DAPI/Antifade (Millipore, Billerica, MA). The signal patterns from a minimum of 30 cells were scored for each core. Cutoffs to determine positive samples were established for each probe based on the negative controls included on each array together with our laboratory’s experience (U.S.) with clinical specimens. A case was considered positive if at least one of the cores exceeded the threshold for positivity. Additional cells were studied from borderline samples. Inconclusive TMA results were further assessed either with whole section FISH when sufficient material was available or based on the classical cytogenetic findings. The number of cases evaluated by whole section FISH are as follows: BCL6 breakapart (1 case); CEP12 (1); CEP18 (3); EGR1/D5S23 (5); CEPX (5).
Classical cytogenetic studies
Results of available classical cytogenetic studies, performed using standard trypsin-Giemsa banded karyotyping (17) on spleen or involved bone marrow specimens, were reviewed.
Statistical analysis
Statistical analyses were performed using the Prism software package, version 3.03 (GraphPad Software, Inc., La Jolla, CA). Categorical data was evaluated using Chi square or Fisher’s exact test. Numerical data was evaluated with a one-way ANOVA with a Tukey post test.
RESULTS
Gross and microscopic pathology
The spleens ranged from 150–3784 g (44 cases) without significant differences between the BP, MP and UC groups (mean weights 1342g, 1786g and 1409g respectively). The UC group was significantly less likely than the BP or MP groups to have grossly prominent white pulp (65%, 67% and 27%, p < 0.02). 31/47 cases fulfilled the 2008 WHO criteria for SMZL, including 17 BP cases with classic biphasic WP nodules (Fig. 1A, B) and 14 MP cases with >90% of the WP nodules composed entirely of lymphocytes with MZ-like morphology (Fig. 1C, D). Sixteen UC cases did not clearly meet the WHO criteria for SMZL or any other specific small B-cell neoplasm (pathologic findings in Table 1). A diffuse or micronodular RP proliferation of small non-MZ-like lymphocytes with atrophic WP, focally prominent WP nodules (Fig 1E, F) or more extensive WP involvement was found in 9 of these cases. Some of the remaining UC cases raised the possibility of a lymphoplasmacytic lymphoma (LPL), a difficult to diagnose or atypical SMZL or another type of MZ lymphoma.
Figure 1.

A. Biphasic SMZL with expanded WP nodules consisting of a central core of small lymphocytes surrounded by an outer zone of marginal zone-like cells The lower nodule may demonstrate remnants of a follicular center. B. In contrast to the small lymphocytes in the central core of the WP nodules (top), the cells in the marginal zone are larger, have more dispersed chromatin including some with nucleoli and have more abundant pale cytoplasm. (A&B, case 39) C. Monophasic SMZL with large WP nodule consisting entirely of marginal zone-like cells with no inner core of small lymphocytes. D. The relatively monotonous neoplastic cells resemble a normal splenic marginal zone. (C&D, case 9). E. Unclassifiable splenic small B-cell lymphoma with small lymphocytes diffusely infiltrating the RP. Some WP nodules composed of small lymphocytes were also present. F. The numerous small lymphocytes appear somewhat plasmacytoid. (E&F, case 43). (H&E, A, C, E, 10×; B, D, F, 100×)
Table 1.
Pathologic Features of Unclassifiable Splenic Small B-cell Lymphomas
| Case | CD5 | CD23 | CD10 | CD43 | IgD | Histologic Findings |
|---|---|---|---|---|---|---|
| 3 | + (flow) | + | − | NE | + | Diffuse RP involvement by small lymphocytes, atrophic WP (λ-bright, CD20-dim+, FMC7−, CD103, CD11c−, CD25+ by flow cytometry) |
| 6 | + | − | − | + | + | WP predominantly CD5 positive small cells with attenuated rim of CD5 negative MZ-type cells |
| 12 | − | − | − | − | − | Diffuse RP involvement by small lymphocytes, atrophic WP |
| 13 | − | − | − | − | − | Non-MZ-like RP involvement, no WP expansion, light chain restricted plasma cells, lymph node involvement most consistent with MZ lymphoma, also chronic myeloproliferative neoplasm |
| 15 | − | − | − | NE | − | Micronodular RP infiltrate of small lymphocytes, atrophic WP |
| 16 | + | + | − | + | + | Morphology like SMZL, phenotype more like CLL (κ-intermediate intensity, CD20-dim, FMC7−, CD38+ by flow cytometry) |
| 23 | − | − | − | − | + | Mostly MZ-like cells but extensive RP involvement, WP also involved but not expanded |
| 24 | − | − | − | − | NE | All small lymphs with both WP and RP involvement |
| 28 | − | − | − | − | − | All small lymphs with both WP and RP involvement |
| 32 | − | − | − | NE | NE | Lymphoplasmacytic RP nodules with epithelioid histiocyte clusters, no WP involvement, LN consistent with lymphoplasmacytic lymphoma - favor lymphoplasmacytic lymphoma* |
| 36 | − | − | − | − | + | Plasmacytoid cells with no MZ-like cells but expanded WP |
| 37 | − | − | − | − | − | All small lymphs with both WP and RP involvement |
| 38 | − | − | − | + | + | WP with large cells including MZ-like cells, transformed cells, and angulated cells surrounded by a rim of small cells |
| 40 | − | − | − | NE | + | All small lymphs with both WP and RP involvement |
| 43 | − | − | − | − | + | Small lymphs with extensive RP involvement but focally prominent WP |
| 44 | − | − | − | − | − | Small lymphs with extensive RP involvement but focally prominent WP |
NE = Not evaluable, WP= white pulp, RP=red pulp, lymphs=lymphocytes
This patient had an IgM kappa paraprotein, cold agglutinin disease and marrow involvement.
Light chain restricted plasma cells (Fig. 2A and B) were present in 41% of cases, with a similar proportion in the BP, MP and UC groups (44%, 36% and 44%). All three groups included cases with prominent RP nodules (8/47), cases with epithelioid histiocyte clusters that were sometimes numerous (15/47) and cases with a prominent ring of T-cells surrounding the WP nodules (11/47) (Fig. 2C and D) without significant differences between the groups. Extramedullary hematopoiesis was present in 6/47 cases.
Figure 2.

Kappa (A) and lambda (B) staining in a SMZL with plasmacytic differentiation demonstrates kappa light chain restricted plasma cells predominantly within the white pulp nodules (case 2, 100×). CD43 (C) and CD3 (D) from the same biphasic SMZL showing CD43 negativity in the lymphoma (case 39, 40×). Note the ring of T cells surrounding the neoplastic white pulp nodules. E. Biphasic SMZL showing IRF4/MUM1 negativity. The inset demonstrates few scattered positive lymphocytes, including some that were large (case 47, 40×, inset 500×). F. Biphasic SMZL showing diffuse IgD positivity (case 39, 40×). (immunohistochemical stains with hematoxylin counterstain)
All splenic hilar lymph nodes available to review were involved by lymphoma. The BP (4/4) and MP (8/8) cases as well as 8/9 UC cases had intact sinuses. All BP cases and 7/8 MP cases had a follicular (nodular) growth pattern. The remaining MP case had a diffuse pattern. The clearly involved areas in the lymph nodes from the MP cases included mostly a monotonous proliferation of MZ-like cells. The BP cases had variable numbers of MZ-like cells and small lymphocytes. Some BP cases also demonstrated ill-defined zonation with a central area of small lymphocytes surrounded by MZ-like cells, but this finding was focal and much less pronounced than in the spleen. The growth pattern in the lymph nodes from the UC group was follicular in 5 cases, diffuse in 2, interfollicular in 1 and not assessable due to insufficient tissue in 1.
Immunohistochemical phenotype
All cases were CD20+, BCL2+, CD10− and cyclin D1− and all but 1 BCL6− (Table 2). A minority of cases from each group was IRF4/MUM1+ although all but 2 of the positive cases had only weak staining. IRF4/MUM1− cases often had scattered positive plasma cells and a few scattered positive lymphocytes that often included some large cells (Fig. 2E). IgD positivity was found in 93% of the BP cases (Fig. 2F), but in fewer MP and UC cases. In one of the BP cases, there were fewer and more weakly IgD+ cells in the marginal zones than elsewhere. No significant differences were identified for any of the other individual immunohistochemical markers, although there was a trend for more CD43+ cases in the MP and UC groups.
Table 2.
Immunohistochemistry (% positive)
| Number of Patients (BP/MP/UC) | Biphasic | Monophasic | Unclassifiable | |
|---|---|---|---|---|
| CD5 | 16/14/15 | 6 | 7 | 13 |
| CD23 | 15/14/16 | 0 | 14 | 13 |
| CD10 | 15/14/16 | 0 | 0 | 0 |
| CD43 | 16/14/12 | 0 | 21^ | 25^^ |
| Bcl-2 | 16/14/14 | 100 | 100 | 100 |
| Bcl-6 | 16/14/16 | 0 | 7 | 0 |
| IRF4/MUM1 | 15/14/16 | 27 | 29 | 31 |
| IgD | 15/14/14 | 93 | 64# | 57* |
| Cyclin D1 | 16/14/16 | 0 | 0 | 0 |
| CD5(−), CD10(−), CD43(−), CD23(−) | 15/14/13 | 94 | 64# | 69 |
| IgD(+), CD5(−), CD10(−), CD43(−), CD23(−) | 15/14/13 | 87 | 29** | 23** |
p < 0.04 vs. BP
p < 0.003 vs. BP
p = 0.08 vs. BP
p = 0.09 vs. BP
p = 0.07 vs BP
BP-biphasic, MP-monophasic, UC-unclassifiable
All but two BP cases had a typical SMZL phenotype (IgD+, CD5−, CD23−, CD10−, CD43−) with the exceptions either CD5+ (1 case) or IgD− (1 case). In contrast, only 29% and 23% of the MP and UC cases respectively had a typical SMZL phenotype (p < 0.003 vs. BP). All but 1 of the MP cases with an atypical phenotype differed by just 1 marker. The most frequent aberrant marker was IgD (5 cases); 2 cases were CD43+; 1 CD5+; and 1 CD23+. The one MP case with 2 phenotypic aberrancies was CD23+, CD43+ but staining for both was weak. In the UC group, 10 cases had an atypical SMZL phenotype including 7 with only 1 aberrancy (6 IgD− and 1 CD43+). Three cases had at least 2 aberrancies; one CD5+, CD43+ but CD23− and the other 2 CD5+, CD23+ (1 CD5+ by flow cytometry, both dim CD20+). One CD5+, CD23+ case with intermediate intensity κ staining by flow cytometry appeared to closely resemble SMZL (case 16, lymphocyte count 2.0×109/L, no smear available to review). The other had a diffuse growth pattern, plasmacytic differentiation, lacked proliferation centers, had bright λ staining by flow cytometry and an absolute lymphocyte count of only 1.2×109/L (case 3). A peripheral blood smear from 2 years prior to the splenectomy showed occasional lymphocytes with possible short villous projections. Three cases were p53+ by immunohistochemistry (one BP and two UC). The BP case had del(17)(p13) by classical cytogenetics. One UC case had cytogenetic abnormalities not involving 17p13. The third case lacked available cytogenetic data.
Cytogenetic studies
17/20 cases (85%) with classical cytogenetic studies had an abnormal karyotype. The normal cases included one UC case with only two metaphases and with one cell having a −Y (Table 3). Six cases had a single cytogenetic abnormality, 5 two abnormalities and 6 ≥3 abnormalities. Only 2 cases, both BP, had loss of the long arm of chromosome 7 (1 had a del(7) (q21q31) and 1 had monosomy 7). Four cases showed +3, four +12 and one partial +12q, three +X, two +5, and two del(6q). Chromosome abnormalities involving 17p13 were seen in 5 cases (4 BP). One case had add(14)(q32) with an IGH/BCL3 translocation by FISH.(15)
Table 3.
Cytogenetic Findings
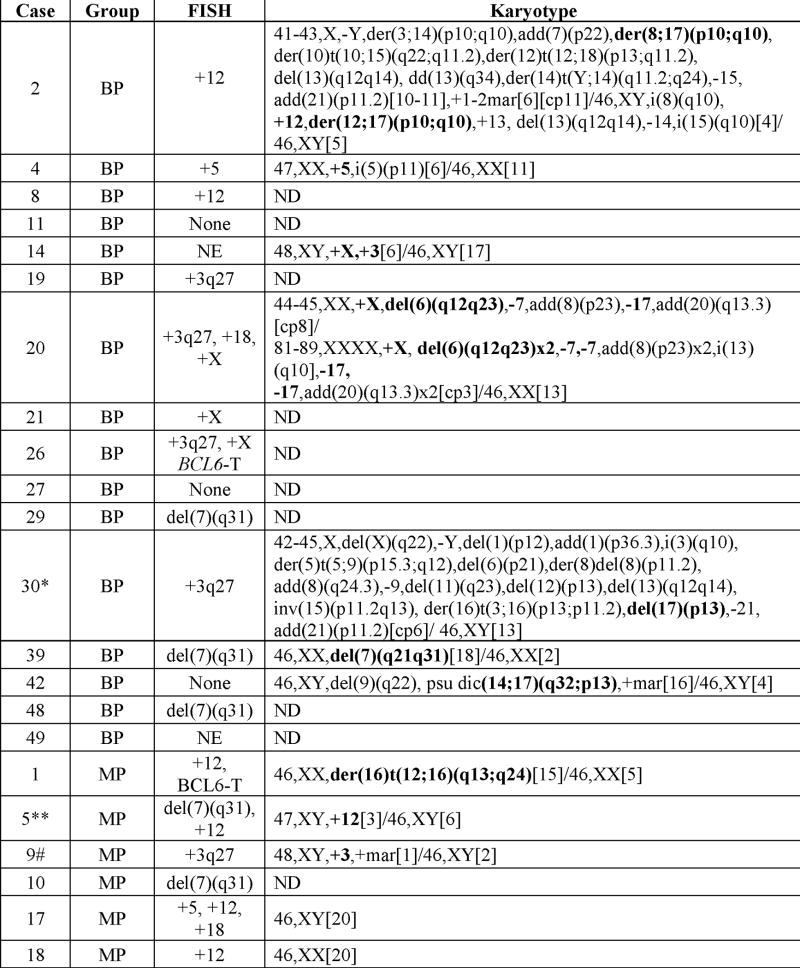 |
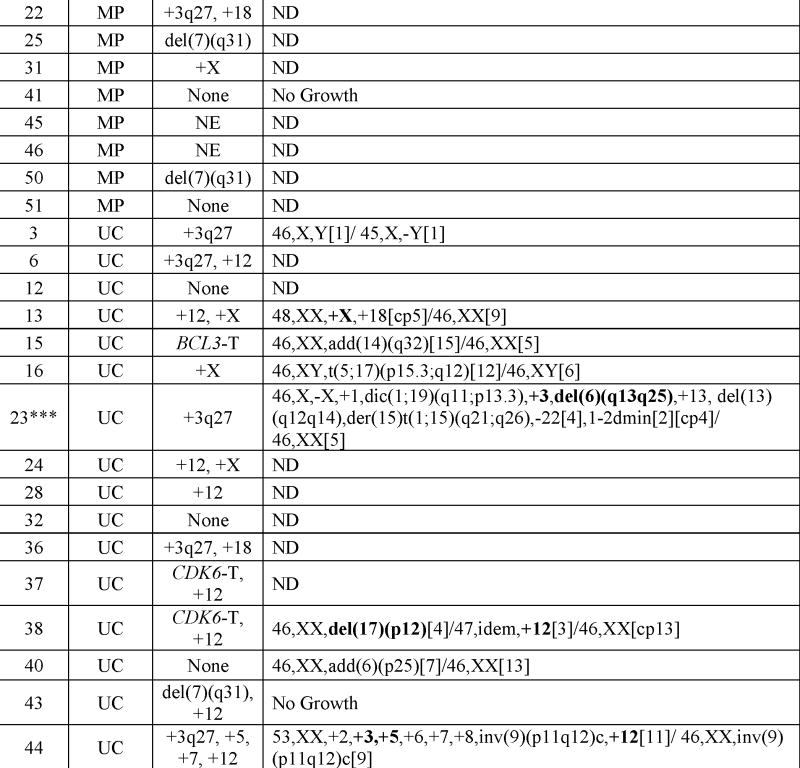 |
Except where indicated, karyotypes were performed on splenectomy specimens. The recurrent abnormalities (or related recurrent abnormalities) are in bold.
T = Translocation; NE = Not evaluable; ND = Not Done
Karyotype obtained from involved bone marrow.
Karyotype obtained from involved lymph node.
Obtained from involved peripheral blood.
This case was included among the cases with +3 even though the abnormality was found in one cell in this very limited study. The resolution of the chromosomes in this case precluded identification of the second additional chromosome present in the single abnormal cell.
34/42 cases (81%) showed FISH abnormalities with a similar proportion in the 3 groups (79–83%) (Table 4). Of the cases without demonstrable abnormalities, four had all probes successfully studied, two were missing just BCL6 and two had 3–4 missing probes (one case that was negative for del(7)(q31) but lacked FISH data for the other probes was not included in the denominator). The most frequent FISH abnormalities among the SMZL cases (including both BP and MP cases) were del(7)(q31) (26%) (Fig. 3), +12 (25%) and +3q27 (27%) and among the UC cases, +12 (50%) and +3q27 (36%) without significant differences between any of the three groups for any of the abnormalities. Only one UC case demonstrated del(7)(q31), however, this case also had an extra normal copy of chromosome 7. CDK6 (2 cases, both with +12) and BCL3 (1 case) translocations were only seen in the UC group. 5/6 UC cases composed entirely of small lymphocytes without MZ-like cells and having both WP and RP involvement had +12 compared to 9/34 of all remaining cases (p<0.02). The lymphocyte counts in the 6 cases were 0.72, 1.32, 4.68, 44.5, 64.7×109/L and unknown in one (no smears available to review). None of the FISH abnormalities correlated with any of the immunophenotypic findings (Table 4).
Table 4.
FISH (% positive)
| Histologic Group |
Immunohistochemistry (% Positive) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Abnormality | Number of Patients (BP/MP/UC) | Biphasic | Monophasic | Unclassifiable | CD5 | CD23 | CD43 | IRF4/MUM1 | IgD | Typical SMZL Phenotype* |
| del(7)(q31) (n=8) | 15/12/16 | 20 | 33 | 6# | 13 | 13 | 25 | 38 | 88 | 50 |
| +18 (n=5) | 14/12/16 | 7 | 17 | 13 | 0 | 20 | 0 | 60 | 60 | 40 |
| +12 (n=14) | 12/12/16 | 17 | 33 | 50 | 7 | 0 | 21 | 29 | 62 | 38 |
| +5 (n=3) | 13/10/15 | 8 | 10 | 7 | 0 | 0 | 0 | 0 | 33 | 33 |
| +X (n=7) | 13/11/16 | 23 | 9 | 19 | 29 | 14 | 14 | 29 | 67 | 33 |
| +3q27 (n=11) | 13/9/14 | 31 | 22 | 36 | 20 | 18 | 10 | 45 | 91 | 60 |
| CDK6-R (n=2) | 11/10/15 | 0 | 0 | 13 | 0 | 0 | 50 | 0 | 50 | 0 |
| BCL3-R (n=1) | 12/10/15 | 0 | 0 | 7 | 0 | 0 | NE | 0 | 0 | NE |
| BCL2-R (n=0) | 11/10/15 | 0 | 0 | 0 | - | - | - | - | - | - |
| MALT1-R (n=0) | 14/11/16 | 0 | 0 | 0 | - | - | - | - | - | - |
| BCL6-R (n=2) | 13/9/14 | 8 | 11 | 0 | 50 | 0 | 0 | 50 | 100 | 50 |
BP = biphasic, MP = monophasic, UC = unclassifiable
R = Rearrangement
This represented one case that had three signals for the centromere of chromosome 7 and two signals for 7q31.
Typical SMZL phenotype considered to be CD5−, CD23−, CD10−, CD43−, and IgD+.
NE = Not evaluable
The following cutoffs were used for FISH positivity: 15% for del(7)(q31), BCL6-R, +3(q27), BCL3-R, +18, +12, +5, and +X; 20% for CDK6-R and MALT1-R; 30% for BCL2-R.
Figure 3.

FISH analysis for del(7)(q31.2) A. Representative case without del(7)(q31.2) showing two signals for CEP7 (green) and two signals for D7S486 (red). B. Representative case with del(7)(q31.2) showing two signals for CEP7 (green), but only one signal for D7S486 (red).
Clinical and Laboratory Characteristics
Bone marrow involvement by lymphoma was documented in 13/14 BP, 12/12 MP, and 7/11 UC cases (BP/MP vs UC, p<0.03)(Table 5). The mean absolute lymphocyte count was higher in the UC group due to 3 cases with a count >44 × 109/L (cases 23, 28 and 43). However, the proportion of cases in each group with a lymphocytosis did not show significant differences. Circulating villous lymphocytes were seen in 2/8 BP and 2/6 MP cases. One UC case lacked villous lymphocytes (case 13) and the other had occasional possible villous lymphocytes (case 3). MP cases were more likely to have adenopathy than BP cases, although few SMZL cases had any peripheral adenopathy. Overall survival was similar for each of the groups (Fig. 4).
Table 5.
Clinical and Laboratory Characteristics
| Number of Patients (BP/MP/UC) | Biphasic | Monophasic | Unclassifiable | |
|---|---|---|---|---|
| Age, years (mean ± sd) | 17/14/16 | 60.9 ± 12.5 | 68.8 ± 10.8 | 66.6 ± 7.8 |
| Gender | 17/14/16 | 12F/5M | 6F/8M | 9F/7M |
| Adenopathy‡ | 15/12/11 | 40 | 83* | 64 |
| Central & Peripheral (%) | 13 | 8 | 36 | |
| Central only (%) | 27 | 75 | 27 | |
| None (%) | 60 | 17 | 36 | |
| Fever (%) | 6/8/7 | 50 | 50 | 14 |
| Weight loss (%) | 6/8/6 | 33.3 | 75 | 0 |
| Stage at diagnosis (Number of patients) | ||||
| Stage 1 | 1 | 0 | 1 | |
| Stage 2 | 0 | 0 | 0 | |
| Stage 3 | 0 | 0 | 3 | |
| Stage 4 | 12 | 12 | 6 | |
| Marrow involvement (% positive) | 14/12/11 | 93 | 100 | 64# |
| CBC data (mean ± sd) | ||||
| Hemoglobin | 15/13/12 | 9.2 ± 2.7 | 9.3 ± 2.4 | 9.5 ± 2.6 |
| WBC (× 109/L) | 15/13/12 | 13.4 ± 10.7 | 10.5 ± 13.1 | 26.4 ± 40.2 |
| % Lymphocytes | 12/12/11 | 20.4 ± 13.2 | 26.8 ± 12.9 | 39.1 ± 26^ |
| Absolute Lymphocyte Count (× 109/L) | 12/12/11 | 2.8 ± 3.2 | 1.7 ± 1.1 | 18.1 ± 29.5@ |
| Platelets (× 109/L) | 14/13/12 | 156 ± 113 | 137 ± 78 | 141 ± 101 |
| Treatment (No. of cases) | ||||
| Splenectomy alone | 1 | 4 | 3 | |
| Rituximab (R) only | 1 | 3 | 3 | |
| Anthracycline based chemotherapy | 0 | 0 | 2 | |
| Anthracycline based chemotherapy + R | 4 | 1 | 0 | |
| Non-anthracycline chemotherapy | 2 | 0 | 0 | |
| Non-anthracycline chemotherapy + R | 1 | 2 | 2 | |
| Unknown | 8 | 4 | 6 |
BP-biphasic, MP-monophasic, UC-unclassifiable
Central lymphadenopathy is defined as adenopathy limited to the abdomen, retroperitoneum and/or mediastinum (data included only for patients who had CT and/or PET). No cases had lymphadenopathy limited to peripheral sites.
p<0.02 vs. biphasic
p<0.03 vs. biphasic and monophasic groups pooled
p<0.03 vs. biphasic and monophasic groups pooled, p<0.05 vs. biphasic
p<0.02 vs. biphasic and monophasic groups pooled
Figure 4.

Overall survival curves for biphasic, monophasic and unclassifiable groups (p=not significant).
DISCUSSION
Defining the borders of SMZL is complex.(18–21) The 2001 WHO classification defined SMZL as a CD5, CD10, CD43, and CD23-negative, but IgD+ small B-cell neoplasm that involves the RP and WP forming BP WP nodules with a central zone of small lymphocytes with or without a central germinal center surrounded by a peripheral zone of larger MZ-like cells.(1) The 2008 WHO classification expanded the definition to include previously described cases that consist entirely of MZ-like cells (2) and to allow for more variation in the immunophenotype.(3) Some splenic lymphomas have morphologic and/or phenotypic features that do not fit even this expanded definition of SMZL. Because of this, the 2008 WHO classification includes a new category, splenic B-cell lymphoma/leukemia, unclassifiable.(13) Some of these UC splenic lymphomas are included in studies of “SMZL,” leading some to emphasize the “considerable histologic, immunohistochemical, and molecular heterogeneity of SMZL,” but also causing confusion.(6) Additional confusion arises from the belief by some that SMZL are not actually neoplasms of splenic MZ cells.(3, 23) These authors emphasize the dissimilarities between SMZL cells and normal splenic MZ cells (23); however, others stress their similarities including their similar ultrastructural features.(2, 6, 24)
Approximately two-thirds of the splenic small B-cell lymphomas reported here fulfill the 2008 WHO criteria for SMZL, although almost half of these cases had a MP pattern, rather that the classic BP pattern required by the 2001 WHO criteria. The MP cases also had predominantly MZ-like cells in the involved areas of the lymph nodes examined. In spite of these morphological differences, the recent inclusion of the MP cases as SMZL is supported by their similarity to the BP cases in terms of their other clinicopathologic features. In addition, some cases also had infrequent BP white pulp nodules. Near universal involvement of bone marrow, in both the BP and MP, but not the UC cases, is another characteristic, although nonspecific feature of SMZL.(25) In contrast to the BP cases, however, the MP cases were more likely to have adenopathy (usually not peripheral) and an immunophenotype atypical for SMZL. The most common phenotypic aberrancy was IgD-negativity followed by CD43-positivity. Other phenotypic aberrancies, also reported by others, include infrequent CD5 and CD23 expression. (19, 26) All but one of the BP cases were IgD+ and all were CD43− and CD23−. Another recent study of SMZL also found a higher proportion of IgD+ cases among the BP rather than MP cases with MZ cells.(6).
These findings suggest that, although the BP and MP cases have many similarities, the lack of a significant non-MZ-like component in the MP cases may have meaningful biologic correlates. These cases might be more of a true MZ lymphoma, whereas the more classic BP cases may be a neoplasm of the essentially undefined small B-cells that lack the morphologic features of classical splenic MZ cells, but which either acquire a MZ morphologic appearance when present in the MZ (as seen with some other B-cell neoplasms)(21) or show actual MZ differentiation. Although most of the cases that predominantly involved the RP were composed of lymphocytes or plasmacytoid cells that did not closely resemble classic splenic MZ-type cells, one case included in the UC group was composed of MP MZ-type cells. This case did show WP involvement but it was not expanded. Further supporting that cases such as this are best considered SMZL, one of the early descriptions of SMZL in which the neoplastic cells were all considered to resemble MZ cells, reported 4/14 cases had diffuse splenic architectural effacement including one case that also had a small focus of nodular growth.(2)
IgD-negativity is also a frequent feature of SDRPSBL, or what has also been reported as “splenic red pulp lymphoma with numerous basophilic villous lymphocytes,” a B-cell neoplasm consisting of relatively small lymphocytes diffusely involving the splenic RP.(13, 14, 20) The cytology of the neoplastic cells has been variably described. Two cases included in the UC group had many features of this provisional entity although they lacked classic MZ-like cells and one had a phenotype most like CLL. The latter case, however lacked WP nodules and the sometimes subtle proliferation centers usually seen with CLL/SLL in the spleen(27). The patient did not have a lymphocytosis and did have some circulating lymphocytes with possible short villous projections. It may be that SDRPSBL includes some cases that are most like a diffuse proliferation of the MZ-component of SMZL(2) and others that are more like a diffuse red pulp variant of the small lymphocytic cases described below with frequent trisomy 12. Unfortunately, rather limited data were available regarding the detailed peripheral blood and bone marrow findings in many of the patients included in this study. These two components are important in the ultimate categorization of some of the splenic small B-cell lymphomas.
The reported incidence of plasmacytic differentiation in SMZL ranges from 21 to 74%.(2, 19, 28) Compared to non-plasmacytic SMZL, those with plasmacytic differentiation have been reported to demonstrate more frequent monoclonal paraproteins and autoimmune hemolytic anemia but otherwise to be clinically similar.(28) In our series, plasmacytic differentiation was present in 41% of cases with an approximately equal distribution among the three groups. In some cases there was a prominent, although not usually exclusive, accumulation of light chain restricted plasma cells within the WP nodules, a characteristic but not specific pattern for SMZL. (6, 19) One of the UC cases probably represents a lymphoplasmacytic lymphoma (LPL) which, when it involves the spleen, grows diffusely or forms small RP nodules.(29) However, because LPL remains a diagnosis of exclusion, the case was retained in this study.
Cytogenetic abnormalities were found in slightly > 80% of all cases studied, similar to the results of others (20, 30) and without clear-cut differences between the 3 groups of cases. Cytogenetic abnormalities have been reported to be more common in SMZL than in splenic red pulp lymphoma with numerous basophilic villous lymphocytes.(20) Although no significant differences were documented between the three groups of patients, based on the FISH studies all but one of the cases with del(7)(q31) were in one of the two SMZL groups with the one UC case being the difficult to interpret case that also had an extra copy of a normal chromosome 7. The case with −7 by classical cytogenetics was also a SMZL. These findings are consistent with del(7)(q31) being relatively specific for SMZL.(4, 10, 11) Overall del(7)(q31) was present in 26% of SMZL cases in the current study which is comparable to the 16–36% reported in previous FISH studies.(11, 30) A study that evaluated loss of heterozygosity reported allelic loss on 7q in 40% of SMZL. (9) Although sometimes present with other trisomies, del(7)(q31) was never found with +3q27, consistent with previous reports that these are usually, but not always, mutually exclusive abnormalities.(4, 30) A similar proportion of SMZL cases (27%) demonstrated +3q27, implying probable partial or complete trisomy 3. This is also consistent with the most commonly reported frequencies of +3 in SMZL (approximately 10–36%)(10, 11), although much lower (4%) and higher (55%) proportions have been reported.(31, 32)
Trisomy 12 was seen 25% of SMZL in the current study by FISH including both BP and MP cases. This is somewhat more frequent than seen in previous FISH studies of SMZL where it has been reported in 6–18% of cases (12, 30) but is comparable to the percentage of cases with gains of 12q by comparative genomic hybridization (CGH).(33) Trisomy 12 was also present in 50% of the UC cases in the current study, including all but one of the 6 UC cases with RP and WP involvement by exclusively small non-MZ type lymphocytes. This is significantly more frequent than was found in the other UC cases or all other cases overall. One of these cases also had a CDK6 rearrangement. Although often used to support the diagnosis of atypical CLL, +12 is not a specific finding, even being found in some classic SMZL. These 6 cases were all CD5 and CD23 negative with very variable lymphocyte counts. This observation lends support to the need at the current time for the category of UC splenic B-cell lymphomas, since at least this subset of non-MZ-like cases are clearly distinctive from any type of SMZL.
The UC category also included a heterogenous group of other cases including a small subset of cases that may be best defined by their cytogenetic abnormalities. One previously reported case included in this series had a micronodular proliferation of small lymphocytes in the RP, atrophic white pulp, a phenotype most like SDRPSBL, and an IGH/BCL3 translocation.(15) The IGH/BCL3 translocation is a rare recurrent cytogenetic abnormality seen in a variety of B-cell neoplasms, often described as CLL/SLL with an atypical morphology and/or phenotype.(34) Rearrangements of CDK6, as found here in 2 cases, have also been found in a minority of small B-cell lymphomas including “SMZL.”(16) Although cases with CDK6 rearrangements have been reported to be CD5+ and to be associated with a marked lymphocytosis and generalized lymphadenopathy (16), the two cases in this series were CD5−, adenopathy was absent in at least one of the patients and the lymphocyte count, known only in one patient, was 4.68×109/L.
An extra X chromosome was found in 18% of the SMZL. Data on +X in SMZL is limited, however one study demonstrated gains in X in 30% of cases by CGH.(4) Other less frequent FISH abnormalities seen in our SMZL cases and in previously reported series include +18, +5, and rearrangement of BCL6.(4, 11, 30) Trisomy 18 and +5 were also seen in a small number of UC cases but not BCL6 rearrangement. Five cases, 4 of which were in the BP group, had abnormalities involving the chromosomal band 17p13. Two additional UC cases showed strong p53 protein positivity by IHC. While not specific for SMZL, abnormalities in the TP53 gene located at this site have been associated with a poor prognosis in SMZL.(35)
In summary, this study illustrates the heterogeneity of splenic small B-cell lymphomas. The findings support the current inclusion of splenic B-cell lymphomas with a MP pattern and predominantly or exclusively MZ-like cells as a morphologic variant of SMZL; however, they suggest that these cases may have some underlying biologic differences. They may be closer to a lymphoma truly derived from splenic MZ cells and may rarely predominantly or exclusively involve the RP. There are also a significant proportion of unclassifiable splenic small B-cell lymphomas that show overlapping features with the SMZL and a similar indolent course. The relationship of SMZL these to other splenic-based, not otherwise categorizable, small B-cell lymphomas that lack a morphologically identifiable MZ component and which have a pure RP distribution or involve both the RP and WP, remains unclear, and argues for retention at the current time of the WHO category, splenic B-cell lymphoma/leukemia, unclassifiable. Some may be related to the non-MZ component of otherwise classical SMZL. Finally, some of the unclassifiable cases may belong to potential entities perhaps best defined by their cytogenetic abnormalities, such as BCL3 or CDK6 translocations.
Acknowledgments
The authors thank Mr. Dale Lewis for his contribution to the FISH analysis and for reviewing the related methodology section of this manuscript. Some of the cytogenetic FISH studies were carried out in the University of Pittsburgh Cancer Institute Cytogenetics Facility, supported in part by NIH Cancer Center Support Grant P30CA047904 to Ronald B. Herberman/SMG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Isaacson PGPM, Catovsky D, Swerdlow S, Montserrat E, Berger F, Muller-Hermelink HK, Nathwani B, Harris NL. Splenic marginal zone lymphoma. In: Jaffe ESHN, Stein H, Vardiman JW, editors. Pathology and Genetics of Tumors of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2001. pp. 135–137. [Google Scholar]
- 2.Hammer RD, Glick AD, Greer JP, Collins RD, Cousar JB. Splenic marginal zone lymphoma. A distinct B-cell neoplasm. Am J Surg Pathol. 1996;20:613–626. doi: 10.1097/00000478-199605000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Isaacson PGPM, Berger F, Swerdlow SH, Thieblemont C, Pittaluga S, Harris NL. Splenic Marginal Zone Lymphoma. In: Swerdlow SHCE, Harris NL, Jaffe ES, Pileri SA, Stein H, Jurgen T, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008. pp. 185–187. [Google Scholar]
- 4.Boonstra R, Bosga-Bouwer A, van Imhoff GW, Krause V, Palmer M, Coupland RW, Dabbagh L, van den Berg E, van den Berg A, Poppema S. Splenic marginal zone lymphomas presenting with splenomegaly and typical immunophenotype are characterized by allelic loss in 7q31-32. Mod Pathol. 2003;16:1210–1217. doi: 10.1097/01.MP.0000095895.19756.77. [DOI] [PubMed] [Google Scholar]
- 5.Matutes E, Oscier D, Montalban C, Berger F, Callet-Bauchu E, Dogan A, Felman P, Franco V, Iannitto E, Mollejo M, Papadaki T, Remstein ED, Salar A, Sole F, Stamatopoulos K, Thieblemont C, Traverse-Glehen A, Wotherspoon A, Coiffier B, Piris MA. Splenic marginal zone lymphoma proposals for a revision of diagnostic, staging and therapeutic criteria. Leukemia. 2008;22:487–495. doi: 10.1038/sj.leu.2405068. [DOI] [PubMed] [Google Scholar]
- 6.Papadaki T, Stamatopoulos K, Belessi C, Pouliou E, Parasi A, Douka V, Laoutaris N, Fassas A, Anagnostopoulos A, Anagnostou D. Splenic marginal-zone lymphoma: one or more entities? A histologic, immunohistochemical, and molecular study of 42 cases. Am J Surg Pathol. 2007;31:438–446. doi: 10.1097/01.pas.0000213419.08009.b0. [DOI] [PubMed] [Google Scholar]
- 7.Callet-Bauchu E, Baseggio L, Felman P, Traverse-Glehen A, Berger F, Morel D, Gazzo S, Poncet C, Thieblemont C, Coiffier B, Magaud JP, Salles G. Cytogenetic analysis delineates a spectrum of chromosomal changes that can distinguish non-MALT marginal zone B-cell lymphomas among mature B-cell entities: a description of 103 cases. Leukemia. 2005;19:1818–1823. doi: 10.1038/sj.leu.2403909. [DOI] [PubMed] [Google Scholar]
- 8.Corcoran MM, Mould SJ, Orchard JA, Ibbotson RE, Chapman RM, Boright AP, Platt C, Tsui LC, Scherer SW, Oscier DG. Dysregulation of cyclin dependent kinase 6 expression in splenic marginal zone lymphoma through chromosome 7q translocations. Oncogene. 1999;18:6271–6277. doi: 10.1038/sj.onc.1203033. [DOI] [PubMed] [Google Scholar]
- 9.Mateo M, Mollejo M, Villuendas R, Algara P, Sanchez-Beato M, Martinez P, Piris MA. 7q31-32 allelic loss is a frequent finding in splenic marginal zone lymphoma. Am J Pathol. 1999;154:1583–1589. doi: 10.1016/S0002-9440(10)65411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sole F, Woessner S, Florensa L, Espinet B, Mollejo M, Martin P, Piris MA. Frequent involvement of chromosomes 1, 3, 7 and 8 in splenic marginal zone B-cell lymphoma. British Journal of Haematology. 1997;98:446–449. doi: 10.1046/j.1365-2141.1997.2163033.x. [DOI] [PubMed] [Google Scholar]
- 11.Remstein ED, Law M, Mollejo M, Piris MA, Kurtin PJ, Dogan A. The prevalence of IG translocations and 7q32 deletions in splenic marginal zone lymphoma. Leukemia. 2008;22:1268–1272. doi: 10.1038/sj.leu.2405027. [DOI] [PubMed] [Google Scholar]
- 12.Wotherspoon AC, Finn TM, Isaacson PG. Trisomy 3 in low-grade B-cell lymphomas of mucosa-associated lymphoid tissue. Blood. 1995;85:2000–2004. [PubMed] [Google Scholar]
- 13.Piris M, Foucar K, Mollejo M, Campo E, Falini B. Splenic B-cell lymphoma/leukaemia, unclassifiable. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008. pp. 191–193. [Google Scholar]
- 14.Mollejo M, Algara P, Mateo MS, Sanchez-Beato M, Lloret E, Medina MT, Piris MA. Splenic small B-cell lymphoma with predominant red pulp involvement: a diffuse variant of splenic marginal zone lymphoma? Histopathology. 2002;40:22–30. doi: 10.1046/j.1365-2559.2002.01314.x. [DOI] [PubMed] [Google Scholar]
- 15.Soma LA, Gollin SM, Remstein ED, Ketterling RP, Flynn HC, Rajasenan KK, Swerdlow SH. Splenic small B-cell lymphoma with IGH/BCL3 translocation. Human Pathology. 2006;37:218–230. doi: 10.1016/j.humpath.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 16.Chen D, Law ME, Theis JD, Gamez JD, Caron LB, Vrana JA, Dogan A. Clinicopathologic features of CDK6 translocation-associated B-cell lymphoproliferative disorders. Am J Surg Pathol. 2009;33:720–729. doi: 10.1097/PAS.0b013e3181934244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherer ME, Shekhter-Levin S, Krause JR, Joyce RA, Gollin SM. Atypical (7;19) translocation in acute myelomonocytic leukemia. Cancer Genet Cytogenet. 1991;57:169–173. doi: 10.1016/0165-4608(91)90148-n. [DOI] [PubMed] [Google Scholar]
- 18.Isaacson PG, Matutes E, Burke M, Catovsky D. The histopathology of splenic lymphoma with villous lymphocytes. Blood. 1994;84:3828–3834. [PubMed] [Google Scholar]
- 19.Mollejo M, Menarguez J, Lloret E, Sanchez A, Campo E, Algara P, Cristobal E, Sanchez E, Piris MA. Splenic marginal zone lymphoma: a distinctive type of low-grade B-cell lymphoma. A clinicopathological study of 13 cases. Am J Surg Pathol. 1995;19:1146–1157. [PubMed] [Google Scholar]
- 20.Traverse-Glehen A, Baseggio L, Bauchu EC, Morel D, Gazzo S, Ffrench M, Verney A, Rolland D, Thieblemont C, Magaud JP, Salles G, Coiffier B, Berger F, Felman P. Splenic red pulp lymphoma with numerous basophilic villous lymphocytes: a distinct clinicopathologic and molecular entity? Blood. 2008;111:2253–2260. doi: 10.1182/blood-2007-07-098848. [DOI] [PubMed] [Google Scholar]
- 21.Piris MA, Mollejo M, Campo E, Menarguez J, Flores T, Isaacson PG. A marginal zone pattern may be found in different varieties of non-Hodgkin’s lymphoma: the morphology and immunohistology of splenic involvement by B-cell lymphomas simulating splenic marginal zone lymphoma. Histopathology. 1998;33:230–239. doi: 10.1046/j.1365-2559.1998.00478.x. [DOI] [PubMed] [Google Scholar]
- 22.Huh YO, Abruzzo LV, Rassidakis GZ, Parry-Jones N, Schlette E, Brito-Bapabulle V, Matutes E, Wotherspoon A, Keating MJ, Medeiros LJ, Catovsky D. The t(14;19) (q32;q13)-positive small B-cell leukaemia: a clinicopathologic and cytogenetic study of seven cases. British Journal of Haematology. 2007;136:220–228. doi: 10.1111/j.1365-2141.2006.06416.x. [DOI] [PubMed] [Google Scholar]
- 23.Isaacson PG. Splenic marginal zone lymphoma. Blood. 1996;88:751–752. [PubMed] [Google Scholar]
- 24.Steiniger B, Timphus EM, Barth PJ. The splenic marginal zone in humans and rodents: an enigmatic compartment and its inhabitants. Histochem Cell Biol. 2006;126:641–648. doi: 10.1007/s00418-006-0210-5. [DOI] [PubMed] [Google Scholar]
- 25.Audouin J, Le Tourneau A, Molina T, Camilleri-Broet S, Adida C, Comperat E, Benattar L, Delmer A, Devidas A, Rio B, Diebold J. Patterns of bone marrow involvement in 58 patients presenting primary splenic marginal zone lymphoma with or without circulating villous lymphocytes. British Journal of Haematology. 2003;122:404–412. doi: 10.1046/j.1365-2141.2003.04449.x. [DOI] [PubMed] [Google Scholar]
- 26.Matutes E, Morilla R, Owusu-Ankomah K, Houlihan A, Catovsky D. The immunophenotype of splenic lymphoma with villous lymphocytes and its relevance to the differential diagnosis with other B-cell disorders. Blood. 1994;83:1558–1562. [PubMed] [Google Scholar]
- 27.Swerdlow SH, Murray LJ, Habeshaw JA, Stansfeld AG. Lymphocytic lymphoma/B-chronic lymphocytic leukaemia--an immunohistopathological study of peripheral B lymphocyte neoplasia. Br J Cancer. 1984;50:587–599. doi: 10.1038/bjc.1984.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Huyen JP, Molina T, Delmer A, Audouin J, Le Tourneau A, Zittoun R, Bernadou A, Diebold J. Splenic marginal zone lymphoma with or without plasmacytic differentiation. Am J Surg Pathol. 2000;24:1581–1592. doi: 10.1097/00000478-200012000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Swerdlow SH, Berger F, Pileri SA, Harris NL, Jaffe ES, Stein H. Lymphoplasmacytic lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. pp. 194–195. [Google Scholar]
- 30.Sole F, Salido M, Espinet B, Garcia JL, Martinez Climent JA, Granada I, Hernandez JM, Benet I, Piris MA, Mollejo M, Martinez P, Vallespi T, Domingo A, Serrano S, Woessner S, Florensa L. Splenic marginal zone B-cell lymphomas: two cytogenetic subtypes, one with gain of 3q and the other with loss of 7q. Haematologica. 2001;86:71–77. [PubMed] [Google Scholar]
- 31.Dierlamm J, Rosenberg C, Stul M, Pittaluga S, Wlodarska I, Michaux L, Dehaen M, Verhoef G, Thomas J, de Kelver W, Bakker-Schut T, Cassiman JJ, Raap AK, De Wolf-Peeters C, Van den Berghe H, Hagemeijer A. Characteristic pattern of chromosomal gains and losses in marginal zone B cell lymphoma detected by comparative genomic hybridization. Leukemia. 1997;11:747–758. doi: 10.1038/sj.leu.2400635. [DOI] [PubMed] [Google Scholar]
- 32.Oscier DG, Matutes E, Gardiner A, Glide S, Mould S, Brito-Babapulle V, Ellis J, Catovsky D. Cytogenetic studies in splenic lymphoma with villous lymphocytes. British Journal of Haematology. 1993;85:487–491. doi: 10.1111/j.1365-2141.1993.tb03337.x. [DOI] [PubMed] [Google Scholar]
- 33.Hernandez JM, Garcia JL, Gutierrez NC, Mollejo M, Martinez-Climent JA, Flores T, Gonzalez MB, Piris MA, San Miguel JF. Novel genomic imbalances in B-cell splenic marginal zone lymphomas revealed by comparative genomic hybridization and cytogenetics. Am J Pathol. 2001;158:1843–1850. doi: 10.1016/S0002-9440(10)64140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin-Subero JI, Ibbotson R, Klapper W, Michaux L, Callet-Bauchu E, Berger F, Calasanz MJ, De Wolf-Peeters C, Dyer MJ, Felman P, Gardiner A, Gascoyne RD, Gesk S, Harder L, Horsman DE, Kneba M, Kuppers R, Majid A, Parry-Jones N, Ritgen M, Salido M, Sole F, Thiel G, Wacker HH, Oscier D, Wlodarska I, Siebert R. A comprehensive genetic and histopathologic analysis identifies two subgroups of B-cell malignancies carrying a t(14;19)(q32;q13) or variant BCL3-translocation. Leukemia. 2007;21:1532–1544. doi: 10.1038/sj.leu.2404695. [DOI] [PubMed] [Google Scholar]
- 35.Chacon JI, Mollejo M, Munoz E, Algara P, Mateo M, Lopez L, Andrade J, Carbonero IG, Martinez B, Piris MA, Cruz MA. Splenic marginal zone lymphoma: clinical characteristics and prognostic factors in a series of 60 patients. Blood. 2002;100:1648–1654. [PubMed] [Google Scholar]